

Abstract
DNA double-strand breaks are highly toxic DNA lesions that cause genomic instability, if not efficiently repaired. RecQ helicases are a family of highly conserved proteins that maintain genomic stability through their important roles in several DNA repair pathways, including DNA double-strand break repair. Double-strand breaks can be repaired by homologous recombination (HR) using sister chromatids as templates to facilitate precise DNA repair, or by an HR-independent mechanism known as non-homologous end-joining (NHEJ) (error-prone). NHEJ is a non-templated DNA repair process, in which DNA termini are directly ligated. Canonical NHEJ requires DNA-PKcs and Ku70/80, while alternative NHEJ pathways are DNA-PKcs and Ku70/80 independent. This review discusses the role of RecQ helicases in NHEJ, alternative (or back-up) NHEJ (B-NHEJ) and microhomology-mediated end-joining (MMEJ) in V(D)J recombination, class switch recombination and telomere maintenance.
Keywords: Alternative end-joining, Ku70/80, microhomology-mediated end-joining, non-homologous end-joining, RecQ helicases, telomere
Introduction
The stability of mitochondrial and genomic DNA is critical for viability of eukaryotic cells, and survival of organisms and species. DNA repair pathways exist to counterbalance the adverse impact of nuclear and mitochondrial DNA damage, generated by exogenous and endogenous DNA damaging agents. Such damage results from exposure to genotoxic chemicals, ultraviolet light (UV), ionizing radiation (IR) and reactive oxygen species (ROS). ROS are a considerable source of endogenous DNA damage, such as DNA strand breaks and oxidized DNA bases. ROS are continuously produced in the cell, particularly because they are a byproduct of mitochondrial oxidative phosphorylation. Defects in DNA repair can lead to an increased load of persistent DNA lesions. Dysfunctional DNA repair is strongly associated with aging and increased risk of age-related disease. Unrepaired DNA damage is thought to contribute to the occurrence of cancer and neurodegenerative disease, including Alzheimer’s disease and Huntington’s disease (Jeppesen et al., 2011).
Eukaryotic organisms possess a sophisticated network of DNA repair pathways, some of which provide redundant or overlapping DNA repair functions. Many DNA repair enzymes are multifunctional, playing roles in DNA repair and DNA replication or recombination, as well as roles in development and gene expression. The main DNA excision repair pathways are base excision repair (BER), mismatch repair (MMR) and nucleotide excision repair (NER). The main DNA double strand-break repair pathways are non-homologous end-joining (NHEJ) and homologous recombination (HR).
The RecQ protein family is a highly conserved group of DNA helicases that play essential roles in transcription, DNA replication, DNA recombination and DNA repair. Escherichia coli RecQ is the only RecQ helicase expressed in the bacterial cell (Bernstein & Keck, 2003). However, five RecQ helicases are expressed in human cells: RECQL1, BLM, WRN, RECQL4 and RECQL5 (Singh et al., 2009). Mutations in BLM, WRN and RECQL4 cause the inherited disorders, such as Bloom syndrome (BS) (Online Mendelian Inheritance in Man (OMIM) #210900), Werner syndrome (WS) (OMIM #277700) and Rothmund–Thomson syndrome (RTS) (OMIM #268400), respectively. Although these diseases are clinically distinct, they share some features, including cancer susceptibility and genomic instability. WS and RTS are segmental progerias, in that affected patients manifest many, but not all, of the typical features of aging. Defects in RECQL1 and RECQL5, which code for the other two main RecQ helicases, have not yet been linked to human disease (Singh et al., 2009). This review focuses on the role of RecQ helicases in NHEJ, including canonical DNA-PK-mediated NHEJ (C-NHEJ), alternative (or back-up) NHEJ (B-NHEJ) and microhomology-mediated end-joining (MMEJ) in V(D)J recombination, class switch recombination and telomere maintenance.
DNA repair, aging and cancer
Accumulation of DNA damage and defects in DNA repair are linked to disease, poor health, premature aging and cancer susceptibility (Bohr, 2008; Brosh, 2013; Maynard et al., 2009, 2014; Wilson et al., 2008). For example, rare inherited progeroid syndromes, that mimic normal physiological aging at an early stage in the life of the patient, are caused by mutations in genes encoding proteins that have roles in DNA repair. Defects in DNA repair cause BS, WS, RTS, Cockayne syndrome (CS) (OMIM #133540) and Trichothiodystrophy (TTD) (OMIM #601675) (Croteau et al., 2014; Kitao et al., 1999; Siitonen et al., 2009; Weeda et al., 1997; Yu et al., 1996). Mutations in the lamin A/C gene (LMNA) cause Hutchinson–Gilford progeria (HGPS OMIM #176670), autosomal dominant Emery–Dreifuss muscular dystrophy (OMIM #7894480), restrictive dermopathy (OMIM #275210) and atypical WS (Bione et al., 1994; Chen et al., 2003b; De Sandre-Giovannoli et al., 2003; Navarro et al., 2004). Mutations in CSA and CSB genes, the products of which promote NER, cause CS, which is characterized by premature aging. Mice depleted in either Ku70 or Ku80 genes, that are both essential in the NHEJ pathway, develop aging-like pathology prematurely (Gu et al., 1997; Nussenzweig et al., 1996). Evidence that HR plays a major role in aging is limited; however, defects in BLM, a RecQ helicase with roles in DSB repair and DNA replication, have been found to be associated with increased cancer risk (Pagon et al., 1993). The molecular and cellular phenotypes in patients with premature aging syndromes have provided considerable insight into mechanisms involved in normal aging.
Deficiencies in BER, MMR, NER and NHEJ are thought to contribute to neurological dysfunction associated with the neurodegenerative diseases, such as Alzheimer’s disease and Huntington’s disease (Jeppesen et al., 2011). Weissman and colleagues (2007) showed that BER capacity is lower in postmortem brain tissue from patients with sporadic Alzheimer’s disease, relative to unaffected controls. In addition, the efficiency of BER appeared lower in differentiated neurons (postmitotic cells), relative to undifferentiated neurons (Sykora et al., 2013). Moreover, some recent evidence suggests that defective BER or MMR may contribute to the pathology of Huntington’s disease (Møllersen et al., 2010; Owen et al., 2005; Wheeler et al., 2003).
Characteristics of human RecQ helicases
Bloom syndrome is caused by mutations in the BLM gene. Patients with BS display short stature, immunodeficiency and predisposition to cancer. BLM is a DNA-stimulated ATPase and ATP-dependent 3′ to 5′ DNA helicase and contains strand-annealing activity. Some mutations linked to BS map to helicase motifs and thus can disable BLM helicase activity (Guo et al., 2007; Rong et al., 2000). BLM interacts with several HR proteins including RAD50, MRE11, RAD51 and BRCA1 (Croteau et al., 2014; Davalos & Campisi, 2003; Ding et al., 2009; Franchitto & Pichierri, 2002; Wang et al., 2000). The single-stranded DNA binding protein, replication protein A (RPA), interacts directly with BLM and stimulates its helicase activity (Brosh et al., 2000). BLM also interacts with WRN, FEN1, EXO1, phosphorylated histone H2AX (γ-H2AX), Fanconi anemia group D2 (FANCD2), Ataxia telangiectasia mutated (ATM), telomeric repeat-binding factor 2 (TRF2), Casp3 and p53 (Freire et al., 2001; Nimonkar et al., 2008; Opresko et al., 2002; Sengupta et al., 2003; von Kobbe et al., 2002; Wang & Bambara, 2005; Wang et al., 2001). BLM binds preferentially to single-stranded DNA, Y-shaped DNA and double Holliday Junctions (dHJ) (Mohaghegh et al., 2001). The yeast BLM ortholog Sgs1 stimulates helicase/exonuclease DNA2 during single-stranded DNA annealing (Zhu et al., 2008). Human BLM does not play a major role in the canonical (C-)NHEJ or V(D)J recombination (Chen et al., 2003a; So et al., 2004).
Werner syndrome is caused by mutations in the WRN gene. Patients with WS display cancer susceptibility, short stature, alopecia, atrophic skin, thin gray hair, type II diabetes, osteoporosis, cataracts, arteriosclerosis and atherosclerosis, indicating that WS is a segmental progeria (Yu et al., 1994). WRN possesses intrinsic 3′ to 5′ DNA helicase activity, DNA single-strand DNA annealing activity and 3′ to 5′ exonuclease activity. WRN binds preferentially to single-stranded DNA, Y-shaped DNA, G-quadruplex and dHJ (Brosh et al., 2001; Croteau et al., 2014). In the presence of single-stranded DNA binding protein (SSB) of E. coli, WRN helicase can unwind short DNA duplexes (<53 base pairs) (Shen et al., 1998), whereas, human SSB protein, RPA, stimulates WRN to catalyze unwinding of long duplex DNA substrates up to 849 base pairs (Brosh et al., 1999; Shen et al., 1998). The 3′ to 5′ exonuclease activity of WRN is stimulated by Ku70/80 (Cooper et al., 2000). WRN contains a nuclear localization signal in its C-terminal region as well as a nucleolar localization signal (Marciniak et al., 1998). In response to DNA damage, WRN translocates out of the nucleolus to perform its enzymatic function (Indig et al., 2012). WRN interacts with RAD52, NBS1, Ku70/80, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Ligase IV/XRCC4 (Baynton et al., 2003; Cheng et al., 2004; Karmakar et al., 2002a, b; Kusumoto et al., 2008), FEN1, PARP1, BLM, TRF1, TRF2 and p53 (Blander et al., 1999; Opresko et al., 2002, 2004; von Kobbe et al., 2002, 2003, 2004a). In cells lacking WRN, the frequency of chromosomal translocations increases (Chen et al., 2003a).
RECQL4 has been less extensively studied than BLM and WRN. Defects in RECQL4 are associated with three distinct clinical diseases, Rothmund–Thomson Syndrome (RTS), Rapadilino (RAPA) (OMIM #266280) and Baller–Gerold syndrome (BGS) (OMIM #218600). Of those patients with RTS, two-thirds have mutations in the RECQL4 gene while the remainder have mutations in gene(s) yet to be identified (Wang et al., 2003b). Patient with either RTS or RAPA are at increased risk for osteosarcomas and lymphomas (Larizza et al., 2010; Siitonen et al., 2009; Wang et al., 2003b).
RECQL4 is a 3′ to 5′ helicase and stimulates DNA strand displacement in the presence of ATP (Macris et al., 2006). RECQL4 function is regulated in part by interaction with RPA and BLM. RPA stimulates the RECQL4 helicase activity by approximately 2-fold on forked substrates (Rossi et al., 2010). RECQL4 and BLM function cooperatively at forked substrates in vitro, and interact physically during S-phase (Singh et al., 2012). WRN, RECQL4 and RECQL5 promote repair of ssDNA breaks and oxidatively damaged DNA bases (Szekely et al., 2005; Tadokoro et al., 2012; von Kobbe et al., 2004b; Werner et al., 2006; Woo et al., 2006). RTS cells show an accumulation of single stranded DNA breaks after peroxide treatment. RECQL4 interacts physically with several BER proteins, stimulates APE1 and FEN1 activities and modulates POLβ ligase activity (Schurman et al., 2009). RECQL4 functions in telomeric maintenance and interacts with the shelterin proteins TRF1 and TRF2. RECQL4 also interacts physically with PARP1, XPA and WRN (Croteau et al., 2012; Fan & Luo, 2008; Ghosh et al., 2012; Petkovic et al., 2005). While RECLQ4 is known to interact with and modulate the activity of repair proteins, it also plays a specialized role in DNA replication initiation (Capp et al., 2009; Collart et al., 2013; Thangavel et al., 2010; Wu et al., 2008a; Xu et al., 2009a,b).
RECQL1 is the most abundant RecQ helicase (Kitao et al., 1998). The RECQL1 and WRN 3′ to 5′ DNA helicase activities unwind the leading strand of forked DNA substrates, while RECQL4 and RECQL5β only unwind the lagging strand (Popuri et al., 2012a). RECQL1 alone is able to unwind short DNA duplexes (<110 base pairs), whereas it requires the presence of RPA to unwind longer substrates (501 base pairs) (Cui et al., 2003, 2004). RECQL1 is thought to stimulate PARP1-mediated DNA replication restart (Berti et al., 2013). Defects in RECQL1 lead to hyper-phosphorylation of RPA32 and activation of CHK1 (Popuri et al., 2012a). DSBs and sister chromatid exchange (SCE) are more pronounced in RECQL1-deficient mouse embryo fibroblasts (Sharma & Brosh, 2007). RECQL1 also appears to be an important component in the cellular response to oxidative damage, as evidenced by the observation that it is rapidly recruited to oxidative DNA lesions and that RECQL1-deficient cells are sensitive to oxidative damage (Sharma et al., 2012).
RECQL5 encodes three RecQ isoforms, RECQL5α, RECQL5β and RECQL5γ. RECQL5α has strand annealing activity (Ren et al., 2008), RECQL5β has 3′ to 5′ DNA helicase activity and ATPase activity, and no biochemical functions have been attributed to RECQL5γ. RPA stimulates RECQL5β helicase activity but inhibits RECQL5β ssDNA-annealing activity (Garcia et al., 2004). RECQL5β is thought to be active during G0 and G1, and may play significant roles in DNA replication, DNA recombination and RNA transcription (Aygün et al., 2008; Kawabe et al., 2000; Kitao et al., 1998; Zheng et al., 2009). RECQL5β may play significant roles in HR, BER and interstrand cross-link repair (Ramamoorthy et al., 2013; Tadokoro et al., 2012; Zheng et al., 2009). RECQL5β directly interacts with RAD51, thereby inhibiting RAD51-mediated D-loop formation (Schwendener et al., 2010). RECQL5β and RECQL1 helicases lack the winged-helix motif, which plays an essential role in DNA unwinding. In addition, the RECQL5β helicase inefficiently resolves dHJ (Croteau et al., 2014; Garcia et al., 2004).
RECQL5β is constitutively expressed in all cell types throughout the cell cycle, while expression of WRN, BLM and RECQL4 is cell cycle-dependent and tissue-specific (Kawabe et al., 2000; Sanz et al., 2000). The cell cycle-independent expression of RECQL5β likely reflects the important role that RECQL5β plays in transcription in resting cells, via its interaction with the C-terminal domain of RNA polymerase II (RNA pol II) (Aygün et al., 2008, 2009; Izumikawa et al., 2008). Interestingly, RECQL5β has two RNA polymerase II interaction motifs, the KIX and SRI domains, which both mediate binding to RNA pol II. A recent crystal structure analysis suggests that the KIX domain on RECQL5 and the transcription elongation factor, TFIIS, share overlapping binding sites on RNA pol II, such that when RECQL5β occupies the site, TFIIS cannot stimulate transcription elongation (Kassube et al., 2013). Thus, the absence of RECQL5 leads to transcriptional stress across the genome, underscoring RECQL5β’s importance during transcription (Saponaro et al., 2014).
Canonical NHEJ
Canonical NHEJ (C-NHEJ) is one of the error-prone DSB repair mechanisms and it is constitutively active throughout the cell cycle (Helleday et al., 2007; Rothkamm & Löbrich, 2003). Core C-NHEJ factors are Ku70/80, DNA-PKcs, XLF and Ligase IV/XRCC4 (Figure 1) (Weterings & Chen, 2007). Ku70/80 has a high affinity for DNA DSBs (Walker et al., 2001) and forms a stable complex with DNA-PKcs that protects DSBs from degradation (Weterings et al., 2003). DNA-PKcs, a PI3 kinase, binds the Ku70/80-DSB complex, which triggers DNA-PKcs autophosphorylation and phosphorylation of additional DSB repair factors (Ding et al., 2003; Reddy et al., 2004). DNA termini are resected and/or extended by DNA polymerases Pol μ and Pol λ (Nick McElhinny et al., 2005), and XLF and the Ligase IV/XRCC4 complex are recruited to the DNA lesion, where XLF interacts with XRCC4 and stimulates re-adenylation of Ligase IV (Ahnesorg et al., 2006; Riballo et al., 2009; Yano et al., 2008). Ligase IV/XRCC4 then ligates the remaining nick. DSBs that require no resection (ligatable DNA ends) are repaired by Ku70/80 and XRCC4/Ligase IV/XLF, independent of DNA-PKcs (Mari et al., 2006; Reynolds et al., 2012). Extensive work on WRN in C-NHEJ, demonstrates that WRN physically and functionally interacts with several C-NHEJ repair proteins (Karmakar et al., 2002b; Kusumoto-Matsuo et al., 2010, 2014). Ku70/80 and Ligase IV/XRCC4 stimulate WRN exonuclease but not WRN helicase activity (Cooper et al., 2000; Kusumoto et al., 2008; Li & Comai, 2001), clear demonstrations of how protein interactions dictate specificity of enzyme activities. Besides WRN, RECQL1 plays a major role in the initial steps of C-NHEJ. This is evidenced by data showing that it physically and functionally interacts with the heterodimer Ku70/80. Moreover, depletion of RECQL1 resulted in reduced end joining in cell-free extracts (Parvathaneni et al., 2013).
Figure 1.
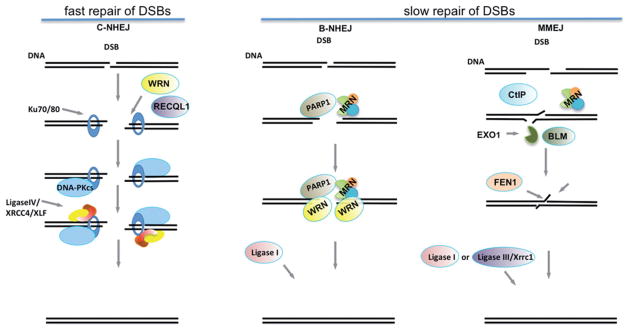
Schematic diagram of DNA end-joining pathways. C-NHEJ, canonical non-homologous end-joining; B-NHEJ, alternative non-homologous end-joining; MMEJ, micro-mediated end-joining; MRN, MRE11-RAD50-NBS1 complex. EXO1/BLM/MRN or BLM/DNA2/MRN carry out CtIP-stimulated end-resection during MMEJ. (see colour version of this figure at www.informahealthcare.com/bmg).
V(D)J recombination
C-NHEJ contributes to V(D)J recombination, the process by which genes encoding immunoglobulin and T-cell receptors are diversified in B and T cells in the bone marrow (Gellert, 2002). The recombination-activating genes 1 and 2 (RAG1 and RAG2) recognize and cleave a signal sequence in these genes, after which the cleaved DNA rearranges into a DNA hairpin that bridges the newly-formed DNA termini. This structure is resolved in a manner that generates diversity at the cleavage site by a process that requires enzymes that catalyze C-NHEJ, namely Artemis and CtIP (Bothmer et al., 2013; Ma et al., 2002; Malu et al., 2012; Weterings et al., 2009). WRN, BLM, RECQL4 and RECQL1 are differentially up-regulated to guarantee genomic stability in proliferating B cells (Kawabe et al., 2000). The different expression levels of the various helicases often reflect ongoing cell cycle-dependent cellular processes. For example, BLM is upregulated during replication to suppress sister chromatid exchange. RECQL1 expression is reduced during G0 phase, due to distinctive roles from BLM and RECQL4 during cellular proliferation and maintenance of chromosomal stability (Kawabe et al., 2000; Popuri et al., 2008; Sharma & Brosh, 2007; Thangavel et al., 2010). In resting B cells, RECQL4 and WRN have a low level of expression, in contrast to their expression levels in proliferating B cells (S-phase). Increased levels of RECQL4 and WRN during S-phase may reflect their important roles in replication and could promote genomic stability by way of their roles in promoting DSB repair (Kawabe et al., 2000; Singh et al., 2010).
Alternative NHEJ
The alternative (i.e. back-up) NHEJ (B-NHEJ), substitutes for C-NHEJ when C-NHEJ is defective (Fattah et al., 2010; Ma et al., 2003; Wang et al., 2003a). WRN is the only RecQ helicase thought to contribute to B-NHEJ (Sallmyr et al., 2008). B-NHEJ is error-prone and independent of DNA homology for templating DSB repair (Mansour et al., 2010). It has been suggested that B-NHEJ is more error-prone than C-NHEJ because it is slower in the repair of DNA (Durante et al., 2013). Consistent with this idea, cells from mice deleted in the genes Ku70, Ku80 or Xrcc4 have higher rates of chromosomal translocations than wild-type control (Difilippantonio et al., 2000; Ferguson et al., 2000; Simsek & Jasin, 2010). B-NHEJ is active throughout the cell cycle, although it appears to be higher during G2 in cells that lack C-NHEJ (Wu et al., 2008b). C-NHEJ and B-NHEJ may be simultaneous/parallel pathways in normal cells; it is not clear whether the pathways work in tandem, or whether B-NHEJ primarily acts as a backup system when C-NHEJ is compromised. B-NHEJ enzymes include MRE11 of the MRE11-RAD50-NBS1 (MRN) complex, PARP1, WRN, Ligase I and DNA polymerase θ (Pol θ), a polymerase of the Y family (Figure 1) (Parsons et al., 2005; Paull & Gellert, 1998; Sallmyr et al., 2008; Simsek et al., 2011). WRN helicase, exonuclease and single-stranded DNA annealing activities may contribute to B-NHEJ. Pol θ extends mismatched primer termini and can bypass abasic sites (Chan et al., 2010; Hogg et al., 2012). The contribution of the enzyme CtBP-interacting protein (CtIP) to B-NHEJ is still not understood, while its role in microhomology-mediated end-joining (MMEJ) is more clear (Durante et al., 2013). A number of studies have used expression of oncogenic BCR-ABL kinase, a fusion protein linked to chronic myeloid leukemia (CML), as a tool to inhibit C-NHEJ and study B-NHEJ (Sallmyr et al., 2008; Sattler et al., 2000). For example, CML BCR-ABL cells show a higher expression levels of WRN and ligase III, as well as residual DNA breaks and large deletions, which indicates active roles for these enzymes in B-NHEJ (Sallmyr et al., 2008; Slupianek et al., 2011). Moreover, downregulation of WRN and Ligase III in CML cell lines resulted in accumulation of unrepaired DNA.
Microhomology-mediated end-joining
Microhomology-mediated end-joining (MMEJ) is an error-prone DNA repair mechanism targeted to long stretches of ssDNA, and thought to be more mutagenic than C-NHEJ and B-NHEJ (Ma et al., 2003). MMEJ does not require Ku70/80, DNA-PKcs or Ligase IV/XRCC4, but does require BLM (Nussenzweig & Nussenzweig, 2007; Yu & Gabriel, 2003; Yu & McVey, 2010). An important role of BLM is to inhibit CtIP/MRE11 (Grabarz et al., 2013). MMEJ occurs during G2-S or G1 (Truong et al., 2013). It processes ssDNA gaps of 2 to 25 nucleotides in length (McVey & Lee, 2008), while ssDNA gaps >30 nucleotides are repaired by ssDNA annealing (SSA). Proteins involved in MMEJ include BLM/MRN, EXO1 or DNA2, FEN1, DNA polymerase β, λ or μ, Ligase I or Ligase III/XRCC1 and MMR proteins (Figure 1) (Crespan et al., 2012; Lee-Theilen et al., 2011; Nimonkar et al., 2011; Paul et al., 2013). After ssDNA is annealed in the gap region, mismatched bases are corrected by MMR. DNA polymerase β, λ or μ fills gaps and FEN1 processes the overhangs.
53BP1 and γH2AX are DNA damage sensors that also play a major role in class switch recombination (CSR). Lymphocytes depleted of 53BP1 or γH2AX display impaired CSR, due to the lack of protection of broken DNA ends, which results in enhanced resection-associated alternative end joining (MMEJ) and inhibition of C-NHEJ repair (Bothmer et al., 2013). It has been reported that phosphorylated 53BP1 and BLM interact directly during DSB repair (Sengupta et al., 2004; Tripathi et al., 2008). Moreover, 53BP1 and TopIIIα have been shown to regulate BLM in cell cycle-dependent manner (Grabarz et al., 2013). In cells absent of 53BP1 or RIF, BLM promotes long deleterious resection mediated by CtIP/MRN (Grabarz et al., 2013). It has been proposed that RECQL5β can substitute for BLM in DSB repair (Branzei & Foiani, 2007), and that RECQL1 or RECQL5β can substitute for BLM to suppress SCE. Mouse ES cells with knockout of one allele of Blm and Recql5 had a higher rate of SCE after UV-induced DNA damage than cells with knockout of both alleles of Blm or Recql5 (i.e. single gene KO) (Hu et al., 2005). Recql5-deficient mouse ES and MEF cells show a profound reduction in DNA replication after the treatment with, topoisomerase I inhibitor, camptothecin (Hu et al., 2009). However, BLM and RECQL5β do not interact directly, and there is no evidence to date that RECQL5β plays a role in B-NHEJ or MMEJ. For a more extensive discussion of the role of RECQL5β in HR, see Popuri et al. (2013).
Class switch recombination
Immature B-cells express B-cell receptors (BCR) on the surface and undergo maturation via class switch recombination (CSR) after migration from the bone marrow to spleen and lymph nodes. During BCR maturation, targeted recombination is carried out in the switch (S) region of immunoglobulin heavy chain genes. CSR takes place in B-cells during G1 and is initiated by activation-induced deaminase (AID). AID deaminates cytosine to uracil in the switch region, leading to a mutagenic process mediated by uracil-DNA glycosylase, and enzymes of MMR, C-NHEJ and/or MMEJ (reviewed in Kotnis et al. 2009). Xrcc4−/− mice show increased resection at DNA termini in the cleaved S region, which can promote oncogenic translocations (Boboila et al., 2010; Simsek & Jasin, 2010), but CSR still occurs. A recent study by Bothmer and co-workers (2013) investigated the contribution of WRN and BLM to CSR. Their results suggest that resection depends on both CtIP and EXO1. Additionally, inhibition of CtIP partially rescues the CSR defect in 53BP1-and H2AX-deficient lymphocytes as does interference with the RecQ helicases BLM and WRN. They concluded that BLM and WRN may contribute to the repair of AID-mediated DNA lesions that occur within the repetitive G-rich and highly transcribed switch regions in B lymphocytes, and that minimizing resection favors efficient CSR (Bothmer et al., 2013; Stavnezer et al., 2008). In addition, Babbe and colleagues (2009) showed that BLM-deficient lymphocytes show altered CSR in Iga1, Iga2 and Iga3 genes in splenic B cells, and BLM-deficient B cells have a mild shift towards MMEJ. Additionally, they found that p53-deficient conditional Blm mice showed an increase in propensity for B cell lymphoma development, and that the cells from these mice showed impaired cell cycle progression and survival and high rates of chromosomal structure abnormalities. Their data suggests that BLM and p53 cooperate in avoiding lymphoma development and in maintaining chromosomal stability (Babbe et al., 2009).
C-NHEJ in human disease
Mice depleted in Ku70, Ku80, Xrcc4 or DNA-PKcs show premature aging. The Ku70−/− and Ku80−/− mice are defective in NHEJ, and display premature aging characterized by osteoporosis, growth failure, incomplete plate closure, atopic skin disease, liver pathology, sepsis, cancer and short life span (Gu et al., 1997; Nussenzweig et al., 1996, 1997). In Ku70−/− and Ku80−/− mice, V(D)J rearrangement is defective (Gu et al., 1997; Ouyang et al., 1997). In humans, a mutant allele of DNA-PKcs causes severe combined immune disease (SCID), by way of its inability to activate Artemis endonuclease (van der Burg et al., 2009). Recent reports suggest that impairment of Ku70 in DNA repair is associated with Huntington’s disease (Enokido et al., 2010; Tamura et al., 2011). In humans, no disease has been directly linked to defects in Ku70 or Ku80.
Telomere maintenance
DNA repair enzymes involved in telomere maintenance include Ku70/80, DNA-PKcs, MRN, BLM, WRN and RECQL4. When telomere length or structure is improperly maintained, uncapped telomeres are recognized by DNA damage response proteins, such as 53BP1 and γ-H2AX. If telomeres are not restored, genetic rearrangements and mutations can accumulate. Cancerous cells, which are deficient in telomere shortening, have activated telomerase or activate the pathway of alternative lengthening of telomeres (ALT) resulting in the so called ALT cells (Bechter et al., 2004; Blasco, 2005). Cells that are immortalized by Simian Virus 40 (SV40) or Epstein Barr Virus (EBV) infection activate the ALT pathway or disregulate the shelterin complex, leading in both cases to the formation of ALT cells (Bechter et al., 2004; Kamranvar et al., 2013). The frequency of telomere fusion is higher in ALT cells than in control cells, and it has been suggested that Ku70/80 plays a major role in telomere fusion in ALT cells (Espejel et al., 2002). However, telomere fusion also occurs in Ku70−/−/80−/− MEFs depleted in shelterin proteins TRF1/2 (Rai et al., 2010), suggesting that telomere fusion is supported by an alternative end joining pathway (Indiviglio & Bertuch, 2009; Sfeir & de Lange, 2012). PARP1 promotes B-NHEJ. This is evidenced by the fact that Parp1−/− MEFs show an increase in chromosome end-to-end fusions or chromosome ends without detectable telomeric DNA after induction of DNA damage (Gomez et al., 2006). PARP1 associates with TRF2, and is capable of poly(ADP-ribosyl)ation of TRF2, which affects its binding to telomeric DNA (Gomez et al., 2006). This demonstrates that neither C- nor B-NHEJ, but MMEJ has a role in the telomeric fusion (Sfeir & de Lange, 2012). Furthermore, 53bp1−/− MEFs depleted in TRF1/2 or Ku70/80 show increased resection of 5′ telomere ends, likely mediated by CtIP/BLM/EXO1 (Sfeir & de Lange, 2012). One interpretation of these data is that the shelterin complex plays a primary role in protecting 5′ telomeric ends, while Ku70/80 and 53BP1 play a secondary role by suppressing B-NHEJ (Rai et al., 2010; Sfeir & de Lange, 2012).
In ALT cells, WRN and BLM helicases have clear roles in telomere maintenance. WRN unwinds G-quaduplex DNA during telomere replication, and facilitates formation and resolution of T-loops (Paeschke et al., 2010). WRN also promotes repair of oxidative lesions in D-loops (Ghosh et al., 2009). In WRN−/− ALT cells, BLM suppresses SCE in telomeric DNA, which suggests that BLM does not need WRN to suppress SCE (Mendez-Bermudez et al., 2012). Recently, it was shown that ALT cell induction is positively regulated by the proteins RAD17 and BLM, and negatively regulated by EXO1 and DNA2 (O’Sullivan et al., 2014). DNA-PKcs cooperates with WRN in D-loops and prevents shortening of the telomeric G-strands in vivo (Kusumoto-Matsuo et al., 2010). Cells from RTS patients accumulate DNA fragile sites in telomeres (Ghosh et al., 2012), suggesting a significant role for RECQL4. In telomere maintenance, WRN and RECQL4 may both have specialized roles during telomere replication (Ghosh et al., 2012). In vitro assays suggest that RECQL4 may be stimulated by either TRF1/2 or by WRN, and this may play a major role during unwinding of D-loops during telomere replication (Ghosh et al., 2012).
Conclusion and perspectives
This review summarizes the roles of human RecQ helicases in C-NHEJ, B-NHEJ and other important DNA end-joining processes in human cells (Figure 2). The data from several publications demonstrate that RecQ helicases are dynamic, multifunctional proteins, playing numerous roles in DNA metabolism. There is significant crosstalk among the pathways, as RecQ helicases cooperate and/or complement each other during DNA end-joining. MMEJ is less well characterized than other DNA end-joining pathways; however, the role of BLM in stimulating the exonuclease EXO1 and DNA2 during MMEJ is well-documented. The roles for human RecQ helicases in CSR are not yet well understood. Future studies should focus on RECQL1, RECQL4 and RECQL5β, whose biological roles are less well characterized than BLM and WRN. While not the subject of this review, RECQL4 has been implicated in BER (Kumata et al., 2007; Schurman et al., 2009), and RECQL5β may play a major role in HR-dependent DSB repair, especially after replication fork collapse (Popuri et al., 2012b, 2013; Schwendener et al., 2010).
Figure 2.

BLM, WRN, RECQL4, RECQL1 and RECQL5β in DNA end-joining, DNA homologous recombination and DNA replication. Only BLM is known to play a major role in MMEJ, SSA and CSR. (see colour version of this figure at www.informahealthcare.com/bmg).
Given the critical roles of RecQ helicases in the response to genotoxic stress, it appears that they may be master regulators of the response to genomic instability. The abundant literature in this regard highlights the importance of DNA damage and DNA repair in aging. A corollary of this fact is that DNA repair enzymes, including RecQ helicases, prevent aging-associated disease at all life stages.
Acknowledgments
The authors thank Drs Miriam Sander and Tomasz Kulikowicz for critical reading of the review.
This work was supported in part by the Intramural Research Program of the NIH, National Institute on Aging, as well as a grant from the Nordea Foundation.
Footnotes
Declaration of interest
The authors declare no conflict of interest.
References
- Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–13. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Aygün O, Svejstrup J, Liu Y. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc Natl Acad Sci USA. 2008;105:8580–4. doi: 10.1073/pnas.0804424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aygün O, Xu X, Liu Y, et al. Direct inhibition of RNA polymerase II transcription by RECQL5. J Biol Chem. 2009;284:23197–203. doi: 10.1074/jbc.M109.015750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbe H, McMenamin J, Hobeika E, et al. Genomic instability resulting from Blm deficiency compromises development, maintenance, and function of the B cell lineage. J Immunol. 2009;182:347–60. doi: 10.4049/jimmunol.182.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baynton K, Otterlei M, Bjørås M, et al. WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem. 2003;278:36476–86. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- Bechter OE, Zou Y, Walker W, et al. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004;64:3444–51. doi: 10.1158/0008-5472.CAN-04-0323. [DOI] [PubMed] [Google Scholar]
- Bernstein DA, Keck JL. Domain mapping of Escherichia coli RecQ defines the roles of conserved N- and C-terminal regions in the RecQ family. Nucleic Acids Res. 2003;31:2778–85. doi: 10.1093/nar/gkg376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347–54. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–27. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- Blander G, Kipnis J, Leal JF, et al. Physical and functional interaction between p53 and the Werner’s syndrome protein. J Biol Chem. 1999;274:29463–9. doi: 10.1074/jbc.274.41.29463. [DOI] [PubMed] [Google Scholar]
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6:611–22. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- Boboila C, Yan C, Wesemann DR, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med. 2010;207:417–27. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–20. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Rommel PC, Gazumyan A, et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J Exp Med. 2013;210:115–23. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev. 2007;21:3019–26. doi: 10.1101/gad.1624707. [DOI] [PubMed] [Google Scholar]
- Brosh RM. DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013;13:542–58. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Li JL, Kenny MK, et al. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem. 2000;275:23500–8. doi: 10.1074/jbc.M001557200. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Majumdar A, Desai S, et al. Unwinding of a DNA triple helix by the Werner and Bloom syndrome helicases. J Biol Chem. 2001;276:3024–30. doi: 10.1074/jbc.M006784200. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Orren DK, Nehlin JO, et al. Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem. 1999;274:18341–50. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- Capp C, Wu J, Hsieh T-S. Drosophila RecQ4 has a 3″–5″ DNA helicase activity that is essential for viability. J Biol Chem. 2009;284:30845–52. doi: 10.1074/jbc.M109.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Yu AM, McVey M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet. 2010;6:e1001005. doi: 10.1371/journal.pgen.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang S, Lee L, et al. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003a;2:191–9. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atypical Werner’s syndrome. Lancet. 2003b;362:440–5. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- Cheng W-H, von Kobbe C, Opresko PL, et al. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem. 2004;279:21169–76. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- Collart C, Allen GE, Bradshaw CR, et al. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science. 2013;341:893–6. doi: 10.1126/science.1241530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MP, Machwe A, Orren DK, et al. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–12. [PMC free article] [PubMed] [Google Scholar]
- Crespan E, Czabany T, Maga G, Hübscher U. Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases λ and β on normal and repetitive DNA sequences. Nucleic Acids Res. 2012;40:5577–90. doi: 10.1093/nar/gks186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–52. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Singh DK, Hoh Ferrarelli L, et al. RECQL4 in genomic instability and aging. Trends Genet. 2012;28:624–31. doi: 10.1016/j.tig.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Arosio D, Doherty KM, et al. Analysis of the unwinding activity of the dimeric RECQ1 helicase in the presence of human replication protein A. Nucleic Acids Res. 2004;32:2158–70. doi: 10.1093/nar/gkh540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Klima R, Ochem A, et al. Characterization of the DNA-unwinding activity of human RECQ1, a helicase specifically stimulated by human replication protein A. J Biol Chem. 2003;278:1424–32. doi: 10.1074/jbc.M209407200. [DOI] [PubMed] [Google Scholar]
- Davalos AR, Campisi J. Bloom syndrome cells undergo p53-dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J Biol Chem. 2003;162:1197–209. doi: 10.1083/jcb.200304016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Difilippantonio MJ, Zhu J, Chen HT, et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404:510–14. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Reddy YVR, Wang W, et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol. 2003;23:5836–48. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S-L, Yu J-C, Chen S-T, et al. Genetic variants of BLM interact with RAD51 to increase breast cancer susceptibility. Carcinogenesis. 2009;30:43–9. doi: 10.1093/carcin/bgn233. [DOI] [PubMed] [Google Scholar]
- Durante M, Bedford JS, Chen DJ, et al. From DNA damage to chromosome aberrations: joining the break. Mut Res. 2013;756:5–13. doi: 10.1016/j.mrgentox.2013.05.014. [DOI] [PubMed] [Google Scholar]
- Enokido Y, Tamura T, Ito H, et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol. 2010;189:425–43. doi: 10.1083/jcb.200905138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejel S, Franco S, Rodríguez-Perales S, et al. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J. 2002;21:2207–19. doi: 10.1093/emboj/21.9.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Luo J. RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA) J Biol Chem. 2008;283:29037–44. doi: 10.1074/jbc.M801928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattah F, Lee EH, Weisensel N, et al. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson DO, Sekiguchi JM, Chang S, et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci USA. 2000;97:6630–3. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchitto A, Pichierri P. Bloom’s syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J Cell Biol. 2002;157:19–30. doi: 10.1083/jcb.200110009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire R, d’Adda Di Fagagna F, Wu L, et al. Cleavage of the Bloom’s syndrome gene product during apoptosis by caspase-3 results in an impaired interaction with topoisomerase III alpha. Nucleic Acids Res. 2001;29:3172–80. doi: 10.1093/nar/29.15.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia PL, Liu Y, Jiricny J, et al. Human RECQ5beta, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J. 2004;23:2882–91. doi: 10.1038/sj.emboj.7600301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem. 2002;71:101–32. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Rossi ML, Aulds J, et al. Telomeric D-loops containing 8-oxo-2′-deoxyguanosine are preferred substrates for Werner and Bloom syndrome helicases and are bound by POT1. J Biol Chem. 2009;284:31074–84. doi: 10.1074/jbc.M109.027532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Rossi ML, Singh DK, et al. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. doi: 10.1074/jbc.M111.295063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez M, Wu J, Schreiber V, et al. PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol Biol Cell. 2006;17:1686–96. doi: 10.1091/mbc.E05-07-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabarz A, Guirouilh-Barbat J, Barascu A, et al. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013;5:21–8. doi: 10.1016/j.celrep.2013.08.034. [DOI] [PubMed] [Google Scholar]
- Gu Y, Seidl KJ, Rathbun GA, et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–65. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- Guo R-B, Rigolet P, Ren H, et al. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35:6297–310. doi: 10.1093/nar/gkm536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Lo J, van Gent DC, Engelward BP. DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 2007;6:923–35. doi: 10.1016/j.dnarep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Hogg M, Sauer-Eriksson AE, Johansson E. Promiscuous DNA synthesis by human DNA polymerase θ. Nucleic Acids Res. 2012;40:2611–22. doi: 10.1093/nar/gkr1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Lu X, Barnes E, et al. Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol Cell Biol. 2005;25:3431–42. doi: 10.1128/MCB.25.9.3431-3442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Lu X, Zhou G, et al. Recql5 plays an important role in DNA replication and cell survival after camptothecin treatment. Mol Biol Cell. 2009;20:114–23. doi: 10.1091/mbc.E08-06-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indig FE, Rybanska I, Karmakar P, et al. Nucleolin inhibits G4 oligonucleotide unwinding by Werner helicase. PLoS One. 2012;7:e35229. doi: 10.1371/journal.pone.0035229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indiviglio SM, Bertuch AA. Ku’s essential role in keeping telomeres intact. Proc Natl Acad Sci USA. 2009;106:12217–18. doi: 10.1073/pnas.0906427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumikawa K, Yanagida M, Hayano T, et al. Association of human DNA helicase RecQ5beta with RNA polymerase II and its possible role in transcription. Biochem J. 2008;413:505–16. doi: 10.1042/BJ20071392. [DOI] [PubMed] [Google Scholar]
- Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 2011;94:166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamranvar SA, Chen X, Masucci MG. Telomere dysfunction and activation of alternative lengthening of telomeres in B-lymphocytes infected by Epstein-Barr virus. Oncogene. 2013;32:5522–30. doi: 10.1038/onc.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar P, Piotrowski J, Brosh RM, et al. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J Biol Chem. 2002a;277:18291–302. doi: 10.1074/jbc.M111523200. [DOI] [PubMed] [Google Scholar]
- Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 2002b;30:3583–91. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassube SA, Jinek M, Fang J, et al. Structural mimicry in transcription regulation of human RNA polymerase II by the DNA helicase RECQL5. Nat Struct Mol Biol. 2013;20:892–9. doi: 10.1038/nsmb.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe T, Tsuyama N, Kitao S, et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene. 2000;19:4764–72. doi: 10.1038/sj.onc.1203841. [DOI] [PubMed] [Google Scholar]
- Kitao S, Lindor NM, Shiratori M, et al. Rothmund-Thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999;61:268–76. doi: 10.1006/geno.1999.5959. [DOI] [PubMed] [Google Scholar]
- Kitao S, Ohsugi I, Ichikawa K, et al. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics. 1998;54:443–52. doi: 10.1006/geno.1998.5595. [DOI] [PubMed] [Google Scholar]
- Kotnis A, Du L, Liu C, et al. Non-homologous end joining in class switch recombination: the beginning of the end. Philos Trans R Soc Lond B Biol Sci. 2009;364:653–65. doi: 10.1098/rstb.2008.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumata Y, Tada S, Yamanada Y, et al. Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochim Biophys Acta. 2007;1773:556–64. doi: 10.1016/j.bbamcr.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Kusumoto R, Dawut L, Marchetti C, et al. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry. 2008;47:7548–56. doi: 10.1021/bi702325t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumoto-Matsuo R, Ghosh D, Karmakar P, et al. Serines 440 and 467 in the Werner syndrome protein are phosphorylated by DNA-PK and affects its dynamics in response to DNA double strand breaks. Aging (Albany NY) 2014;6:70–81. doi: 10.18632/aging.100629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumoto-Matsuo R, Opresko PL, Ramsden D, et al. Cooperation of DNA-PKcs and WRN helicase in the maintenance of telomeric D-loops. Aging (Albany NY) 2010;2:274–84. doi: 10.18632/aging.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Theilen M, Matthews AJ, Kelly D, et al. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol. 2011;18:75–9. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Comai L. Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-Ku70/80 complex. J Biol Chem. 2001;276:9896–902. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- Ma J-L, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol. 2003;23:8820–8. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–94. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- Macris MA, Krejci L, Bussen W, et al. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA Repair (Amst) 2006;5:172–80. doi: 10.1016/j.dnarep.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Malu S, De Ioannes P, Kozlov M, et al. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J Exp Med. 2012;209:955–63. doi: 10.1084/jem.20111437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour WY, Rhein T, Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010;38:6065–77. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak RA, Lombard DB, Johnson FB, Guarente L. Nucleolar localization of the Werner syndrome protein in human cells. Proc Natl Acad Sci USA. 1998;95:6887–92. doi: 10.1073/pnas.95.12.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari P-O, Florea BI, Persengiev SP, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci USA. 2006;103:18597–602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard S, Keijzers G, Hansen A-M, et al. Associations of subjective vitality with DNA damage, cardiovascular risk factors and physical performance. Acta Physiol (Oxf) 2014 doi: 10.1111/apha.12296. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard S, Schurman SH, Harboe C, et al. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009;30:2–10. doi: 10.1093/carcin/bgn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Bermudez A, Hidalgo-Bravo A, Cotton VE, et al. The roles of WRN and BLM RecQ helicases in the alternative lengthening of telomeres. Nucleic Acids Res. 2012;40:10809–20. doi: 10.1093/nar/gks862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh P, Karow JK, Brosh RM, et al. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–9. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møllersen L, Rowe AD, Larsen E, et al. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 2010;6:e1001242. doi: 10.1371/journal.pgen.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny SA, Havener JM, Garcia-Diaz M, et al. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol Cell. 2005;19:357–66. doi: 10.1016/j.molcel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Ozsoy AZ, Genschel J, et al. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–11. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, Chen C, da Costa Soares V, et al. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature. 1996;382:551–5. doi: 10.1038/382551a0. [DOI] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131:223–5. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Nussenzweig A, Sokol K, Burgman P, et al. Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: the effects of ionizing radiation on growth, survival, and development. Proc Natl Acad Sci USA. 1997;94:13588–93. doi: 10.1073/pnas.94.25.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, Arnoult N, Lackner DH, et al. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol. 2014;21:167–74. doi: 10.1038/nsmb.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko PL, Otterlei M, Graakjaer J, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–74. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Opresko PL, von Kobbe C, Laine J-P, et al. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem. 2002;277:41110–19. doi: 10.1074/jbc.M205396200. [DOI] [PubMed] [Google Scholar]
- Ouyang H, Nussenzweig A, Kurimasa A, et al. Ku70 is required for DNA repair but not for T cell antigen receptor gene recombination in vivo. J Exp Med. 1997;186:921–9. doi: 10.1084/jem.186.6.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen BAL, Yang Z, Lai M, et al. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. 2005;12:663–70. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- Paeschke K, McDonald KR, Zakian VA. Telomeres: structures in need of unwinding. FEBS Lett. 2010;584:3760–72. doi: 10.1016/j.febslet.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagon RA, Adam MP, Bird TD, et al. Bloom’s syndrome. Seattle, WA: University of Washington; 1993. [Google Scholar]
- Parsons JL, Dianova II, Allinson SL, Dianov GL. Poly(ADP-ribose) polymerase-1 protects excessive DNA strand breaks from deterioration during repair in human cell extracts. FEBS J. 2005;272:2012–21. doi: 10.1111/j.1742-4658.2005.04628.x. [DOI] [PubMed] [Google Scholar]
- Parvathaneni S, Stortchevoi A, Sommers JA, et al. Human RECQ1 interacts with Ku70/80 and modulates DNA end-joining of double-strand breaks. PLoS One. 2013;8:e62481. doi: 10.1371/journal.pone.0062481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul K, Wang M, Mladenov E, et al. DNA ligases I and III cooperate in alternative non-homologous end-joining in vertebrates. PLoS One. 2013;8:e59505. doi: 10.1371/journal.pone.0059505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M. The 3″ to 5″ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–79. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- Petkovic M, Dietschy T, Freire R, et al. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci. 2005;118:4261–9. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- Popuri V, Bachrati CZ, Muzzolini L, et al. The human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J Biol Chem. 2008;283:17766–76. doi: 10.1074/jbc.M709749200. [DOI] [PubMed] [Google Scholar]
- Popuri V, Croteau DL, Brosh RM, Bohr VA. RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle. 2012a;11:4252–65. doi: 10.4161/cc.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popuri V, Ramamoorthy M, Tadokoro T, et al. Recruitment and retention dynamics of RECQL5 at DNA double strand break sites. DNA Repair (Amst) 2012b;11:624–35. doi: 10.1016/j.dnarep.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popuri V, Tadokoro T, Croteau DL, Bohr VA. Human RECQL5: guarding the crossroads of DNA replication and transcription and providing backup capability. Crit Rev Biochem Mol Biol. 2013;48:289–99. doi: 10.3109/10409238.2013.792770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R, Zheng H, He H, et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010;29:2598–610. doi: 10.1038/emboj.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy M, May A, Tadokoro T, et al. The RecQ helicase RECQL5 participates in psoralen-induced interstrand cross-link repair. Carcinogenesis. 2013;34:2218–30. doi: 10.1093/carcin/bgt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy YVR, Ding Q, Lees-Miller SP, et al. Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J Biol Chem. 2004;279:39408–13. doi: 10.1074/jbc.M406432200. [DOI] [PubMed] [Google Scholar]
- Ren H, Dou S-X, Zhang X-D, et al. The zinc-binding motif of human RECQ5beta suppresses the intrinsic strand-annealing activity of its DExH helicase domain and is essential for the helicase activity of the enzyme. Biochem J. 2008;412:425–33. doi: 10.1042/BJ20071150. [DOI] [PubMed] [Google Scholar]
- Reynolds P, Anderson JA, Harper JV, et al. The dynamics of Ku70/80 and DNA-PKcs at DSBs induced by ionizing radiation is dependent on the complexity of damage. Nucleic Acids Res. 2012;40:10821–31. doi: 10.1093/nar/gks879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E, Woodbine L, Stiff T, et al. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res. 2009;37:482–92. doi: 10.1093/nar/gkn957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong SB, Väliaho J, Vihinen M. Structural basis of Bloom syndrome (BS) causing mutations in the BLM helicase domain. Mol Med. 2000;6:155–64. [PMC free article] [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Kulikowicz T, et al. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair (Amst) 2010;9:796–804. doi: 10.1016/j.dnarep.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K, Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low X-ray doses. Proc Natl Acad Sci USA. 2003;100:5057–62. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008;112:1413–23. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz MM, Proytcheva M, Ellis NA, et al. BLM, the Bloom’s syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteins. Cytogenet Cell Genet. 2000;91:217–23. doi: 10.1159/000056848. [DOI] [PubMed] [Google Scholar]
- Saponaro M, Kantidakis T, Mitter R, et al. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell. 2014;157:1037–49. doi: 10.1016/j.cell.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Verma S, Shrikhande G, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–8. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- Schurman SH, Hedayati M, Wang Z, et al. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum Mol Genet. 2009;18:3470–83. doi: 10.1093/hmg/ddp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwendener S, Raynard S, Paliwal S, et al. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J Biol Chem. 2010;285:15739–45. doi: 10.1074/jbc.M110.110478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Linke SP, Pedeux R, et al. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J. 2003;22:1210–22. doi: 10.1093/emboj/cdg114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Robles AI, Linke SP, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol. 2004;166:801–13. doi: 10.1083/jcb.200405128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–7. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Brosh RM. Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS One. 2007;2:e1297. doi: 10.1371/journal.pone.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Phatak P, Stortchevoi A, et al. RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair (Amst) 2012;11:537–49. doi: 10.1016/j.dnarep.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JC, Gray MD, Oshima J, Loeb LA. Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence and stimulation by replication protein A. Nucleic Acids Res. 1998;26:2879–85. doi: 10.1093/nar/26.12.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siitonen HA, Sotkasiira J, Biervliet M, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–8. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Brunet E, Wong SY-W, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–16. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Ahn B, Bohr VA. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009;10:235–52. doi: 10.1007/s10522-008-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Karmakar P, Aamann M, et al. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–71. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Popuri V, Kulikowicz T, et al. The human RecQ helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 2012;40:6632–48. doi: 10.1093/nar/gks349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupianek A, Poplawski T, Jozwiakowski SK, et al. BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res. 2011;71:842–51. doi: 10.1158/0008-5472.CAN-10-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So S, Adachi N, Lieber MR, Koyama H. Genetic interactions between BLM and DNA ligase IV in human cells. J Biol Chem. 2004;279:55433–42. doi: 10.1074/jbc.M409827200. [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Yang J-L, Ferrarelli LK, et al. Modulation of DNA base excision repair during neuronal differentiation. Neurobiol Aging. 2013;34:1717–27. doi: 10.1016/j.neurobiolaging.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely AM, Bleichert F, Nümann A, et al. Werner protein protects nonproliferating cells from oxidative DNA damage. Mol Cell Biol. 2005;25:10492–506. doi: 10.1128/MCB.25.23.10492-10506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro T, Ramamoorthy M, Popuri V, et al. Human RECQL5 participates in the removal of endogenous DNA damage. Mol Biol Cell. 2012;23:4273–85. doi: 10.1091/mbc.E12-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Sone M, Iwatsubo T, et al. Ku70 alleviates neurodegeneration in Drosophila models of Huntington’s disease. PLoS One. 2011;6:e27408. doi: 10.1371/journal.pone.0027408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Mendoza-Maldonado R, Tissino E, et al. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010;30:1382–96. doi: 10.1128/MCB.01290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Kaur S, Sengupta S. Phosphorylation-dependent interactions of BLM and 53BP1 are required for their anti-recombinogenic roles during homologous recombination. Carcinogenesis. 2008;29:52–61. doi: 10.1093/carcin/bgm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci USA. 2013;110:7720–5. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, Ijspeert H, Verkaik NS, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–8. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Harrigan JA, May A, et al. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol. 2003;23:8601–13. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Harrigan JA, Schreiber V, et al. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004a;32:4003–14. doi: 10.1093/nar/gkh721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Karmakar P, Dawut L, et al. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. J Biol Chem. 2002;277:22035–44. doi: 10.1074/jbc.M200914200. [DOI] [PubMed] [Google Scholar]
- von Kobbe C, May A, Grandori C, Bohr VA. Werner syndrome cells escape hydrogen peroxide-induced cell proliferation arrest. FASEB J. 2004b;18:1970–2. doi: 10.1096/fj.04-1895fje. [DOI] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–14. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- Wang H, Perrault AR, Takeda Y, et al. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003a;31:5377–88. doi: 10.1093/nar/gkg728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LL, Gannavarapu A, Kozinetz CA, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003b;95:669–74. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- Wang W, Bambara RA. Human Bloom protein stimulates flap endonuclease 1 activity by resolving DNA secondary structure. J Biol Chem. 2005;280:5391–9. doi: 10.1074/jbc.M412359200. [DOI] [PubMed] [Google Scholar]
- Wang XW, Tseng A, Ellis NA, et al. Functional interaction of p53 and BLM DNA helicase in apoptosis. J Biol Chem. 2001;276:32948–55. doi: 10.1074/jbc.M103298200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–39. [PMC free article] [PubMed] [Google Scholar]
- Weeda G, Eveno E, Donker I, et al. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60:320–9. [PMC free article] [PubMed] [Google Scholar]
- Weissman L, Jo D-G, Sørensen MM, et al. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–55. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner SR, Prahalad AK, Yang J, Hock JM. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun. 2006;345:403–9. doi: 10.1016/j.bbrc.2006.04.093. [DOI] [PubMed] [Google Scholar]
- Weterings E, Chen DJ. DNA-dependent protein kinase in nonhomologous end joining: a lock with multiple keys? J Cell Biol. 2007;179:183–6. doi: 10.1083/jcb.200705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterings E, Verkaik NS, Brüggenwirth HT, et al. The role of DNA dependent protein kinase in synapsis of DNA ends. Nucleic Acids Res. 2003;31:7238–46. doi: 10.1093/nar/gkg889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterings E, Verkaik NS, Keijzers G, et al. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol Cell Biol. 2009;29:1134–42. doi: 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, Lebel L-A, Vrbanac V, et al. Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum Mol Genet. 2003;12:273–81. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Bohr VA, McKinnon PJ. DNA damage, DNA repair, ageing and age-related disease. Mech Ageing Dev. 2008;129:349–52. doi: 10.1016/j.mad.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo LL, Futami K, Shimamoto A, et al. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res. 2006;312:3443–57. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Wu J, Capp C, Feng L, Hsieh T-S. Drosophila homologue of the Rothmund-Thomson syndrome gene: essential function in DNA replication during development. Dev Biol. 2008a;323:130–42. doi: 10.1016/j.ydbio.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Wang M, Wu W, et al. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair (Amst) 2008b;7:329–38. doi: 10.1016/j.dnarep.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Xu X, Rochette PJ, Feyissa EA, et al. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009a;28:3005–14. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lei Z, Huang H, et al. dRecQ4 is required for DNA synthesis and essential for cell proliferation in Drosophila. PLoS One. 2009b;4:e6107. doi: 10.1371/journal.pone.0006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano K-I, Morotomi-Yano K, Wang S-Y, et al. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9:91–6. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010;38:5706–17. doi: 10.1093/nar/gkq379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–62. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Goddard KA, et al. Linkage disequilibrium and haplotype studies of chromosome 8p 11.1–21.1 markers and Werner syndrome. Am J Hum Genet. 1994;55:356–64. [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gabriel A. Ku-dependent and Ku-independent end-joining pathways lead to chromosomal rearrangements during double-strand break repair in Saccharomyces cerevisiae. Genetics. 2003;163:843–56. doi: 10.1093/genetics/163.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Kanagaraj R, Mihaljevic B, et al. MRE11 complex links RECQ5 helicase to sites of DNA damage. Nucleic Acids Res. 2009;37:2645–57. doi: 10.1093/nar/gkp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung W-H, Shim EY, et al. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]