

This is the first study to show the direct effects of α-synuclein on synaptic vesicle trafficking and to elucidate the underlying structural mechanisms. Acutely increasing α-synuclein severely inhibits synaptic vesicle recycling from the plasma membrane. The endocytic defects require a properly folded N-terminal α-helical domain of α-synuclein.
Abstract
Parkinson's disease is associated with multiplication of the α-synuclein gene and abnormal accumulation of the protein. In animal models, α-synuclein overexpression broadly impairs synaptic vesicle trafficking. However, the exact steps of the vesicle trafficking pathway affected by excess α-synuclein and the underlying molecular mechanisms remain unknown. Therefore we acutely increased synuclein levels at a vertebrate synapse and performed a detailed ultrastructural analysis of the effects on presynaptic membranes. At stimulated synapses (20 Hz), excess synuclein caused a loss of synaptic vesicles and an expansion of the plasma membrane, indicating an impairment of vesicle recycling. The N-terminal domain (NTD) of synuclein, which folds into an α-helix, was sufficient to reproduce these effects. In contrast, α-synuclein mutants with a disrupted N-terminal α-helix (T6K and A30P) had little effect under identical conditions. Further supporting this model, another α-synuclein mutant (A53T) with a properly folded NTD phenocopied the synaptic vesicle recycling defects observed with wild type. Interestingly, the vesicle recycling defects were not observed when the stimulation frequency was reduced (5 Hz). Thus excess α-synuclein impairs synaptic vesicle recycling evoked during intense stimulation via a mechanism that requires a properly folded N-terminal α-helix.
INTRODUCTION
Parkinson's disease (PD) is an age-related movement disorder associated with the aberrant aggregation of α-synuclein and formation of Lewy bodies (Spillantini et al., 1998; Lee and Trojanowski, 2006). Familial PD is linked to α-synuclein overexpression, caused by multiplication of the α-synuclein gene (SNCA), and to several missense mutations, including A30P and A53T (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003; Zarranz et al., 2004; Dawson et al., 2010). Sporadic PD is also associated with increased α-synuclein expression (Chiba-Falek et al., 2006; Dumitriu et al., 2012), as is dementia with Lewy bodies (DLB; Surguchov, 2008). Thus examining the function of α-synuclein and the consequences of its overexpression is a means for understanding the pathobiology underlying several neurological diseases.
α-Synuclein is a small protein (aa 140) found at neuronal synapses (Maroteaux et al., 1988; Murphy et al., 2000; Bendor et al., 2013). It is naturally disordered in an aqueous environment (Davidson et al., 1998; Eliezer et al., 2001). However, when presented with negatively charged acidic phospholipids in small vesicles, the N-terminal domain (NTD) of α-synuclein (aa 1–110) binds and folds into an amphipathic α-helix (Davidson et al., 1998; Perrin et al., 2000; Chandra et al., 2003; Ulmer et al., 2005). Immunostaining of the endogenous protein reveals that α-synuclein is abundant at synapses, where it colocalizes with presynaptic vesicle clusters (Maroteaux et al., 1988; Murphy et al., 2000).
Because α-synuclein is found at synapses under physiological conditions and because its overexpression is linked to several diseases, there is a growing interest in understanding how α-synuclein affects synaptic structure and function. In mouse models of PD and in DLB patients, a large fraction of α-synuclein aggregates occur in presynaptic boutons (Kramer and Schulz-Schaeffer, 2007; Scott et al., 2010; Spinelli et al., 2014). Furthermore, synaptic defects caused by the overexpression of α-synuclein can precede α-synuclein aggregation in somata and the formation of Lewy bodies, potentially implicating the synaptic impairments in early stages of disease pathogenesis (Scott et al., 2010; Volpicelli-Daley et al., 2011). Chronic overexpression of human α-synuclein broadly impairs synaptic vesicle trafficking and results in the down-regulation or loss of several other presynaptic proteins (Larsen et al., 2006; Nemani et al., 2010; Scott et al., 2010; Scott and Roy, 2012; Gaugler et al., 2012). These growing connections between α-synuclein and synaptic dysfunction indicate a need for gaining a deeper understanding of the underlying mechanisms.
Although it is has been shown that chronic overexpression of α-synuclein impairs synaptic vesicle trafficking (Nemani et al., 2010; Scott et al., 2010), there are several outstanding questions that warrant further investigation. First, the direct effects of excess α-synuclein on synapses remain unclear, because chronic overexpression decreased the expression levels of several other presynaptic proteins, including those that regulate synaptic vesicle exocytosis (e.g., VAMP and complexin), endocytosis (e.g., amphiphysin), and vesicle clustering and reclustering (e.g., synapsin) (Nemani et al., 2010; Scott et al., 2010). As a consequence, overexpression of α-synuclein may lead to compensatory phenotypes that mask its direct effects on vesicle trafficking or cause a complex phenotype that is only partially related to α-synuclein. Overcoming these issues requires that excess synuclein be delivered acutely to synapses and the effects examined after a short time and before compensatory changes take place. Second, the precise steps of the vesicle trafficking pathway affected by excess α-synuclein are unclear. After exocytosis and neurotransmitter release, synaptic vesicles are locally recycled by endocytosis from the plasma membrane (Schweizer and Ryan, 2006; Morgan et al., 2013a). One pathway for recycling vesicles occurs via clathrin-mediated endocytosis, which proceeds through several morphologically and molecularly distinct stages (Heuser and Reese, 1973; Slepnev and De Camilli, 2000; Saheki and De Camilli, 2012). At high levels of stimulation, synaptic vesicles can also be recycled via activity-dependent bulk endocytosis (Cousin, 2009; Morgan et al., 2013a). SynaptopHluorin assays revealed that α-synuclein overexpression reduced the recycling pool of synaptic vesicles, suggesting endocytic defects (Nemani et al., 2010). However, whether α-synuclein is affecting early or late endocytosis, clathrin-mediated or bulk endocytosis or both, remains an open question that requires detailed ultrastructural analyses of presynaptic membranes under controlled stimulation conditions. Finally, the structural mechanism by which excess α-synuclein impairs synapses is unclear. Which domain of synuclein is responsible for causing the phenotypes? Is it the disordered or α-helical conformation of the protein that causes deleterious effects at synapses? Answering these questions requires the ability to perform a structure–function analysis of multiple α-synuclein mutants. These types of acute perturbations, combined with ultrastructural and mutational analyses, have not yet been performed, due to limitations in most experimental models.
We therefore took advantage of a model vertebrate synapse, the lamprey giant reticulospinal (RS) synapse, which is uniquely suited for these experiments and meets all of the above criteria. Lamprey RS synapses allow for acute perturbations of presynaptic proteins, detailed high-resolution ultrastructural analyses of presynaptic membranes, and rapid investigation of mutants through axonal injection of recombinant proteins. Furthermore, over the past two decades, studies performed at the RS synapses have yielded many fundamental insights into the molecular mechanisms of synaptic vesicle trafficking (Pieribone et al., 1995; Shupliakov et al., 1997; Brodin and Shupliakov, 2006). In this paper, we report that excess α-synuclein and a PD-linked mutant, A53T, inhibit synaptic vesicle recycling from the plasma membrane during high-frequency stimulation, and we provide evidence for the underlying structural mechanisms.
RESULTS
Synucleins are highly conserved from lampreys to humans
Before beginning the synapse studies, we characterized the synucleins in sea lamprey, Petromyzon marinus. We cloned three synuclein isoforms from lamprey: two γ-synucleins and a third isoform that has not yet been assigned (Busch and Morgan, 2012). Lamprey giant RS neurons, which give rise to the synapses of interest, abundantly expressed only one of the γ-synuclein isoforms (GenBank: JN544525), while the other isoforms were expressed at very low levels (Busch and Morgan, 2012). Multiple sequence alignment reveals that full-length lamprey γ-synuclein shares 56, 54, and 55% identity at the amino acid level with human α-, β-, and γ-synucleins, respectively (Figure 1A). The first 90 amino acids at the N-termini of lamprey γ-synuclein and human α-synuclein share even greater homology at 67% identity and 90% similarity, as this is the most conserved region among all synucleins (Figure 1A; George, 2002). In contrast, the C-termini of all synucleins are notably less conserved, such that human β- and γ-synuclein share only 61 and 47% identity with α-synuclein. Thus lamprey γ-synuclein shares a degree of homology with human α-synuclein that is comparable to the homology between human synuclein orthologues.
FIGURE 1:

Lamprey γ-synuclein is structurally similar to human α-synuclein. (A) Multiple sequence alignment of human synucleins (α, β, γ) and lamprey γ-synuclein. Black highlighted residues are those that are identical to human α-synuclein. Note the higher degree of similarity within the NTDs of all synucleins (aa 1–90; backlit in gray). Black lines indicate the 11-amino-acid repeats that characterize all synucleins and that fold into an α-helix. Epitope of the pan-synuclein antibody used in this study is indicated. (B) Top, solution NMR structure of human α-synuclein bound to detergent micelles (Ulmer et al., 2005); bottom, the predicted structure of lamprey γ-synuclein. Ribbon models were generated using University of California–San Francisco Chimera modeling software. (C and D) A pan-synuclein antibody generated against human α-synuclein recognizes all three recombinant human synucleins and GST-tagged lamprey γ-synuclein, indicating structural conservation. The antibody does not recognize GST alone. Top panels are the Western blots against synuclein (WB:Syn); bottom panels are Coomassie-stained (Coom) gels of the same proteins. (E and F) In rat brain lysates and in lamprey spinal cord (SC) and brain lysates, the pan-synuclein antibody recognizes an endogenous 17-kDa protein—the expected size for monomeric synuclein. (G) GST-tagged lamprey γ-synuclein and its NTD (Nterm) bind to suvs containing PC and PA in a 3:1 M ratio, but not to PC alone (n = 2). Neither the C-terminal domain (Cterm) of lamprey synuclein nor GST alone binds PC/PA, but instead remain as free protein. No binding to multilamellar vesicles (mlv) was observed.
Comparative sequence analysis software (3D-JIGSAW, version 2.0) predicts that the NTD of lamprey γ-synuclein folds into an extended α-helix, followed by a less structured, random coil at the C-terminus, similar to the structure of human α-synuclein bound to lipid micelles (Figure 1B; Ulmer et al., 2005). As another indicator of structural conservation, recombinant human and lamprey synucleins were all recognized by the same pan-synuclein antibody raised against a highly conserved epitope within the N-terminus of human α-synuclein (aa 19–41; Figure 1, A, C, and D). This antibody also recognized endogenous rat and lamprey synucleins (Figure 1, E and F). Similar to human α-synuclein (Davidson et al., 1998; Perrin et al., 2000), full-length lamprey γ-synuclein and its NTD (aa 1–89) bound to small unilamellar vesicles (suvs) containing a mixture of phosphatidylcholine (PC) and phosphatidic acid (PA), but remained as free protein in the presence of suvs containing only PC (Figure 1G; n = 2). Lamprey γ-synuclein did not bind to large, multilamellar vesicles (Figure 1G). Thus, as with human α-synuclein, lamprey γ-synuclein demonstrates a preference for binding to acidic phospholipids, but only when they are presented in highly curved surfaces such as small vesicles. These data demonstrate that lamprey γ-synuclein and human α-synuclein are highly conserved, which predicts that they will cause similar synaptic phenotypes.
Synuclein is expressed at lamprey RS synapses
Lamprey RS synapses are large, en passant glutamatergic synapses formed between the descending giant RS axons in the ventromedial tract of the spinal cord and several types of intraspinal neurons (Figure 2A; Rovainen, 1974; Wickelgren et al., 1985; Buchanan, 2001). Before studying the acute effects of excess synuclein on synaptic vesicle trafficking, we wanted to examine the endogenous expression of synuclein in the lamprey spinal cord. Cross-sections of spinal cord were double labeled with antibodies against synuclein and synaptic vesicle glycoprotein 2 (SV2), a reliable marker of presynaptic vesicle clusters in all vertebrates examined, including lampreys (Buckley and Kelly, 1985; Lau et al., 2011). Synuclein colocalized with SV2 in synaptic puncta throughout the lamprey spinal cord (Figure 2, B–G). The Pearson's correlation coefficient for the immunofluorescence signals was 0.78 ± 0.04, indicating robust colocalization of synuclein with SV2 at synapses (n = 5 images, four animals). Furthermore, synuclein colocalized with SV2 at the giant RS synapses, which contain ∼1000 synaptic vesicles per cluster and are located at the periphery of the giant axons (Figure 2, E–G). Thus synuclein is endogenously expressed at lamprey giant RS synapses, providing a unique opportunity to investigate the acute effects of excess synuclein on synaptic vesicle trafficking.
FIGURE 2:
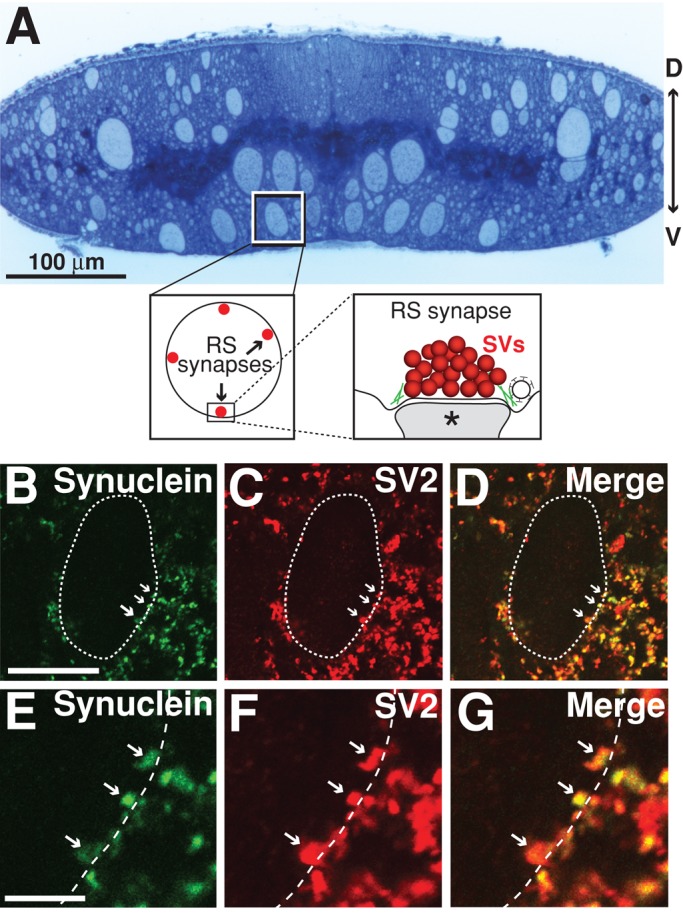
Synuclein is localized to synaptic vesicle clusters at lamprey giant RS synapses. (A) Top, cross-section of a lamprey spinal cord stained with toluidine blue. Note the giant RS axons in the ventromedial tract. The box marks a single giant RS axon and the view shown in B–D. D, dorsal; V, ventral. Bottom, insets show the location of giant RS synapses along the periphery of the giant axons. SVs, synaptic vesicles. Asterisk marks postsynaptic dendrite. (B–G) Immunolabeling of synuclein (green) and SV2 (red) in the lamprey spinal cord. Merged image reveals strong colocalization between synuclein and SV2, indicating synuclein's localization at synaptic vesicle clusters (Pearson's colocalization coefficient = 0.78 ± 0.04; n = 5). Dotted line indicates the border of the giant RS axon. Arrows indicate RS synapses, which are shown in high magnification in panels E–G. Puncta outside the dotted line are synapses formed by smaller intraspinal axons within the neuropil. Scale bar in B is 20 μm and applies to panels B–D. Scale bar in E is 5 μm and applies to panels E–G.
Excess lamprey γ-synuclein has no effect on vesicle clustering at unstimulated synapses
The vesicle trafficking defects observed at mammalian synapses after overexpression of human α-synuclein could be explained, in theory, by primary disruptions in synaptic vesicle clustering. For example, if excess synuclein caused the synaptic vesicles to become declustered and move away from the synaptic contact, then this would reduce the total number of vesicles immediately available for exocytosis and endocytosis, resulting in impaired vesicle trafficking. To determine this unequivocally requires examining the effects of excess synuclein on vesicle clusters when the synapses are inactive. Here the lamprey RS synapses provide an advantage, because within the isolated spinal cord, giant RS axons do not fire action potentials unless they are stimulated; furthermore, giant RS synapses have low spontaneous release rates, making the resting synapses relatively quiescent (Brodin et al., 1994).
To determine the effect of excess synuclein on unstimulated synapses, we microinjected glutathione S-transferase (GST)-tagged full-length lamprey γ-synuclein into giant RS axons, thereby introducing it directly to the presynapse (Figure 3, A and B). We also examined the effects of lamprey γ-synuclein NTD, since this is the most highly conserved region of all synucleins (Figure 3B). We estimate the axonal concentration of the injected proteins to be in the range of 6–20 μM (see Material and Methods), which is ∼2–5 times greater than the estimated amount of endogenous synuclein at synapses and is commensurate with synuclein overexpression in animal models of PD (Nemani et al., 2010; Westphal and Chandra, 2013). At unstimulated synapses, excess GST, full-length lamprey γ-synuclein, and NTD did not cause any obvious structural changes to presynaptic vesicle clusters when compared with synapses from uninjected control axons (Figure 3, C–F). In all cases, the synaptic vesicle clusters remained large and tightly clustered around the active zone, the electron-dense contact between the presynaptic axon and postsynaptic dendrite where neurotransmitter release occurs. A quantitative analysis across synapses revealed no significant difference in the size of the synaptic vesicle (SV) clusters between these conditions (Figure 3G; uninjected controls: 131 ± 12 SVs/section, n = 12 synapses, 4 axons; GST: 103 ± 9 SVs/section, n = 13 synapses, 2 axons; GST-synuclein: 139 ± 22 SVs/section, n = 11 synapses, 2 axons; GST-synuclein NTD: 101 ± 13 SVs/section, n = 8 synapses, 2 axons; analysis of variance [ANOVA] p = 0.18). Thus excess synuclein did not affect synaptic vesicle clustering at resting, unstimulated synapses, indicating that vesicle declustering is not the mechanism by which synuclein disrupts synaptic vesicle trafficking.
FIGURE 3:
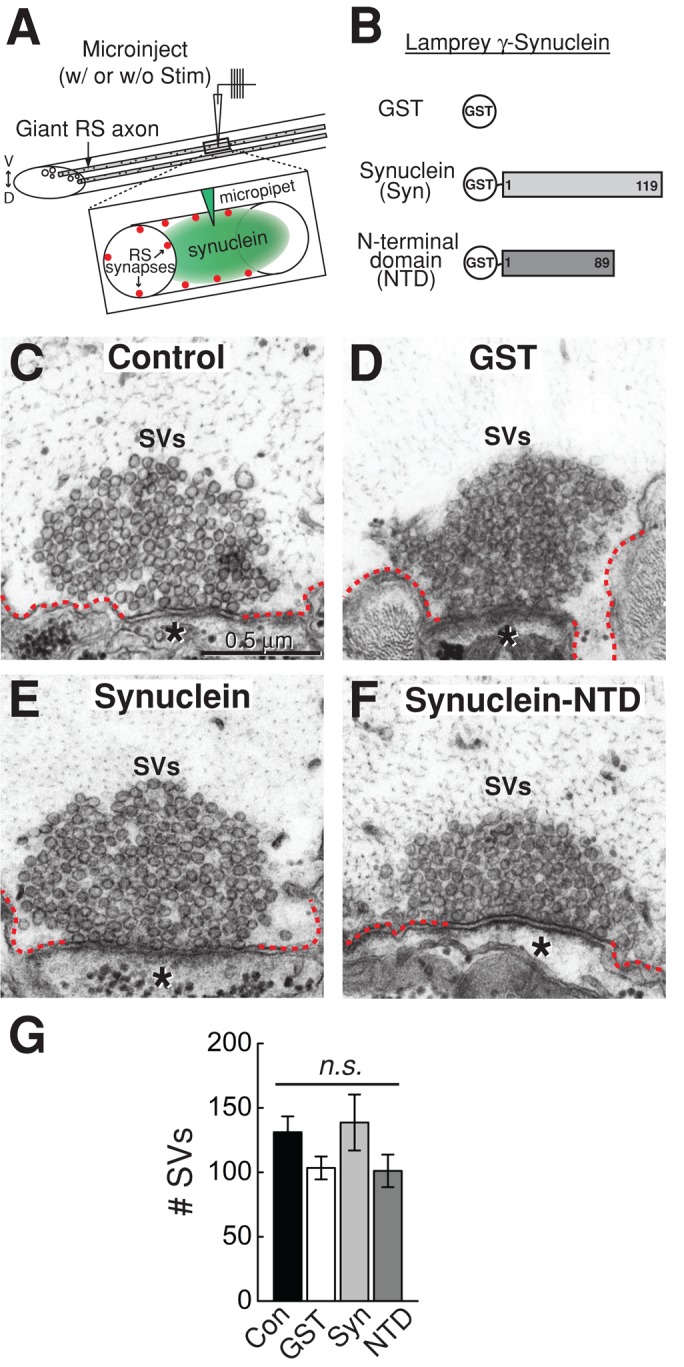
Excess synuclein does not affect synaptic vesicle clustering at unstimulated synapses. (A) Experimental paradigm used in this study. Giant RS axons were microinjected with excess recombinant synuclein. This was followed by immediate fixation either without stimulation or after stimulation with action potentials. Inset shows how the proteins gain access to the presynaptic vesicle clusters. (B) Diagram of the recombinant proteins that were injected. (C–F) Electron micrographs of unstimulated giant RS synapses after no injection (Control) or after injection of GST (GST), full-length lamprey γ-synuclein (Synuclein), or its NTD (Synuclein-NTD). In all cases, the synaptic vesicle (SV) clusters are large and appear normal. Asterisks mark the postsynaptic dendrites. Dotted lines indicate the plasma membrane. Scale bar in C applies to panels C–F. (G) The number of SVs was not significantly different across any of these conditions. Bars represent mean ± SEM from n = 8–13 synapses, 2 axons. n.s., not significant by ANOVA.
Excess lamprey γ-synuclein impairs synaptic vesicle recycling evoked during intense stimulation
Next we examined whether excess synuclein affects synaptic vesicle trafficking at stimulated synapses. By combining the acute introduction of excess synuclein with controlled stimulation conditions and ultrastructural analyses, we could determine for the first time the exact stages of synaptic vesicle trafficking directly affected by increased levels of synuclein. We repeated the injections of GST, lamprey γ-synuclein, and NTD. But this time we stimulated the axons with action potentials at high frequency (20 Hz, 5 min) to induce synaptic vesicle exocytosis and endocytosis. Though this is a strong stimulus, the RS neurons can fire steadily or burst in vivo at 10–20 Hz (Di Prisco et al., 2000; Dubuc et al., 2008). After 20-Hz stimulation, GST-treated synapses still exhibited large synaptic vesicle clusters, and the plasma membrane adjacent to the active zone became slightly evaginated (Figure 4A). Plasma membrane evaginations occur at stimulated synapses, because vesicle endocytosis from the plasma membrane is slower than exocytosis (Morgan et al., 2004; von Kleist et al., 2011). Thus, with GST, endocytosis is sufficient for maintaining large synaptic vesicle clusters.
FIGURE 4:

Excess lamprey γ-synuclein inhibits synaptic vesicle recycling during intense stimulation. (A–C) Electron micrographs of stimulated (20 Hz, 5 min) RS synapses after treatment with GST alone, full-length γ-synuclein, or the NTD. In the GST controls, large synaptic vesicle (SV) clusters were visible, and plasma membrane extensions (dotted lines) were modest, owing to efficient SV recycling. In contrast, after treatment with synuclein or NTD, the number of vesicles was greatly reduced, and large cisternae (C) were apparent, indicating a defect in SV recycling. Cisternae often had clathrin-coated pits emanating from them (circles), suggesting they derived from the plasma membrane. Asterisks indicate postsynaptic dendrites. Scale bar in panel A applies to A–C. (D) Examples of cisternae after treatment with excess lamprey γ-synuclein. (E–J) Quantification of the endocytic defects produced by lamprey synuclein. Synuclein reduced the number of SVs at synapses (E) and increased the amount of membrane trapped in cisternae (F–H). Synuclein NTD additionally increased the size of plasma membrane evaginations (I). Changes in clathrin coats were not significant (J). Bars represent mean ± SEM from n = 13–35 synapses, 2–3 axons. Asterisks indicate statistical significance from GST controls by ANOVA (Tukey's post hoc p < 0.05).
However, when stimulated synapses were treated with full-length lamprey γ-synuclein, there was a dramatic effect on presynaptic morphology. Specifically, there was a massive loss of synaptic vesicles and an increase in large, atypical, irregularly shaped membrane compartments, which we refer to as “cisternae,” in keeping with previous studies (Figure 4B; Heuser and Reese, 1973; Andersson et al., 2008). The most common cisternae were irregular and vesicular in nature (Figure 4D, top). However, cisternae sometimes appeared as flattened sacs or tubules (Figure 4D, bottom), which is noteworthy, because α-synuclein was recently shown to have the ability to tubulate lipid membranes (Westphal and Chandra, 2013). Clathrin-coated pits were often observed emanating from the cisternae (Figure 4, B and D). Interestingly, the NTD of lamprey γ-synuclein produced a similar phenotype (Figure 4C). Quantitative analysis revealed that both full-length lamprey γ-synuclein and NTD caused a significant reduction in the number of synaptic vesicles per synapse compared with GST controls (Figure 4E; GST: 82 ± 20 SVs/section, n = 13 synapses, 2 axons; GST-synuclein: 33 ± 4 SVs/section, n = 35 synapses, 3 axons; GST-synuclein NTD: 8 ± 1 SVs, n = 13 synapses, 2 axons; ANOVA p < 0.00005). The remaining synaptic vesicles had the same diameter as in controls (GST: 50.2 ± 0.5 nm, n = 200 SVs; GST-synuclein: 48.5 ± 0.6 nm, n = 200 SVs; GST-synuclein NTD: 50.3 ± 1.0 nm, n = 94 vesicles; ANOVA p > 0.05). Excess γ-synuclein and NTD also increased the number of cisternal structures compared with controls (Figure 4F; GST: 1.5 ± 0.4 cisternae, n = 13 synapses, 2 axons; GST-synuclein: 4.1 ± 0.6 cisternae, n = 35 synapses, 3 axons; GST-synuclein NTD: 2.7 ± 0.5 cisternae, n = 13 synapses, 2 axons; ANOVA p < 0.05). Both γ-synuclein and NTD induced the appearance of unusually large cisternae (>1000 nm), which were not observed at control synapses (Figure 4G). As a result of inducing more and larger cisternae, full length γ-synuclein and NTD caused a significant fourfold increase in the total amount of membrane trapped within cisternae (Figure 4H; GST: 0.5 ± 0.1 μm, n = 13 synapses, 2 axons; GST-synuclein: 2.2 ± 0.3 μm, n = 35 synapses, 3 axons; GST-synuclein NTD: 2.2 ± 0.5 μm, n = 13 synapses, 2 axons; ANOVA p < 0.005). Thus, at synapses stimulated at high frequency, lamprey γ-synuclein and NTD induced a dramatic depletion of synaptic vesicle membrane, paralleled by an increase in membrane trapped within atypical cisternae.
During stimulation, synaptic vesicles fuse with the plasma membrane before being locally recycled via one of several endocytic pathways (Heuser and Reese, 1973; Slepnev and De Camilli, 2000; Clayton and Cousin, 2009; Wenzel et al., 2012; Morgan et al., 2013a). The phenotype observed with excess γ-synuclein and NTD is consistent with a disruption in synaptic vesicle recycling, which results in a loss of synaptic vesicles concomitant with an expansion of the plasma membrane and/or an increase in endocytic intermediates (Shupliakov et al., 1997; Morgan et al., 2000, 2004; Andersson et al., 2008; von Kleist et al., 2011). To better understand the nature of the endocytic defect, we needed to determine the origin of the cisternae. The fact that there were clathrin-coated pits budding from the cisternae suggested that they might be derived from the plasma membrane (Figure 4, B–D). To test this, we systematically traced 36 cisternae through serial sections of synapses treated with excess full-length lamprey γ-synuclein (n = 20 synapses). Of the cisternae that could be reliably traced back to their origins, 83% (30/36) were still connected to the plasma membrane (Figure 5), while only 17% (6/36) were separated from the plasma membrane. Thus, at the time of fixation, the vast majority of cisternae were still connected to the plasma membrane, indicating that they are plasma membrane extensions. Lending further support to this idea, plasma membrane evaginations were 45% larger at synapses treated with NTD, which is consistent with the greater loss of synaptic vesicles observed in this condition (Figure 4I; GST: 2.0 ± 0.1 μm, n = 13 synapses, 2 axons; GST-synuclein: 2.2 ± 0.1 μm, n = 34 synapses, 3 axons; GST-synuclein NTD: 2.9 ± 0.3 μm, n = 12 synapses, 2 axons; ANOVA p < 0.01). The number of clathrin-coated pits and vesicles observed at synapses with excess synuclein or NTD was not significantly different from controls (Figure 4J; GST: 3.1 ± 0.8 clathrin coats, n = 13 synapses, 2 axons; GST-synuclein: 1.6 ± 0.4 clathrin coats, n = 35 synapses, 3 axons; GST-synuclein NTD: 5.2 ± 0.8 clathrin coats, n = 13 synapses, 2 axons; ANOVA p < 0.0005). Taken together, these data indicate that excess lamprey γ-synuclein inhibits synaptic vesicle recycling from the plasma membrane during intense stimulation.
FIGURE 5:

Cisternae caused by excess lamprey γ-synuclein are often connected to the plasma membrane. (A–D) Serial electron micrographs through a stimulated RS synapse treated with lamprey γ-synuclein. In A, a single cisterna (C) appears to be disconnected from the plasma membrane. However, in B, the cisterna is contiguous with the plasma membrane (arrows), indicating that it is an extension of the plasma membrane. In C and D, the cisterna is no longer distinguishable from the plasmalemma. SVs, synaptic vesicles. Asterisks mark the postsynaptic dendrite. Scale bar in panel A applies to A–D. (E) Left, a large cisterna that has several clathrin-coated pits budding from it is shown, along with its connection to the plasma membrane; right, inset shows the neck of the bulk endocytic structure (arrows).
Excess human α-synuclein also impairs synaptic vesicle recycling, indicating functional conservation
The fact that the highly conserved NTD of lamprey γ-synuclein was sufficient to reproduce the endocytic phenotype suggested that other synuclein orthologues would have similar effects. We therefore tested the effects of excess human α-synuclein on synaptic vesicle trafficking. Buffer was injected as a negative control. Excess human α-synuclein also reduced the number of synaptic vesicles and increased cisternae and plasma membrane evaginations at synapses during high-frequency stimulation (20 Hz, 5 min; Figure 6, A, B, H, and I). The number of synaptic vesicles was significantly reduced by 78% relative to controls (see Figure 8F later in this article) (control: 136 ± 10 SVs/section, n = 44 synapses, 4 axons; human α-synuclein: 30 ± 4 SVs/section, n = 22 synapses, 2 axons; ANOVA p = 5.4 × 10−11). The diameter of the remaining synaptic vesicles was not significantly changed (control: 52.4 ± 0.4 nm, n = 200 vesicles; α-synuclein: 51.3 ± 0.6 nm, n = 200 vesicles; t test p = 0.12). Large, irregularly shaped cisternae, both vesicular and tubular, were also observed with excess human α-synuclein (Figure 6, B and C). Excess α-synuclein caused a significant twofold increase in the number of cisternae at synapses (see Figure 8G), and nearly 25% of them were > 1000 nm in circumference (see Figure 8H). Furthermore, the total amount of membrane trapped within cisternae increased 3.4-fold (see Figure 8I; control: 0.7 ± 0.1 μm, n = 45 synapses, 4 axons; α-synuclein: 2.2 ± 0.4 μm, n = 22 synapses, 2 axons; ANOVA p < 0.00005). With α-synuclein, 92% of the cisternae were still connected to the plasma membrane (34/37; n = 18 synapses; Figure 6, D–G). Reconstructions of serial electron micrographs showed that the cisternae induced by α-synuclein were larger than those at control synapses and that they were still connected to the plasma membrane in at least one section, once again indicating that these structures are predominantly extensions of the plasma membrane (Figure 6, H and I, arrowheads). In addition, plasma membrane evaginations increased by 55% in the presence of α-synuclein, relative to controls (Figure 6, H and I; see Figure 8J; control: 2.0 ± 0.08 μm, n = 44 synapses, 4 axons; α-synuclein: 3.1 ± 0.17 μm, n = 21 synapses, 2 axons; ANOVA p < 0.001). Interestingly, human α-synuclein also caused a significant twofold increase in the total number of clathrin-coated pits and vesicles, suggesting effects on clathrin-mediated synaptic vesicle recycling (see Figure 8K; control: 1.7 ± 0.3 clathrin coats, n = 44 synapses, 4 axons; α-synuclein: 3.5 ± 0.4 clathrin coats, n = 22 synapses, 2 axons; ANOVA p < 0.0001). Thus, as with lamprey γ-synuclein, excess human α-synuclein inhibited synaptic vesicle recycling, indicating functional conservation between the synuclein orthologues.
FIGURE 6:

Excess human α-synuclein also inhibits synaptic vesicle recycling during intense stimulation. (A and B) Electron micrographs of stimulated RS synapses (20 Hz, 5 min) treated with buffer only (Control) or wild-type human α-synuclein. At control synapses, large synaptic vesicle (SV) clusters were visible, and plasma membrane evaginations (dotted lines) were modest because of efficient synaptic vesicle recycling. In contrast, after treatment of synapses with human α-synuclein, there were fewer SVs, cisternae were apparent, and plasma membrane evaginations were greatly expanded. Asterisks mark postsynaptic dendrites. C, cisterna. Circles, clathrin-coated pits or vesicles. Scale bar in A applies to B. (C) Cisternae caused by excess human α-synuclein. (D–G) Electron micrographs showing the connections between the cisternae and plasma membrane (arrows). (H and I) Three-dimensional reconstructions of stimulated RS synapses treated with buffer (control) or with excess human α-synuclein. The control synapse exhibits a large cluster of synaptic vesicles (blue) at the active zone (red), a relatively flat presynaptic plasma membrane (green), and small, infrequent cisternae (magenta). In contrast, after excess human α-synuclein, there were only a few synaptic vesicles, very large plasma membrane evaginations, and several larger, atypical cisternae. Most large cisternae were connected to the plasma membrane in at least one section (arrowheads). Scale bar in H applies to I.
FIGURE 8:

Endocytic defects caused by excess human α-synuclein require proper folding of the N-terminal α-helix. (A and B) T6K and A30P greatly reduced the synaptic vesicle recycling defects after 20-Hz, 5-min stimulation. Synaptic vesicle (SV) clusters were larger, and the cisternae were less prevalent than with WT α-synuclein (see Figure 6). Asterisks mark the postsynaptic dendrites. Dotted lines indicate the plasma membrane. C, cisterna; circles, clathrin-coated pits or vesicles. Scale bar in A applies to B. (C) The cisternae observed with excess T6K (top) or A30P (bottom) were smaller and discontinuous with the plasma membrane (see D and E). (D and E) Three-dimensional reconstructions of stimulated RS synapses treated with T6K and A30P do not exhibit major endocytic defects but instead appear more similar to controls (compare with Figure 6H). Scale bar in D applies to E. (F–K) Quantification of the ultrastructural analyses. Consistent with an inhibition in synaptic vesicle recycling, wild-type α-synuclein decreased the number of synaptic vesicles (F), while increasing the number of cisternae (G), the frequency of large cisternae (H), the amount of membrane trapped within cisternae (I), and the size of plasma membrane evaginations (J). The increase in clathrin-coated pits and vesicles with α-synuclein also indicates vesicle recycling defects (K). In contrast, the T6K and A30P mutations reduced all aspects of the vesicle recycling defects associated with α-synuclein. Bars represent mean ± SEM from n = 22–44 synapses, 2–4 axons. Asterisks indicate statistical significance by ANOVA (Tukey's post hoc p < 0.05).
Mutations that disrupt the α-helix of α-synuclein greatly reduce the endocytic defects
Our results here and in a prior study suggest that the highly conserved NTD is responsible for the synaptic vesicle–trafficking defects (Nemani et al., 2010). However, no structure–function analysis has been performed to determine which feature of the NTD is causing the defects, and we therefore set out to determine this. It is well established that the NTDs of synucleins fold into an amphipathic α-helix upon binding to acidic phospholipids within small vesicles (Davidson et al., 1998; Perrin et al., 2000; Chandra et al., 2003). Using circular dichroism and nuclear magnetic resonance (NMR) analyses, several mutations in the NTD have been identified that significantly reduce the α-helical content of the protein (Perrin et al., 2000; Ulmer and Bax, 2005). One is “A30P,” a missense mutation in which A30 is mutated to a proline, which is found in some familial PD patients (Figure 7A; Perrin et al., 2000; Ulmer and Bax, 2005). Though only a point mutation, A30P causes one turn of the α-helix to adopt an extended conformation and in doing so destabilizes the surrounding turns and induces a large stretch of the amphipathic α-helix to fall out of register (Ulmer and Bax, 2005). The other is a synthetic mutant, “T6K,” in which six threonines lying along the hydrophobic face of the amphipathic α-helix are mutated to lysines (i.e., T22K, T33K, T44K, T59K, T81K, T92K; Figure 7A; Perrin et al., 2000). With both mutants, the α-helical content is only about half of that observed for wild-type α-synuclein (Perrin et al., 2000). We used a liposome flotation assay to determine how well these mutants still bind to the acidic phospholipid, PA (Burre et al., 2010, 2012). In this assay, small, sonicated liposomes (25–30 nm) primarily float up into the top two fractions of an Accudenz gradient upon centrifugation. Thus any liposome-bound protein will appear in these fractions, while unbound, free protein will remain in the lower fractions (Figure 7B). As expected, wild-type α-synuclein bound strongly to liposomes containing PC/PA, but not to those containing only PC (Figure 7, C and D; n = 3). In comparison, A30P and T6K still bound robustly to PC/PA liposomes, but with reduced affinity (Figure 7, C and D; n = 3). Thus T6K and A30P disrupt the α-helical conformation of α-synuclein but retain significant lipid-binding capacity, corroborating previous observations (Perrin et al., 2000; Bussell and Eliezer, 2004; Burre et al., 2012).
FIGURE 7:

α-Synuclein mutations A30P and T6K alter proper folding of the N-terminal α-helix. (A) Wild-type (WT) human α-synuclein and mutants. A30P and T6K exhibit significantly reduced α-helical content in the NTD, based on circular dichroism analysis (Perrin et al., 2000). Asterisks mark the locations of the mutations. (B) Liposome flotation assay. Liposomes (circles), and thus bound protein (bars), are predominantly in the top two fractions after centrifugation, while unbound protein is in the lower fractions, where the percentage of liposomes is low. The data in the graph show an example of fluorescence measurements from NBD-labeled PC liposomes. (C) WT α-synuclein binds to PC/PA liposomes but not to PC alone. T6K and A30P also bind to PC/PA liposomes, but with reduced affinity, confirming previous observations (Perrin et al., 2000). (D) Quantification of data from liposome-binding assays showing α-synuclein and mutants binding to PC and PC/PA. Data points represent mean ± SEM from n = 3 independent experiments.
To determine the extent to which the α-synuclein–associated endocytic defects are influenced by changes in the α-helical content of the protein, we tested the effects of T6K and A30P on vesicle trafficking. Surprisingly, stimulated synapses treated with either T6K or A30P appeared more similar to control synapses than to synapses treated with wild-type α-synuclein (Figure 8, A–E). With both mutants, the synaptic vesicle clusters were much larger than at synapses treated with wild-type α-synuclein, and in the case of A30P, the vesicle clusters were indistinguishable from controls (Figure 8F; control: 136 ± 10 SVs/section, n = 44 synapses, 4 axons; α-synuclein: 30 ± 4 SVs/synapse, n = 22 synapses, 2 axons; T6K: 75 ± 9 SVs/synapse, n = 28 synapses, 2 axons; A30P: 148 ± 16 SVs, n = 24 synapses, 2 axons; ANOVA p = 5.4 × 10–11). The number of cisternal structures was still increased with T6K and A30P (Figure 8G; control: 1.6 ± 0.2 cisternae, n = 44 synapses, 4 axons; α-synuclein: 3.1 ± 0.6 cisternae, n = 22 synapses, 2 axons; T6K: 3.0 ± 0.5 cisternae, n = 28 synapses, 2 axons; A30P: 2.6 ± 0.5 cisternae, n = 24 synapses, 2 axons; ANOVA p < 0.05). However, the cisternae observed in the presence of T6K and A30P were the small, vesicular type normally observed in controls, and they did not exhibit obvious connections to the plasma membrane (Figure 8, C–E and H). The total amount of membrane within cisternae after T6K and A30P was significantly reduced in comparison with wild-type α-synuclein (Figure 8I; control: 0.7 ± 0.1 μm, n = 45 synapses, 4 axons; α-synuclein: 2.2 ± 0.4 μm, n = 22 synapses, 2 axons; T6K: 1.1 ± 0.2 μm, n = 28 synapses, 2 axons; A30P: 1.2 ± 0.3 μm, n = 24 synapses, 2 axons; ANOVA p < 0.00005). Paralleling the recovery of synaptic vesicles and cisternae, the plasma membrane evaginations were also progressively smaller with T6K and A30P (Figure 8J; control: 2.0 ± 0.1 μm, n = 44 synapses, 4 axons; α-synuclein: 3.1 ± 0.2 μm, n = 21 synapses, 2 axons; T6K: 2.9 ± 0.2 μm, n = 28 synapses, 2 axons; A30P: 2.5 ± 0.2 μm, n = 24 synapses, 2 axons; ANOVA p < 0.001). Clathrin-coated pits and vesicles were still elevated with T6K, but were similar to control values with A30P (Figure 8K; control: 1.7 ± 0.3 clathrin coats, n = 44 synapses, 4 axons; α-synuclein: 3.5 ± 0.4 clathrin coats, n = 22 synapses, 2 axons; T6K: 3.8 ± 0.5 clathrin coats, n = 28 synapses, 2 axons; A30P: 1.8 ± 0.4 clathrin coats, n = 24 synapses, 2 axons; ANOVA p < 0.0001). Thus the T6K and A30P mutations within the NTD greatly reduced the endocytic defects caused by excess human α-synuclein, indicating that a properly folded α-helix is a major structural determinant underlying the synaptic vesicle trafficking defects induced by excess α-synuclein.
The A53T α-synuclein mutant also impairs synaptic vesicle recycling
If a properly folded α-helix is responsible for causing the synaptic vesicle recycling defects observed with excess α-synuclein, then other mutants that retain the α-helical conformation should similarly affect vesicle recycling. One such mutant is A53T, another PD-linked mutation in α-synuclein, which exhibits normal lipid-induced α-helicity (Perrin et al., 2000). As predicted by the model, excess A53T caused a phenotype that was nearly identical to that induced by wild-type α-synuclein (Figure 9). The number of synaptic vesicles was significantly reduced (Figure 9, A–F; control: 87 ± 7 SVs/section, n = 35 synapses, 2 axons; A53T: 35 ± 8 SVs/section, n = 25 synapses, 2 axons; Student's t test p < 0.001). The number of cisternae increased (Figure 9G; control: 0.4 ± 0.1 cisternae, n = 35 synapses, 2 axons; A53T: 2.9 ± 0.4 cisternae, n = 25 synapses, 2 axons; Student's t test p < 0.5 × 10−8). As with wild-type α-synuclein, A53T shifted the distribution of cisternae toward larger sizes such that ∼25% of the cisternae were >1000 nm in circumference (Figure 9H). With A53T, the total amount of membrane within cisternae increased significantly (Figure 9I; control: 0.2 ± 0.1 μm, n = 35 synapses, 2 axons; A53T: 3.0 ± 0.5 μm, n = 25 synapses, 2 axons; Student's t test p < 0.05 × 10−6). Furthermore, the size of plasma membrane evaginations was increased (Figure 9J; control: 2.1 ± 0.1 μm, n = 35 synapses, 2 axons; A53T: 3.0 ± 0.3 μm, n = 25 synapses, 2 axons; Student's t test p < 0.001). A53T also increased the number of clathrin-coated pits and vesicles ∼twofold (Figure 9K; control: 2.3 ± 0.9 clathrin coats, n = 35 synapses, 2 axons; A53T: 5.4 ± 1.9 clathrin coats, n = 25 synapses, 2 axons; Student's t test p < 0.0005). Thus A53T phenocopied the endocytic defects observed with excess wild-type α-synuclein, further corroborating that a properly folded α-helix is the major structural determinant underlying the synaptic vesicle recycling defects.
FIGURE 9:

A53T also impairs synaptic vesicle recycling evoked by high-frequency stimulation. (A and B) Compared with control synapses, A53T caused a reduction in the number of synaptic vesicles (SVs) and expansion of the plasma membrane (dotted lines) after 20-Hz, 5-min stimulation. Large cisternae were also apparent. C, cisterna; circles, clathrin-coated pits or vesicles. Scale bar in A applies to B. (C) Cisternae caused by A53T. (D and E) Three-dimensional reconstructions show the endocytic phenotype caused by A53T. A large cisterna was connected to the plasma membrane (arrowhead). Scale bar in D applies to E. (F–K) Synapses treated with A53T induced the same phenotype as wild-type α-synuclein: a reduction in synaptic vesicles (F); an increase in the number, size, and amount of membrane within cisternae (G–I); an increase in plasma membrane evaginations (J); and an increase in the number of clathrin-coated pits and vesicles (K). Bars represent mean ± SEM from n = 25–35 synapses, 2 axons. Asterisks indicate statistical significance by Student's t test (p < 0.05).
Excess α-synuclein does not impair synaptic vesicle recycling evoked by lower-frequency stimulation
The high-frequency stimulation used in these experiments is known to evoke activity-dependent bulk endocytosis at synapses, in addition to clathrin-mediated endocytosis (Clayton and Cousin, 2009; Wenzel et al., 2012). In contrast, lower-frequency stimulation (5 Hz or less) appears to evoke predominantly clathrin-mediated synaptic vesicle recycling at lamprey RS synapses (Shupliakov et al., 1997; Ringstad et al., 1999). To further probe the mode(s) of endocytosis being affected by excess α-synuclein, we used a milder stimulation paradigm. After stimulation at 5 Hz for 30 min, synapses treated with excess human α-synuclein appeared very similar to control synapses (Figure 10, A–D). Quantification revealed that there was no significant difference in the number of synaptic vesicles (Figure 10E; control: 154 ± 14 SVs/section, n = 22 synapses, 2 axons; α-synuclein: 131 ± 12 SVs/section, n = 20 synapses, 2 axons; Student's t test p = 0.22). Changes in the number of cisternae and their size distribution were unremarkable (Figure 10, F and G; control: 2.5 ± 0.6 cisternae; α-synuclein: 3.5 ± 0.7 cisternae; Student's t test p = 0.31). In both controls and α-synuclein–treated synapses, the cisternae were generally the small, vesicular type (Figure 10, C and D). The total amount of membrane in the cisternae was not significantly altered (Figure 10H; control: 1.0 ± 0.2 μm; α-synuclein: 1.8 ± 0.5 μm; Student's t test p = 0.09). Neither was the average size of plasma membrane evaginations significantly changed (Figure 10I; control: 1.9 ± 0.1 μm; α-synuclein: 2.2 ± 0.2 μm; Student's t test p = 0.07), and the number of clathrin-coated structures was similar (Figure 10J; control: 3.7 ± 0.6 clathrin coats; α-synuclein: 3.2 ± 0.7 clathrin coats; Student's t test p = 0.56). Thus excess α-synuclein only impairs synaptic vesicle recycling evoked during high-frequency stimulation.
FIGURE 10:

α-Synuclein does not affect synaptic vesicle recycling evoked by lower frequency stimulation. (A and B) Following a lower-frequency stimulation (5 Hz, 30 min), synapses treated with excess human α-synuclein appeared similar to control synapses. No endocytic phenotype was observed. Dotted lines indicate plasma membrane. SVs, synaptic vesicles; circles, clathrin-coated pits or vesicles. Asterisks mark the postsynaptic dendrites. Scale bar in A applies to B. (C and D) Three-dimensional reconstructions of control and α-synuclein treated synapses after 5-Hz, 30-min stimulation. No obvious differences were observed. (E–J) Quantification revealed that there was no significant change in the number of synaptic vesicles (E); the number, size distribution, or amount of membrane within cisternae (F–H); the size of plasma membrane evaginations (I); or the number of clathrin coats (J). Bars represent mean ± SEM from n = 20–22 synapses, 2 axons. n.s., not significant by Student's t test (p > 0.05).
DISCUSSION
To date, there have been conflicting reports about the stages in the synaptic vesicle trafficking pathway that are perturbed by α-synuclein overexpression. While some studies reported primary defects in exocytosis (Larsen et al., 2006; Gaugler et al., 2012; Lundblad et al., 2012), others reported impairment of vesicle reclustering after endocytosis (Nemani et al., 2010) or pleiotropic effects (Scott et al., 2010). With chronic overexpression, it is difficult to assess which vesicle trafficking defects are due to excess α-synuclein because of the decreased expression levels of other presynaptic proteins, including those involved in exocytosis, endocytosis, and vesicle clustering (Nemani et al., 2010; Scott et al., 2010). Thus the direct effects of excess synuclein on synaptic vesicle trafficking remained unclear until now.
By utilizing acute perturbations, we were able to overcome these limitations and examine the effects of excess synuclein on vesicle trafficking within a few minutes of its introduction to synapses, bypassing the types of molecular compensation that occur with chronic overexpression. Our data revealed that excess α-synuclein dramatically impaired synaptic vesicle recycling from the plasma membrane, as evidenced by a loss of synaptic vesicles and an expansion of the plasma membrane. Excess synuclein also increased the amount of membrane trapped within cisternae, the vast majority of which were still connected to the plasma membrane, further corroborating the endocytic defect. The impairment of endocytosis was activity dependent, phenotypically conserved between lamprey γ-synuclein and human α-synuclein, and dependent upon a properly folded α-helix in the NTD. While the acute perturbation strategy is better for revealing direct effects of excess synuclein on synaptic vesicle trafficking, we acknowledge that chronic overexpression of α-synuclein more closely models what occurs in PD. In that respect, it is interesting to note that there are some strong similarities between the synaptic phenotypes observed after acute or chronic overexpression of α-synuclein. Similar to our findings, after overexpression of α-synuclein in mammalian neurons, the density of vesicles is decreased within synaptic boutons (Scott et al., 2010). In addition, abnormally large vesicular structures are apparent (Scott et al., 2010; Boassa et al., 2013). We would now interpret those large vesicular structures to be cisternae, caused by an impairment of endocytosis. Similar phenotypes have been reported at dopaminergic synapses after α-synuclein overexpression (Gaugler et al., 2012).
While we cannot completely rule out any defects in exocytosis, our data are most consistent with excess α-synuclein inhibiting synaptic vesicle recycling. The decrease in synaptic vesicles and expansion of the plasma membrane are classically representative of endocytic defects (Morgan et al., 2000, 2004; Andersson et al., 2008). Furthermore, our data suggest that excess α-synuclein affects several modes of synaptic vesicle recycling via both clathrin-mediated endocytosis and activity-dependent bulk endocytosis (Figure 11). One predominant mechanism for recycling synaptic vesicles is clathrin-mediated endocytosis (Granseth et al., 2006; Heerssen et al., 2008; Saheki and De Camilli, 2012). The increased number of clathrin-coated pits and vesicles we observed after acutely introducing excess α-synuclein is indicative of an effect on clathrin-mediated synaptic vesicle recycling. Why lamprey γ-synuclein did not alter clathrin-coated structures is not clear, but one possibility is that this is due to differences in the amino acid sequence, for example, in the C-terminal domain. Excess α-synuclein also appeared to inhibit activity-dependent bulk endocytosis, which is triggered at high frequencies (>10 Hz; Clayton et al., 2008; Cousin, 2009; Wenzel et al., 2012; Morgan et al., 2013a). In support of this, the endocytic phenotype was unremarkable when the stimulation was lowered to 5 Hz, a condition that normally does not evoke a lot of activity-dependent bulk endocytosis at synapses. Further experiments are needed to determine whether the effects on clathrin-mediated synaptic vesicle recycling are direct, for example, through an interaction between α-synuclein and clathrin coat components, or whether they are an indirect consequence of inhibiting activity-dependent bulk endocytosis. Recent evidence from synuclein triple-knockout mice demonstrated that synucleins participate in early stages of clathrin-mediated synaptic vesicle recycling, suggesting that a direct effect is possible (Vargas et al., 2014).
FIGURE 11:
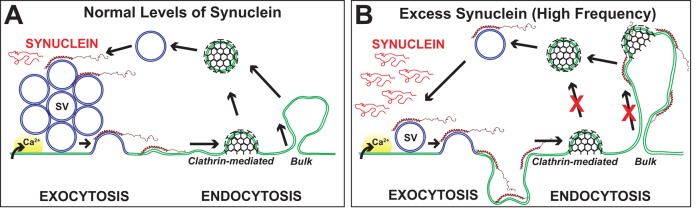
Model for how excess α-synuclein impairs synaptic vesicle recycling. (A) Synaptic vesicle (SV) recycling with endogenous levels of synuclein. After calcium (Ca2+) influx and exocytosis, vesicles are efficiently recycled via local endocytosis from the plasma membrane. (B) With excess α-synuclein, SV recycling is disrupted, as evidenced by a loss of SVs and an expansion of the plasma membrane, which often buckled inward to create apparent cisternae. Excess α-synuclein inhibited at least two modes of synaptic vesicle recycling, clathrin-mediated and bulk endocytosis, but only during high-frequency stimulation. The working model is that excess α-synuclein, in its α-helical conformation, masks or mislocalizes key lipids or proteins necessary for initiating synaptic vesicle recycling.
We estimated the amount of injected α-synuclein to be 6–20 μM, a concentration range that in theory could induce a variety of synaptic phenotypes. However, we did not observe any obvious differences or trends in the phenotypes caused by excess α-synuclein or the mutants across the axonal region studied. That is, an endocytic phenotype was observed at the vast majority of α-synuclein– and A53T-treated synapses, and a drastic reduction of the endocytic phenotype was observed at the A30P- and T6K-treated synapses, regardless of the distance from the injection site. This indicates that a range of phenotypes was not lumped together across a steep dose–response curve. However, it should be mentioned that, with acute or chronic overexpression, it is simply not possible to know the precise concentration of protein that reaches the synapse in a functional state. This would seem particularly true for a protein like α-synuclein, which binds with high affinity to a variety of lipids and cellular membranes (Maroteaux et al., 1988; Davidson et al., 1998; Nakamura, 2013). Recent studies have reported multiple pools of α-synuclein at synapses after overexpression: soluble, vesicle-bound, and aggregated forms (Spinelli et al., 2014), as well as tubule-bound forms (Boassa et al., 2013). In future synaptic studies, it will also be important to distinguish how much of the excess α-synuclein is localized to synaptic vesicles and cytosol versus the plasma membrane, mitochondria, clathrin-coated vesicles, and other synaptic membranes.
In addition to pinpointing the vesicle recycling defects caused by excess α-synuclein, this study is the first to delineate the structural mechanism. The first clue emerged when we discovered that the NTD of lamprey γ-synuclein was sufficient to reproduce the endocytic defects. Similarly, the NTD of human α-synuclein was sufficient to produce the synaptic vesicle trafficking defects observed at mammalian synapses (Nemani et al., 2010). Among all synuclein orthologues, the NTD is the most highly conserved region, predicting that it has an important function (George, 2002; Sun and Gitler, 2008; Busch and Morgan, 2012). A structure–function analysis of α-synuclein's NTD allowed us to determine the underlying features causing the vesicle trafficking defects. The synthetic T6K mutant and the PD-linked A30P mutant both exhibit disrupted folding of the N-terminal α-helix while retaining significant lipid-binding capacity (Perrin et al., 2000; Ulmer and Bax, 2005; Burre et al., 2012). As a consequence of these mutations, excess α-synuclein no longer produced severe endocytic phenotypes and instead allowed synaptic vesicles to proceed fairly efficiently through the recycling pathways. Thus a properly folded α-helical domain within α-synuclein is a critical structural determinant underlying the endocytic defects. Further corroborating this idea, the PD-linked mutant A53T, with a properly folded α-helix, also exhibited the vesicle-recycling defects. How might this work? When in excess, a properly folded α-synuclein may bind and mask lipids that are critical for initiating vesicle reinternalization from the plasma membrane, such as phosphoinositides (Figure 11) (Ford et al., 2001; Di Paolo et al., 2004). Alternatively, excess α-synuclein may bind and mislocalize or otherwise alter the functions of other endocytic proteins. Future experiments will determine the extent to which these mechanisms are in play.
The synaptic vesicle recycling defects produced by excess wild-type α-synuclein and A53T are suggestive of the types of synaptic defects that could occur in patients with PD. At first, it may seem surprising that A30P failed to reproduce the endocytic defects, since A30P is also genetically linked to PD (Kruger et al., 1998). However, this result is consistent with previous studies in which A30P expression in mammalian neurons had no effect on synaptic transmission (Nemani et al., 2010). A30P also did not properly localize to synaptic vesicle clusters (Fortin et al., 2005), likely because of its reduced α-helicity and lipid binding. Though this is speculative, one possibility is that A30P may cause fewer or delayed synaptic defects in humans, since this mutation exhibits incomplete penetrance (Kruger et al., 1998).
Whether excess α-synuclein exacerbates the normal function of synuclein or causes a neurotoxic gain of function at synapses is still an open question. A limiting factor is the lack of deep understanding of α-synuclein's normal function (Bendor et al., 2013). Early studies showed that knockdown of α-synuclein altered synaptic morphology in ways consistent with a role in vesicle trafficking (Murphy et al., 2000). Similarly, single and double synuclein knockouts (α, α/β, α/γ) exhibited defects in synaptic transmission, though the phenotypes reported were mild (Abeliovich et al., 2000; Chandra et al., 2004; Senior et al., 2008). However, some of the single and double knockouts induced an up-regulation of the remaining synuclein isoform(s) (Chandra et al., 2004). Recent studies have reinvestigated the normal roles for synucleins with the generation of synuclein triple-knockout mice (α/β/γ). The triple knockouts exhibited reduced formation of SNARE (Soluble NSF Attachment Protein REceptor) complexes and reduced synaptic transmission, as well as altered presynaptic structure, supporting a normal role for synucleins in synaptic vesicle trafficking (Burre et al., 2010; Greten-Harrison et al., 2010). Consistent with our model (Figure 11), a recent study places α-synuclein's normal function at early stages of synaptic vesicle endocytosis (Vargas et al., 2014). Absence of synucleins (α/β/γ) slowed the kinetics of endocytosis and reduced the number of recycled synaptic vesicles (Vargas et al., 2014). Thus synuclein modulates synaptic vesicle recycling whether it is absent or in excess. This predicts that synaptic levels of α-synuclein should be tightly regulated in order to ensure proper vesicle recycling, which is consistent with the varied levels of synuclein observed at synapses (Fortin et al., 2005; Murphy et al., 2000). Further investigation of synuclein's normal functions is warranted, as is a better understanding of how age-dependent changes in synuclein function at synapses impacts the pathobiology of PD and other synucleinopathies.
MATERIALS AND METHODS
Recombinant proteins
Cloning and expression of GST-tagged lamprey synucleins and untagged human synucleins were as previously described (Perrin et al., 2000; Busch and Morgan, 2012). An additional GST fusion comprising only the NTD (aa 1–89) of lamprey γ-synuclein was also generated (GenBank: JN544525). Recombinant proteins were expressed in BL21-CodonPlus (DE3)-RILP Competent Cells (Agilent Technologies, Santa Clara, CA). GST fusion proteins were then purified using glutathione Sepharose beads (GE Healthcare, Pittsburgh, PA). Human α-synuclein and mutants were precipitated from the soluble protein fractions using ammonium sulfate, and the precipitate was resuspended in 20 mM sodium phosphate buffer (pH 7.4). α-Synuclein and mutants were further purified using a Sepharose hydrophobic interaction column, followed by gel filtration on a Sephacryl S-200 size-exclusion column and lyophilization. Proteins were resuspended or dialyzed into the appropriate buffer before each assay.
Western blotting
Western blotting was performed as previously described (Morgan et al., 2004; Busch and Morgan, 2012). Briefly, 0.5 μg of each recombinant protein was separated on a 10% SDS–polyacrylamide gel. For detecting endogenous proteins, 5 μg rat brain lysates and 20 μg lamprey brain and spinal cord lysates were separated by SDS–PAGE. Proteins were transferred to nitrocellulose membranes, blocked with 5% dry milk, and then incubated with a rabbit polyclonal pan-synuclein antibody (AbCam ab53726; Cambridge, MA) at 1:1000 in TBST (20 mM Tris Base, pH 8.0, 137 mM NaCl, 0.1% Tween 20) for 1 h at room temperature (RT). Secondary antibody used was 1:4000 goat–anti-rabbit immunoglobulin G (IgG; Thermo Scientific, Waltham, MA). Western blots were developed using Thermo Scientific Pierce ECL substrates.
Vesicle binding assays
The vesicle binding assay shown in Figure 1G was performed as described previously for human α-synuclein (Davidson et al., 1998; Perrin et al., 2000). Briefly, suvs were prepared from a mixture of POPC and POPA (3:1) or pure POPC (Avanti Polar Lipids, Alabaster, AL), and incubated with recombinant GST-tagged lamprey synuclein for 2 h at a 20:1 mass ratio of lipid to protein. Samples were separated by gel exclusion chromatography (Superose 6); and peaks representing free protein, suv, and large multilamellar vesicles (mlv) were pooled and subjected to immunoblotting with anti-GST antibody (GeneTex GTX110736; Irvine, CA).
The liposome flotation assay in Figure 7 was performed as described in previous studies (Burre et al., 2010, 2012). Liposomes comprising a mixture of POPC and NBD-labeled POPC (99:1) or a mixture of POPC, POPA, and NBD-POPC (49:50:1) (Avanti Polar Lipids, Alabaster, AL) were prepared and sonicated. Sonicated liposomes were then incubated with 5 μg α-synuclein (WT, T6K, or A30P) in HKE buffer (25 mM HEPES, pH 7.4, 150 mM KCl, 1 mM EDTA) at RT for 2 h. Samples were then centrifuged at 280,000 × g for 3 h at RT in an Accudenz gradient (Accurate Chemical and Scientific, Westbury, NY). Afterward, samples were separated into eight fractions. Fluorescence in each fraction was analyzed using a NanoDrop 3300 fluorospectrometer to determine which fractions contained the liposomes. The fractions were also analyzed by Western blotting to evaluate the bound versus free protein. Western blotting was as above, except that secondary antibody was used at 1:1000. Background-subtracted band intensity was measured using ImageJ. Then the band intensity for each fraction was calculated as a percentage of the sum total for each experiment, and data were averaged from n = 3 independent experiments.
Immunofluorescence
All procedures involving lampreys were approved by the Institutional Animal Care and Use Committees at the University of Texas at Austin (UT-Austin) and Marine Biological Laboratory (MBL) in accordance with standards set by the National Institutes of Health (NIH). Late larval-stage sea lampreys (P. marinus; 11–13 cm) were anesthetized with 0.1 g/l tricaine methanesulfonate (Finquel MS-222; Argent Laboratories; Redmond, WA). Spinal cords were then dissected in oxygenated lamprey ringer containing (in mM) 100 NaCl, 2.1 KCl, 2.6 CaCl2, 1.8 MgCl2, 4 glucose, 0.5 glutamine, 2 HEPES (pH 7.4), as previously described (Oliphint et al., 2010). Dissected spinal cords were fixed, cryoprotected, embedded in TissueTek OCT, cross-sectioned at 14 μm, and immunostained, as described by Oliphint et al. (2010). Primary antibodies used were a rabbit polyclonal pan-synuclein antibody (AbCam ab53726; Cambridge, MA) and a mouse monoclonal antibody against SV2 (DSHB; University of Iowa, Iowa City, IA). The SV2 antibody has been previously characterized in vertebrates, including lampreys (Buckley and Kelly, 1985; Lau et al., 2011; Busch and Morgan, 2012). The synuclein antibody is characterized in the current study (Figures 1–2 and Supplemental Figure 1). Secondary antibodies used were: Alexa Fluor 488–conjugated goat–anti-rabbit IgGs and Alexa Fluor 594–conjugated goat–anti-mouse IgGs (Life Technologies, Grand Island, NY). Confocal images were acquired with a Zeiss LSM Pascal Exciter on an Axioskop 2FS using a 40×, 1.3 NA EC Plan-Neofluar oil immersion objective. For images in Figure 2, E–G, an additional 3× digital zoom was applied. To quantify the colocalization of the immunofluorescence signals for synuclein and SV2, we used the ImageJ plug-in JACoP to calculate the Pearson's correlation coefficients (n = 5 images, 4 spinal cords; Bolte and Cordelieres, 2006; Dunn et al., 2011).
Microinjections and stimulation
Microinjections were performed as previously described (Morgan et al., 2004, 2013b). Briefly, a 2- to 3-cm section of spinal cord was dissected and pinned ventral side up in a Sylgard-lined Petri dish containing oxygenated lamprey ringer (Figure 3A). All recombinant proteins were dialyzed into lamprey internal solution (180 mM KCl, 10 mM HEPES, pH 7.4). Proteins (120–200 μM) were then loaded into glass microelectrodes and injected directly into giant RS axons using small pulses of N2 (5–20 ms, 30–50 psi, 0.1–0.3 Hz) delivered via a Toohey Spritzer. For estimating the axonal protein concentration, all proteins were coinjected with 100 μM fluorescein dextran (10 or 40 kDa, anionic, lysine fixable; Life Technologies). After 10–30 min of injection, the axonal fluorescence reached 1/10th to 1/20th of that in the microelectrode. Therefore the estimated axonal concentration of the proteins was 6–20 μM. After injection, some spinal cords were immediately fixed without stimulation. In other experiments, injected axons were stimulated intracellularly by action potentials, using short, depolarizing current pulses, generated by an Axoclamp 2B amplifier (1 ms, 20–80 nA, 20 Hz for 5 min or 5 Hz for 30 min). With the stimulation continuing, the spinal cords were fixed for electron microscopy using 3% glutaraldehyde, 2% paraformaldehyde in 0.1 M Na cacodylate (pH 7.4). Fixation occurred within 5–10 s, as determined by the disappearance of action potentials.
Electron microscopy and morphometric analysis
Spinal cords were processed for electron microscopy as previously described (Oliphint et al., 2010; Morgan et al., 2013b). Spinal cords were postfixed with 2% osmium tetroxide containing 2% potassium ferrocyanide for 1.5 h on ice, stained en bloc with 2% uranyl acetate, dehydrated in an ethanol series, and embedded in Embed-812. Ultrathin sections (70 nm) were cut and counterstained with 2% uranyl acetate and 0.4% lead citrate, after which individual synapses were imaged at 26,500× or 37,000×, respectively, using a Technai Spirit BioTwin T12 or JEOL JEM 200CX transmission electron microscope. For each experimental condition, images of more than 10 synapses were collected from at least two axons, and they were acquired from the same region of the injected axon (25–150 μm from the injection site), ensuring that the protein concentration was comparable.
Morphometric analyses of presynaptic membranes were performed in ImageJ by an experimenter blinded to the experimental condition. All synaptic vesicles were counted within a 1-μm radius of the active zone. Cisternae were defined as irregularly shaped, intracellular membranous structures larger than 100 nm in diameter. Cisternal size was determined by measuring the distance around the perimeter. “Total cisternae” was calculated by summing the total amount of membrane within individual cisternae per synapse. Plasma membrane evaginations were measured as previously described (Morgan et al., 2004). A straight line (1 μm) was drawn laterally from the edge of the active zone to the nearest point on the axolemma, and then the curved distance between these two points was measured. After obtaining measurements from the right and left sides of the active zone, we recorded the average for each synapse. Clathrin-coated pits and vesicles were distinguished by the presence of an electron-dense coat. All data shown in the graphs are reported as the mean ± SEM per synapse. Graphs and ANOVA statistics were generated using Origin Pro 7.0 (OriginLab, Northampton, MA).
Each of the three-dimensional reconstructions was generated from five serial sections using Reconstruct software (Fiala, 2005). First, the micrographs were aligned to each other using fiduciary markers. Next the cisternae, active zones, and plasma membranes were traced on each micrograph. Finally, the traces were reconstructed as objects into a three-dimensional scene as either a trace slab or Boissonnat surface, and synaptic vesicles were reconstructed as 50-nm-diameter spheres.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH/National Institute of Neurological Disorder and Stroke RO1 NS078165 (to J.R.M.), the Morton Cure Paralysis Fund (to J.R.M.), and the Branfman Family Foundation (to J.M.G.) and by a Dorothea Bennett graduate fellowship (to D.J.B.). We thank Dwight Romanovicz and the ICMB Microscopy Facility at UT-Austin, as well as Louie Kerr and the Central Microscopy Facility at the MBL, for technical assistance with electron microscopy. We also thank Sreeganga Chandra, Pietro De Camilli, and Subhojit Roy for their helpful comments on the manuscript.
Abbreviations used:
- ANOVA
analysis of variance
- DLB
dementia with Lewy bodies
- GST
glutathione S-transferase
- IgG
immunoglobulin G
- mlv
multilamellar vesicles
- NMR
nuclear magnetic resonance
- NTD
N-terminal domain
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PD
Parkinson's disease
- RS
reticulospinal
- RT
room temperature
- suv
small unilamellar vesicles
- SV2
synaptic vesicle glycoprotein 2.
Footnotes
*Present address: Department of Biomedical Engineering, Cockrell School of Engineering, University of Texas at Austin, Austin, TX 78712.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-02-0708) on October 1, 2014.
REFERENCES
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Andersson F, Jakobsson J, Low P, Shupliakov O, Brodin L. Perturbation of syndapin/PACSIN impairs synaptic vesicle recycling evoked by intense stimulation. J Neurosci. 2008;28:3925–3933. doi: 10.1523/JNEUROSCI.1754-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79:1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, Terada M, Hu J, Bushong EA, Hwang M, Masliah E, George JM, Ellisman MH. Mapping the subcellular distribution of alpha-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson's disease pathogenesis. J Neurosci. 2013;33:2605–2615. doi: 10.1523/JNEUROSCI.2898-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brodin L, Shupliakov O. Giant reticulospinal synapse in lamprey: molecular links between active and periactive zones. Cell Tissue Res. 2006;326:301–310. doi: 10.1007/s00441-006-0216-2. [DOI] [PubMed] [Google Scholar]
- Brodin L, Shupliakov O, Pieribone VA, Hellgren J, Hill RH. The reticulospinal glutamate synapse in lamprey: plasticity and presynaptic variability. J Neurophysiol. 1994;72:592–604. doi: 10.1152/jn.1994.72.2.592. [DOI] [PubMed] [Google Scholar]
- Buchanan JT. Contributions of identifiable neurons and neuron classes to lamprey vertebrate neurobiology. Prog Neurobiol. 2001;63:441–466. doi: 10.1016/s0301-0082(00)00050-2. [DOI] [PubMed] [Google Scholar]
- Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Sharma M, Sudhof TC. Systematic mutagenesis of alpha-synuclein reveals distinct sequence requirements for physiological and pathological activities. J Neurosci. 2012;32:15227–15242. doi: 10.1523/JNEUROSCI.3545-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DJ, Morgan JR. Synuclein accumulation is associated with cell-specific neuronal death after spinal cord injury. J Comp Neurol. 2012;520:1751–1771. doi: 10.1002/cne.23011. [DOI] [PubMed] [Google Scholar]
- Bussell R, Jr., Eliezer D. Effects of Parkinson's disease-linked mutations on the structure of lipid-associated alpha-synuclein. Biochem. 2004;43:4810–4818. doi: 10.1021/bi036135+. [DOI] [PubMed] [Google Scholar]
- Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. A broken alpha-helix in folded alpha-synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, et al. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci USA. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba-Falek O, Lopez GJ, Nussbaum RL. Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Mov Disord. 2006;21:1703–1708. doi: 10.1002/mds.21007. [DOI] [PubMed] [Google Scholar]
- Clayton EL, Cousin MA. The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J Neurochem. 2009;111:901–914. doi: 10.1111/j.1471-4159.2009.06384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, Evans GJ, Cousin MA. Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J Neurosci. 2008;28:6627–6632. doi: 10.1523/JNEUROSCI.1445-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA. Activity-dependent bulk synaptic vesicle endocytosis—a fast, high capacity membrane retrieval mechanism. Mol Neurobiol. 2009;39:185–189. doi: 10.1007/s12035-009-8062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- Di Prisco GV, Pearlstein E, Le Ray D, Robitaille R, Dubuc R. A cellular mechanism for the transformation of a sensory input into a motor command. J Neurosci. 2000;20:8169–8176. doi: 10.1523/JNEUROSCI.20-21-08169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuc R, Brocard F, Antri M, Fenelon K, Gariepy JF, Smetana R, Menard A, Le Ray D, Viana Di Prisco G, Pearlstein E, et al. Initiation of locomotion in lampreys. Brain Res Rev. 2008;57:172–182. doi: 10.1016/j.brainresrev.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Dumitriu A, Moser C, Hadzi TC, Williamson SL, Pacheco CD, Hendricks AE, Latourelle JC, Wilk JB, Destefano AL, Myers RH. Postmortem interval influences alpha-synuclein expression in Parkinson disease brain. Parkinsons Dis. 2012;2012:614212. doi: 10.1155/2012/614212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliezer D, Kutluay E, Bussell R, Jr., Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, McMahon HT. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, Cantoni M, Bensadoun JC, Schneggenburger R, Knott GW, et al. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- George JM. The synucleins. Genome Biol. 2002;3:reviews3002. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci USA. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen H, Fetter RD, Davis GW. Clathrin dependence of synaptic-vesicle formation at the Drosophila neuromuscular junction. Curr Biol. 2008;18:401–409. doi: 10.1016/j.cub.2008.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau BY, Foldes AE, Alieva NO, Oliphint PA, Busch DJ, Morgan JR. Increased synapsin expression and neurite sprouting in lamprey brain after spinal cord injury. Exp Neurol. 2011;228:283–293. doi: 10.1016/j.expneurol.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Decressac M, Mattsson B, Bjorklund A. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci USA. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Comstra HS, Cohen M, Faundez V. Presynaptic membrane retrieval and endosome biology: defining molecularly heterogeneous synaptic vesicles. Cold Spring Harb Perspect Biol. 2013a;5:a016915. doi: 10.1101/cshperspect.a016915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Di Paolo G, Werner H, Shchedrina VA, Pypaert M, Pieribone VA, De Camilli P. A role for talin in presynaptic function. J Cell Biol. 2004;167:43–50. doi: 10.1083/jcb.200406020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Jiang J, Oliphint PA, Jin S, Gimenez LE, Busch DJ, Foldes AE, Zhuo Y, Sousa R, Lafer EM. A role for an Hsp70 nucleotide exchange factor in the regulation of synaptic vesicle endocytosis. J Neurosci. 2013b;33:8009–8021. doi: 10.1523/JNEUROSCI.4505-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Prasad K, Hao W, Augustine GJ, Lafer EM. A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J Neurosci. 2000;20:8667–8676. doi: 10.1523/JNEUROSCI.20-23-08667.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K. α-Synuclein and mitochondria: partners in crime. Neurotherapeutics. 2013;10:391–399. doi: 10.1007/s13311-013-0182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphint PA, Alieva N, Foldes AE, Tytell ED, Lau BY, Pariseau JS, Cohen AH, Morgan JR. Regenerated synapses in lamprey spinal cord are sparse and small even after functional recovery from injury. J Comp Neurol. 2010;518:2854–2872. doi: 10.1002/cne.22368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Woods WS, Clayton DF, George JM. Interaction of human alpha-synuclein and Parkinson's disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem. 2000;275:34393–34398. doi: 10.1074/jbc.M004851200. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Ringstad N, Gad H, Low P, Di Paolo G, Brodin L, Shupliakov O, De Camilli P. Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron. 1999;24:143–154. doi: 10.1016/s0896-6273(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Rovainen CM. Synaptic interactions of identified nerve cells in the spinal cord of the sea lamprey. J Comp Neurol. 1974;154:189–206. doi: 10.1002/cne.901540206. [DOI] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P. Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol. 2012;4:a005645. doi: 10.1101/cshperspect.a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer FE, Ryan TA. The synaptic vesicle: cycle of exocytosis and endocytosis. Curr Opin Neurobiol. 2006;16:298–304. doi: 10.1016/j.conb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Scott D, Roy S. α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior SL, Ninkina N, Deacon R, Bannerman D, Buchman VL, Cragg SJ, Wade-Martins R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci. 2008;27:947–957. doi: 10.1111/j.1460-9568.2008.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Low P, Grabs D, Gad H, Chen H, David C, Takei K, De Camilli P, Brodin L. Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science. 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. α-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Slepnev VI, De Camilli P. Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat Rev Neurosci. 2000;1:161–172. doi: 10.1038/35044540. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, Meshul CK, Unni VK. Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson's disease. J Neurosci. 2014;34:2037–2050. doi: 10.1523/JNEUROSCI.2581-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Gitler AD. Discovery and characterization of three novel synuclein genes in zebrafish. Dev Dyn. 2008;237:2490–2495. doi: 10.1002/dvdy.21569. [DOI] [PubMed] [Google Scholar]
- Surguchov A. Molecular and cellular biology of synucleins. Int Rev Cell Mol Biol. 2008;270:225–317. doi: 10.1016/S1937-6448(08)01406-8. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Bax A. Comparison of structure and dynamics of micelle-bound human alpha-synuclein and Parkinson disease variants. J Biol Chem. 2005;280:43179–43187. doi: 10.1074/jbc.M507624200. [DOI] [PubMed] [Google Scholar]
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34:9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomilin N, Pechstein A, Chau N, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011;146:471–484. doi: 10.1016/j.cell.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Wenzel EM, Morton A, Ebert K, Welzel O, Kornhuber J, Cousin MA, Groemer TW. Key physiological parameters dictate triggering of activity-dependent bulk endocytosis in hippocampal synapses. PLoS One. 2012;7:e38188. doi: 10.1371/journal.pone.0038188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal CH, Chandra SS. Monomeric synucleins generate membrane curvature. J Biol Chem. 2013;288:1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickelgren WO, Leonard JP, Grimes MJ, Clark RD. Ultrastructural correlates of transmitter release in presynaptic areas of lamprey reticulospinal axons. J Neurosci. 1985;5:1188–1201. doi: 10.1523/JNEUROSCI.05-05-01188.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.