

Abstract
Added sugars comprising of table sugar, brown sugar, corn syrup, maple syrup, honey, molasses, and other sweeteners in the prepared processed foods and beverages have been implicated in the pathophysiology of cardiovascular diseases. This article deals with the reactive oxygen species (ROS) as a mechanism of sugar-induced cardiovascular diseases. There is an association between the consumption of high levels of serum glucose with cardiovascular diseases. Various sources of sugar-induced generation of ROS, including mitochondria, nicotinamide adenine dinucleotide phosphate-oxidase, advanced glycation end products, insulin, and uric acid have been discussed. The mechanism by which ROS induce the development of atherosclerosis, hypertension, peripheral vascular disease, coronary artery disease, cardiomyopathy, heart failure, and cardiac arrhythmias have been discussed in detail. In conclusion, the data suggest that added sugars induce atherosclerosis, hypertension, peripheral vascular disease, coronary artery disease, cardiomyopathy, heart failure, and cardiac arrhythmias and that these effects of added sugars are mediated through ROS.
Keywords: added sugars, reactive oxygen species, atherosclerosis, hypertension, coronary artery disease, peripheral vascular disease
Added sugars comprise of table sugar, brown sugar, corn syrup, maple syrup, honey, molasses and other sweeteners in the prepared and processed foods and beverages. Sugars (simple carbohydrates) refer to monosaccharides (glucose, galactose, and fructose) and disaccharides (sucrose [glucose + fructose] found in sugarcane, beets, honey, and corn syrup; lactose [glucose + lactose] found in milk products; and maltose [glucose + glucose] found in malt). Complex carbohydrates are glucose-containing polysaccharides, that is, starch. The American Heart Association recommends six teaspoons (24 g) of added sugar per day for women and nine teaspoons (36 g) per day for men or in other words added sugar should provide 100 cal/d for women and 150 cal/d for men, respectively.1 The American Heart Association recommends that not more than 5% of calories should come from sugar.1 High consumption of added sugars has a higher risk of cardiovascular disease and the associated risk factors.2 3 Benn et al,4 have reported that elevation of nonfasting glucose is associated with an increased risk of ischemic heart disease (IHD) and myocardial infarction (MI). A high intake of sugar enhances the risk of coronary heart disease (CHD) in diabetic individuals using diuretics.5 The International Study of Macro/Micronutrients and Blood Pressure reported that daily consumption of every extra sugar-sweetened beverage increased the systolic arterial pressure by 1.6 mm Hg and the diastolic pressure by 0.8 mm Hg.6
The present review deals with the glucose and oxidative stress, cardiovascular disease, serum lipids, and inflammatory mediators, and the glucose-induced oxidative stress as a mechanism of cardiovascular diseases. A recent publication has shown that there is a significant correlation between the added sugar consumption and an increased risk of cardiovascular disease mortality.7
Reactive Oxygen Species
The formation of reactive oxygen species (ROS) is schematically depicted in Fig. 1 and has been described in detail elsewhere.8 The univalent reduction of oxygen (O2), in the presence of a free electron generates superoxide anion (O2 −). Hydrogen peroxide (H2O2) is formed by dismutation of O2 − by superoxide dismutase (SOD). H2O2 is not a free radical and is stable, lipid soluble, crosses membrane barrier, and has a longer half-life as compared with O2 −. H2O2 is converted to H2O and O2 by catalase and glutathione peroxidase. H2O2 and O2 − in the presence of metal (iron or copper) via the Haber–Weiss or Fenton reaction generates hydroxyl radical (•OH), which is very reactive and unlike H2O2 and O2 − which travel some distance from their site production, •OH acts locally at the site of generation. O2 − interacts with nitric oxide to produce peroxynitrite (ONOO−), which at physiological pH generates peroxynitrous acid (ONOO H) and because of its very unstable nature it rapidly gives rise to •OH.
Fig. 1.
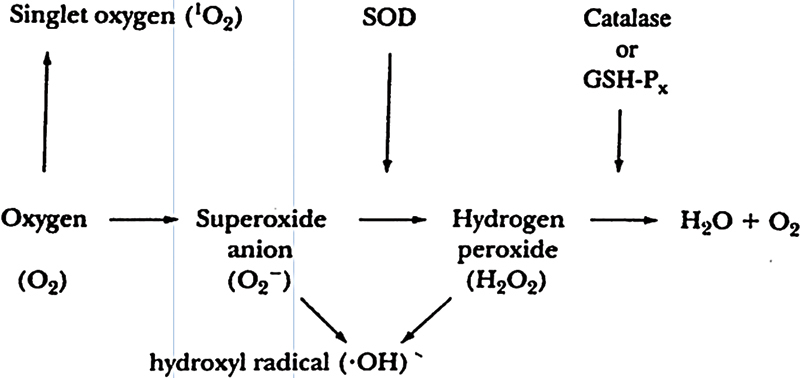
Figure shows the formation of various reactive oxygen species. GSH-Px, glutathione peroxidase; SOD, superoxide dismutase.
Glucose and Generation of Reactive Oxygen Species
Glucose can generate ROS through various ways including mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase, sorbitol pathway, activated glycation, and insulin pathway.
Mitochondrial Generation of Reactive Oxygen Species
The increase in glucose metabolism generates reduced nicotinamide adenine dinucleotide and reduced flavin adenine dinucleotide, which increases the generation of O2 − that is converted to H2O2 and •OH in the mitochondria.9 The increased metabolic flux of glucose via mitochondrial glucose oxidation leads to an increased production of ROS.10 It has also been reported that fission-mediated fragmentation of mitochondrial tubules is associated with an enhanced production of mitochondrial ROS in hyperglycemic condition.11 High glucose level upregulate thioredoxin-interacting protein, which is involved in high glucose-induced ROS production in the mitochondria.12 The above data suggest that mitochondria is partly involved in the generation of ROS in hyperglycemia.
Glucose and Nicotinamide Adenine Dinucleotide Phosphate-Oxidase
The NADPH-oxidase system is a membrane-associated enzyme complex that lies dormant and is present in the vascular endothelium and smooth muscle cells, cardiomyocytes, macrophages, and neutrophils. Activated NADPH-oxidase catalyzes the reduction of O2 to O2 − by way of NADPH derived from the pentose phosphate pathway as shown in the equation below. O2 − is then converted to H2O2 and •OH as described above.
High-glucose concentration activates NADPH-oxidase.13 Balteau et al14 have reported that the high glucose level activates cardiac NADPH-oxidase through a novel cardiac glucose transport system, the sodium-dependent glucose cotransporter, SGLT1 (sodium-glucose linked transporter1). NADPH-oxidase-induced generation of ROS in the heart depends upon NADPH generated by oxidative part of the pentose phosphate pathway.15 16 High glucose-induced NADPH-oxidase activation and subsequent ROS production has been implicated in apoptosis of umbilical endothelial cells.17 High glucose level activates NADPH-oxidase both in vascular smooth muscle cells and endothelial cells.18 NADPH-oxidase of macrophages is activated by hyperglycemia.19 High glucose level increases the activity of NADPH-oxidase in the endothelial microparticles.20 The data suggest that added glucose can generate ROS through activation of NADPH-oxidase.
Glucose, Advanced Glycation End Products, and Reactive Oxygen Species
Advanced glycation end products (AGEs) are a heterogeneous group of molecules formed from nonenzymatic reaction of reducing sugars with amino group of proteins, lipids, and nucleic acids.21 22 23 There are three pathways for the formation of AGEs from glucose: the Millard reaction, oxidation of glucose, and polyol pathway.
Millard Reaction
In Millard reaction glucose attaches to free amino acids of proteins nonenzymatically to form a Schiff base which spontaneously rearranges to form the Amadori product. These initial reactions are reversible. Subsequent reactions lead to the formation of AGEs through oxidation, degradation, and rearrangement. Oxidation of the glycated product is known as glycoxidation or the Millard reaction. Some of the AGE products include Nε-carboxymethyl-lysine, Nε-carboxyethyl-lysine, pyrilline, pentosidine, and argpyramidine. There are other ways by which glucose can form AGEs.
Oxidation of Glucose
Auto-oxidation of glucose and the peroxidation of lipids into dicarbonyl derivatives lead to the formation of AGEs.24 These dicarbonyl derivatives are glyoxal, methylglyoxal (MG), and 3-deoxyglucosone, and interact with monoacids to form AGEs. MG is the product of the glycolytic pathway formed through spontaneous transformation of triose phosphate.25
MG along with 3-deoxyglucosone and glyoxal are the major sources of AGEs.26 MG reacts oxidatively with arginine or lysine residue of proteins to form AGEs.27
Polyol Pathway
The other pathway for the formation of AGEs from glucose is the polyol pathway. In this pathway glucose is converted to sorbitol by aldose reductase and then to fructose by sorbitol dehydrogenase. Fructose-3-phosphate, a metabolite of fructose is converted into α-oxaldehyde which interacts with monoacids to form AGEs.28
AGEs interact with its cellular receptor RAGE (receptor for AGEs) and activates nuclear factor kappa-B (NF-κβ)29 and generate ROS.30 31 NF-κβ increases the gene expression of cytokines (interleukin [IL]-1, IL-6, IL-8, tumor necrosis factor-α) and adhesion molecules (vascular cell adhesion molecules-1 [VCAM-1], intercellular adhesion molecule-1 [ICAM-1]).8 29 32 Cytokines are known to activate NADPH-oxidase, which generates ROS.33 34 35
Circulating AGEs are positively correlated with high sensitivity C-reactive protein (hs-CRP).36 hs-CRP has been reported to increase the generation of ROS by neutrophils.37 These data suggest that AGEs can increase the levels the ROS through activation of RAGE, activation of NF-κβ increases cytokines resulting in activation of NADPH-oxidase, and subsequent formation of ROS. hs-CRP generates ROS from neutrophils.
Insulin and Reactive Oxygen Species
Glucose intake increases the secretion of insulin.38 39 Insulin activates a plasma membrane enzyme system with the properties of NADPH-oxidase resulting in the generation of H2O2.40 41 42 Goldstein et al43 reported that insulin-induced generation of H2O2 is through activation of NOX4, a homologous family of NADPH-oxidase. Espinosa et al44 have also shown that insulin generates H2O2 through increased expression of NOX4 a homologous family of NADPH-oxidase. The data suggest that insulin increases ROS through activation of NADPH-oxidase.
Uric Acid and Reactive Oxygen Species
Fructose in sugar raises uric acid in human45 and rodents.46 Uric acid scavenges carbon centered and peroxyl radicals in hydrophilic environment, but cannot scavenge lipophilic radicals and cannot break the radical chain propagation within the lipid membrane.47 The antioxidant activity of uric acid is in the plasma and body fluids only. Uric acid is a pro-oxidant and generates ROS in various ways48 including interacting with peroxynitrite49 and oxidized lipids.50 The data suggest that fructose produces ROS through various mechanisms.
Sugar, Reactive Oxygen Species, and Cardiovascular Diseases
Atherosclerosis
In the segment described above, we have shown that added sugar is involved in the generation of ROS, and increased expression of cytokines and cell adhesion molecules. ROS have been implicated in the initiation and progression of hypercholesterolemia-induced atherosclerosis.51 52 53 54 55 56 The oxidative hypothesis of the development of atherosclerosis is detailed elsewhere.56 In short, low-density lipoprotein cholesterol (LDL-C) is mildly oxidized to minimally modified LDL (MM-LDL), which stimulates smooth muscle cells and endothelial cells to produce monocyte chemoattractant protein-1 (MCP-1). ROS increases expression of cell adhesion molecules ICAM-1, VCAM-1, and endothelial leukocyte adhesion molecules on the endothelial cells.57 58 59 Monocytes adhere to the endothelial cell surface with the help of cell adhesion molecule. MM-LDL is further oxidized to oxidized LDL (OX-LDL). MCP-1 and OX-LDL helps in the migration of monocyte to the subendothelial area. Monocyte/macrophage gets differentiated by the help of monocyte colony stimulating factor released from endothelial cells by MM-LDL. The differentiated macrophage develops receptor for OX-LDL, which is picked by macrophages to form foam cells. Macrophage generates numerous growth factors leading to the formation of collagen, elastic fiber, and protein and transcription of smooth muscle cells. Smooth muscle cell proliferation, and migration, synthesis of connective tissue and matrix, migration of monocytes and formation of foam cells results in the development and progression of atherosclerosis.
Hypertension
About 25% people with type-1 diabetes and 80% with type-2 diabetes have high blood pressure. Diet rich in high fructose, a key ingredient in corn syrup, can produce high blood pressure. Studies in animals have shown an association between the sugar intake and development of hypertension.60 Fructose leads to hypertension in dogs61 and rats.62 63 A diet containing excess sugar induces hypertension in F344 and F1-hybrid rat.64 Fructose ingestion raises blood pressure in healthy young humans.65 There is enough evidence to show that sugar (fructose) raises blood pressure. Here, we are going to discuss about the role of glucose-induced ROS in the development of hypertension in experimental animals and in humans.
An increased level of ROS precedes the development of hypertension in spontaneously hypertensive rats (SHRs).66 There are increased levels of thiobarbituric acid reactive substances (TBARs) and F2α-isoprostane the markers of oxidative stress, tissue concentrations of H2O2 and O2 − and activation of NADPH-oxidase and xanthine oxidase, decreased levels of nitric oxide and antioxidant enzymes in experimental hypertension.67 68 69 The generation of O2 − is increased in vasculature of SHRs70 and deoxycorticosterone acetate salt sensitive hypertension.71 There is attenuation and prevention of hypertension with antioxidants.72 73
Hypertensive patients have higher serum levels of H2O2 compared with control subjects.74 There is an increased production of ROS by polymorphonuclear leukocytes in hypertensive patients.75 76 Patients with hypertension (essential, salt-sensitive, and renovascular) have increased levels of TBARs, 8-isoprostane.77 78 79 Many experimental models of hypertension have indicated that there is an increased level of ROS and decreased levels of antioxidants or both.80 81 82 It is clear that ROS levels are elevated in hypertension. Now the question is how ROS induces hypertension?
ROS can induce vascular contraction directly or indirectly via endothelin-1 or destruction of nitric oxide (a vasodilator). Let us first consider the direct effect of ROS on vasculature. Bharadwaj and Prasad83 reported that O2 − produces a contraction of the isolated rabbit aorta that is endothelium-dependent. They also reported that H2O2 produced contraction in isolated rabbit aorta.84 In vivo studies Prasad et al85 reported that xanthine plus xanthine oxidase-induced ROS increased systemic and pulmonary vascular resistance in anaesthetized dogs. Polymorphonuclear leukocyte-derived ROS increases the total systemic and pulmonary vascular resistance.86 The other possibility of ROS-induced hypertension is through production of endothelin-1, a vasoconstrictor. ROS increases the production of endothelin-1 in mesangial cells.87 Vascular smooth muscle contraction is dependent upon intracellular calcium. ROS increases intracellular calcium through release from endoplasmic reticulum. O2 − increases intracellular Ca2+ in cultured human myocardial cells.88 Superoxide anion,89 90 H2O2,90 91 and OH92 increase the intracellular Ca2+ in cultured vascular endothelial cells.
The other mechanism of ROS-induced hypertension could be the ROS-induced endothelial dysfunction. Nitric oxide (NO) is endothelial-derived relaxing factor.93 ROS can damage endothelial cells and reduce the production of NO. O2 – can deactivate NO by combining with it and producing other more active radicals such as peroxinitrite and OH.94 95 ROS would reduce the bioavailability of NO and hence vasoconstriction.
ROS by damaging the endothelial would reduce the production of prostacyclin, which is a vasodilator. ROS also reduces the capacity of the cells to convert arachidonic acid to prostacyclin.96 The reduction in the production of vasodilators in the endothelium will allow the circulating catecholamine to produce unopposed constriction of the blood vessels and hence raise the blood pressure. Sucrose consumption increases the sympathetic activity in the rat.97 It has been reported that introduction of starch and sugars into human nutrition increases sympathetic activity.98 Increase in the sympathetic activity would raise the blood pressure. These data suggest that ROS through various mechanisms could induce hypertension.
Coronary Artery Disease
There is an association between elevated fasting plasma glucose and increased risk of IHD and MI in people with or without diabetes.99 100 Benn et al4 using Mendelian randomization approach reported that both observational and genetic lifelong elevated nonfasting or fasting plasma glucose levels are associated with increased risk of IHD and MI. Intensive glycemic control in diabetic patients is associated with a 15% reduction in the risk of IHD.101 High blood sugar is a risk for the heart diseases. It is reported that glycated hemoglobin a measure of long-term blood glucose level, is an independent risk for CHD in both diabetic and nondiabetic subjects.102 Reports from the Nurses Health Study show that women with high glycemic blood had an increased risk for CHD (twofold risk during 10 years follow-up).103 High sugar consumption is associated with an increased cardiovascular disease in both within the country and across countries.104 There is an evidence that consumption of high amount of added sugar especially sugar-sweetened beverages increases the risk of cardiovascular diseases.2 3 There is a significant association between added sugar consumption and increased risk for mortality from cardiovascular diseases.7 A high level of sugars also increases the risk factors for CHD. It is known that diets high in sucrose, glucose, and fructose increase the plasma levels of triglycerides.105 106 107 There is an inverse association between dietary sucrose and high density lipoprotein-cholesterol (HDL-C).107 Consumption of beverages containing high sugar increases inflammation108 and proinflammatory cytokines.109
Buildup of atherosclerotic plaques in the coronary artery reduces blood flow to the myocardium resulting in symptoms ranging from angina to heart attack. Prolonged ischemia leads to cell necrosis. Ischemia can further increase the formation of ROS through xanthine–xanthine oxidase pathway which would further damage the myocardium.
The most important risk factors for CHD include hypercholesterolemia,110 111 diabetes,111 112 cigarette smoking,113 and hypertension.114 Hypercholesterolemia increases the generation of ROS through various ways.56 115 Hypercholesterolemia may increase the activity of oxidant-producing enzyme system: NADPH-oxidase, xanthine oxidase,116 and myeloperoxidase117 resulting in the formation of ROS. Oxidative stress in IHD could also be due to formation of •OH through interaction of O2 − and NO.94 95 According to oxidative hypotheses of atherosclerosis,51 52 53 54 55 56 oxidation of LDL assists in the formation of foam cells and contributes to various proatherogenic processes. Atherosclerosis and rupture of the atherosclerotic plaques of the coronary artery play a major role in the development of IHD. The facts that hypercholesterolemia produces atherosclerosis in animal models,51 52 53 and antioxidants suppress hypercholesterolemic atherosclerosis51 52 53 118 119 suggest that ROS play a role in the development of atherosclerosis. This also suggests that ROS are involved in the development of IHD. If this is the case then IHD should be associated with an increase in levels of the ROS. As such it has been reported that serum levels of malondialdehyde, a parameter of increased levels of ROS are elevated in patients with CHD.120 121 122
Cardiomyopathy
There are very few articles suggesting the role of sugars in cardiomyopathy. It has been reported that consumption of high fructose diet (32% sucrose solution) for 10 weeks reduced the ejection fraction by 8% in the heart of rats.123 Administration of 10% fructose solution for 2 weeks to Wistar rats produces left ventricular dysfunction measured by the left ventricular elastance a measure of left ventricular contractility independent of preloads, afterload, and heart rate.124 Chang et al124 also reported that these rats had increased end-diastolic volume. Davidoff et al125 and Dutta et al126 showed that consumption of high-sucrose diet (68% total energy) reduced the contractility of isolated cardiomyocytes in rats. In vivo studies using echocardiographic assessment of cardiac function in diabetic mice showed that there is systolic and diastolic dysfunction.127 Van den Bergh et al128 assessed the hemodynamic changes in type-2 diabetic db/db mouse and observed a reduction in the cardiac contractility. It is also known that diabetic cardiomyopathy is associated with hyperglycemia.129 130 131 This is also supported by the fact that insulin reverses the diabetic cardiomyopathy in rats.132 Cardiomyopathy in diabetes may be due to increased levels of ROS. There are evidences that the levels of ROS are elevated in diabetes. Paolisso and Giugliano133 reported that plasma free radical concentrations are positively correlated to fasting plasma insulin in patients with type-2 diabetes. Increased serum levels of malondialdehyde (MDA), an index of levels of ROS, have been observed in patients with type-2 diabetes.134 135 There is an increase in the O2 − levels in serum of diabetic patients.136 Increases in the serum levels of MDA have also been reported in type-1 and type-2 diabetic rats.137 138 139 It is evident that sucrose-induced cardiac dysfunction and diabetic cardiomyopathy is associated with hyperglycemia. It is also known that sugar increases the formation of ROS. Is there any evidence that ROS induce cardiac dysfunction and decreases myocardial contractility?
It has been reported that xanthine–xanthine oxidase-generated ROS85 and polymorphonuclear leukocyte-derived ROS86 reduce the myocardial contractility and cardiac function measured hemodynamically in dogs. Prasad et al140 also reported that xanthine–xanthine oxidase-derived ROS reduced the cardiac function and contractility in anesthetized dog and reduced the left ventricular pressure in the isolated perfused rabbit heart. They also reported that SOD, an antioxidant enzyme prevented the xanthine–xanthine oxidase-induced reduction in left ventricular pressure in the isolated perfused rabbit heart. The above data suggest that sugar induces cardiomyopathy through the generation of ROS.
Heart Failure
Added sugars could produce heart failure because it would lead to IHD and cardiomyopathy. The details of sugar and IHD and cardiomyopathy have been described above.
Cardiac Arrhythmias
Although, there are numerous unauthenticated information regarding the role of sugars in the development of cardiac arrhythmias only a few articles are available dealing with this subject. Patients with diabetes have a 40% greater risk of developing atrial fibrillation. Epidemiological studies have reported an association between diabetes or elevated blood glucose and risk of atrial fibrillation.141 142 143 However, there are few studies which reported no association between diabetes or elevated glucose and risk of atrial fibrillation.144 This discrepancy could be due to inclusion of obesity, which produces diabetes and atrial fibrillation. The other reason could be that the studies were not designed to focus on diabetes. Recently, in a population-based case–control study, Dublin et al145 have observed that diabetes was associated with higher risk of atrial fibrillation and the risk was higher with longer duration of treated diabetes and worse glycemic control. They also reported that patients receiving treatment for diabetes had a 40% greater risk of atrial fibrillation as compared with nondiabetics. Beshai J. F. from Heart Rhythm Center at the University of Chicago has stated that fluctuations in blood sugar levels and changes in insulin levels can lead to atrial fibrillation in diabetics.
Lactose causes cardiac arrhythmias in water flea Daphnia pulex.146 Excess sugar intake causes cardiac arrhythmias because sugar causes fluctuation in insulin and adrenalin. Insulin147 and adrenalin148 induce cardiac arrhythmias.
Do ROS induce cardiac arrhythmias? There are some data from animal studies and in humans, which suggest that excess ROS are involved in the pathogenesis of cardiac arrhythmias. Morita et al149 have reported that perfusion of H2O2 in fibrotic rat and rabbit heart in Langendorff preparation induces cardiac arrhythmias. ROS have been implicated in the cardiac arrhythmias.150 151 152 The data suggest that sugar-induced cardiac arrhythmias are mediated through ROS.
Peripheral Arterial Disease
Peripheral arterial disease (PAD) is a circulatory disorder that affects blood vessels outside the heart and brain. PAD is due to atherosclerosis characterized by atherosclerotic occlusive disease of the lower extremities and is a marker of atherothrombotic disease in other vascular beds such as coronary artery, carotid artery, and cerebral artery. Prevalence of PAD is 12 to 14% in the general population, but is 20% in the individuals above the age of 70.153 Overall, 30% of diabetics suffer from PAD, and 20% of symptomatic patients have PAD.154 Diabetes, smoking, hypercholesterolemia, C-reactive protein, which are the risk factors for atherosclerosis are also risk factors for PAD. High intake of sugar increases insulin level in some individuals, but not all, and that these individuals are susceptible to the effect of sucrose for occlusive arterial disease.155
Hyperglycemia in diabetes would produce ROS through various mechanisms already described in the section “glucose and generation of reactive oxygen species” of this review. ROS have been implicated in the development of atherosclerosis, which has been described in the section “atherosclerosis” of this review.
These data suggest that sugars are involved in the development of PAD through the generation of ROS.
Comments
The American Heart Association recommends that not more than 5% of calories should come from sugar.1 It has been recommended that women should not take more than 6 tsp/d and men should not take more than 9 tsp/d of added sugar. The weight varies among men and women. If a man weighing 50 kg and another man weighing 80 kg consume similar amounts of sugar, that is, 9 tsp/d of added sugar, the one with a lower weight would have more adverse effects as compared with the other with a higher weight. There should be a guideline where the normal consumption of sugar should be based on the weight range. Some of the top sources of added sugar diet are shown in Table 1. The population should be advised to reduce the consumption of a diet containing large amounts of sugars.
Table 1. Some of the sources of added sugars.
| Source | Amount of sugar (g) |
|---|---|
| 12 ounce can of regular soda | 36.0 |
| 16 ounce bottle of sugar-sweetened ice tea | 46.0 |
| 20 ounce bottle of lemon-lime soda | 64.0 |
| Plain chocolate (100 g) | 62.0 |
| Chocolate spread (100 g) | 57.10 |
| Cola (100 g) | 10.9 |
| Sweetened fruit juice (100 g) | 9.80 |
| Fruit pastilles (100 g) | 59.30 |
| Iced cakes (100 g) | 54.0 |
| Chocolate-coated biscuits (100 g) | 45.80 |
| Frosted corn flakes (100 g) | 37.0 |
| Fruit yogurt (100 g) | 16.60 |
| Tomato ketchup (100 g) | 27.5 |
| Stir-in sweet and sour sauce (100 g) | 20.20 |
| A slice of apple pie | 20.0 |
| Half cup of ice cream | 14.0 |
Source: 1. Top sources of added sugars in our diet—NHS choices. Available at: http://www.nhs.uk/livewell/goodfood/pages/top-sources-of-added-sugar-in-our-diet.aspx. Accessed February 17, 2014.
2. Van Horn L, Johnson RK, Flickinger BD, Vafiadis DK, Yin-Piazza S; Added Sugars Conference Planning Group. Translation and implementation of added sugars consumption recommendations: a conference report from the American Heart Association Added Sugars Conference 2010. Circulation 2010;122(23):2470–2490
From the literature,155 it appears that sugar consumption increases the levels of insulin in some individuals but not in all and the individuals with increased levels of insulin are prone to develop occlusive arterial disease. In this review, we have discussed that both glucose and insulin increases the levels of ROS, which have been implicated in the development of cardiovascular disease. It is not clear, then why only those people who have high insulin levels with consumption of high sugars develop occlusive arterial disease. The most recent data38 39 suggest that glucose increases the secretion of insulin in all individuals. It appears that both glucose and insulin take part in the development of cardiovascular diseases via an increased generation of ROS.
Conclusions
The data suggest that added sugars are involved in the development of atherosclerosis, hypertension, peripheral vascular disease, coronary artery disease, cardiomyopathy, heart failure, and cardiac arrhythmias. The data also suggest that the cardiovascular diseases are mediated through ROS generated by sugars.
Acknowledgment
This work was supported by a grant from the College of Medicine Research Fund of the University of Saskatchewan, Saskatoon, Canada.
Footnotes
Conflict of Interest The authors have no conflict of interest to disclose.
References
- 1.Johnson R K, Appel L J, Brands M. et al. Dietary sugars intake and cardiovascular health: a scientific statement from the American Heart Association. Circulation. 2009;120(11):1011–1020. doi: 10.1161/CIRCULATIONAHA.109.192627. [DOI] [PubMed] [Google Scholar]
- 2.Malik V S, Popkin B M, Bray G A, Després J P, Hu F B. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation. 2010;121(11):1356–1364. doi: 10.1161/CIRCULATIONAHA.109.876185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Koning L Malik V S Kellogg M D Rimm E B Willett W C Hu F B Sweetened beverage consumption, incident coronary heart disease, and biomarkers of risk in men Circulation 2012125141735–1741., S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benn M, Tyberg-Hensen A, McCarthy M I, Jensen G B, Grande P, Nordestgaard B G. Nonfasting glucose, ischemic heart disease, and myocardial infarction: a Mendelian randomization study. J Am Coll Cardiol. 2012;59(25):2356–2365. doi: 10.1016/j.jacc.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherman W M. Metabolism of sugars and physical performance. Am J Clin Nutr. 1995;62(1):228S–241S. doi: 10.1093/ajcn/62.1.228S. [DOI] [PubMed] [Google Scholar]
- 6.Brown I J, Stamler J, Van Horn L. et al. Sugar-sweetened beverage, sugar intake of individuals, and their blood pressure: international study of macro/micronutrients and blood pressure. Hypertension. 2011;57(4):695–701. doi: 10.1161/HYPERTENSIONAHA.110.165456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Q, Zhang Z, Gregg E W, Flanders W D, Merritt R, Hu F B. Added sugar intake and cardiovascular diseases mortality among US adults. JAMA Intern Med. 2014;174(4):516–524. doi: 10.1001/jamainternmed.2013.13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prasad K. New York, NY: Springer Verlag; 2000. Oxygen free radicals and peripheral vascular disease; pp. 427–438. [Google Scholar]
- 9.Turrens J F. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt 2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 11.Yu T, Sheu S-S, Robotham J L, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79(2):341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah A, Xia L, Goldberg H, Lee K W, Quaggin S E, Fantus I G. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J Biol Chem. 2013;288(10):6835–6848. doi: 10.1074/jbc.M112.419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnefont-Rousselot D. Glucose and reactive oxygen species. Curr Opin Clin Nutr Metab Care. 2002;5(5):561–568. doi: 10.1097/00075197-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 14.Balteau M, Tajeddine N, de Meester C. et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res. 2011;92(2):237–246. doi: 10.1093/cvr/cvr230. [DOI] [PubMed] [Google Scholar]
- 15.Gupte R S, Floyd B C, Kozicky M. et al. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med. 2009;47(3):219–228. doi: 10.1016/j.freeradbiomed.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuurbier C J, Eerbeek O, Goedhart P T. et al. Inhibition of the pentose phosphate pathway decreases ischemia-reperfusion-induced creatine kinase release in the heart. Cardiovasc Res. 2004;62(1):145–153. doi: 10.1016/j.cardiores.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52(11):2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 18.Inoguchi T, Li P, Umeda F. et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 19.Benerjee Banerjee D, Sharma P. Dual effects of glucose on macrophage NADPH-oxidase activity: a possible link between diabetes and tuberculosis. Oxid Antioxid Med Sci. 2012;1:91–96. [Google Scholar]
- 20.Jansen F, Yang X, Franklin B S. et al. High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc Res. 2013;98(1):94–106. doi: 10.1093/cvr/cvt013. [DOI] [PubMed] [Google Scholar]
- 21.Thorpe S R, Baynes J W. Maillard reaction products in tissue proteins: new products and new perspectives. Amino Acids. 2003;25(3-4):275–281. doi: 10.1007/s00726-003-0017-9. [DOI] [PubMed] [Google Scholar]
- 22.Prasad K. Soluble receptor for advanced glycation end products (sRAGE) and cardiovascular disease. Int J Angiol. 2006;15:57–68. [Google Scholar]
- 23.Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol. 1992;23:1–34. doi: 10.1016/s1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 24.Uribarri J, Tuttle K R. Advanced glycation end products and nephrotoxicity of high-protein diets. Clin J Am Soc Nephrol. 2006;1(6):1293–1299. doi: 10.2215/CJN.01270406. [DOI] [PubMed] [Google Scholar]
- 25.Thornalley P J. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification—a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996;27(4):565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 26.Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest. 1994;70(2):138–151. [PubMed] [Google Scholar]
- 27.Jana C K, Das N, Sohal R S. Specificity of age-related carbonylation of plasma proteins in the mouse and rat. Arch Biochem Biophys. 2002;397(2):433–439. doi: 10.1006/abbi.2001.2690. [DOI] [PubMed] [Google Scholar]
- 28.Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res. 2007;2007:61038. doi: 10.1155/2007/61038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann M A, Drury S, Fu C. et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97(7):889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 30.Yan S D, Schmidt A M, Anderson G M. et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269(13):9889–9897. [PubMed] [Google Scholar]
- 31.Wautier M P, Chappey O, Corda S, Stern D M, Schmidt A M, Wautier J L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280(5):E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 32.Reznikov L L, Waksman J, Azam T. et al. Effect of advanced glycation end products on endotoxin-induced TNF-alpha, IL-1beta and IL-8 in human peripheral blood mononuclear cells. Clin Nephrol. 2004;61(5):324–336. doi: 10.5414/cnp61324. [DOI] [PubMed] [Google Scholar]
- 33.Mohammed A M, Syeda K, Hadden T, Kowluru A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem Pharmacol. 2013;85(1):109–114. doi: 10.1016/j.bcp.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braquet P, Hosford D, Braquet M, Bourgain R, Bussolino F. Role of cytokines and platelet-activating factor in microvascular immune injury. Int Arch Allergy Appl Immunol. 1989;88(1-2):88–100. doi: 10.1159/000234755. [DOI] [PubMed] [Google Scholar]
- 35.Paubert-Braquet M, Lonchampt M O, Koltz P, Guilbaud J. Tumor necrosis factor (TNF) primes human neutrophil neutrophil (PMN) platelet-activating factor (PAF)-induced superoxide generation. Consequences in promoting PMN-mediated endothelial cells (EC) damages. Prostaglandins. 1988;35:803. [Google Scholar]
- 36.Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, Vlassara H. et al. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci. 2007;62(4):427–433. doi: 10.1093/gerona/62.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prasad K. C-reactive protein increases oxygen radical generation by neutrophils. J Cardiovasc Pharmacol Ther. 2004;9(3):203–209. doi: 10.1177/107424840400900308. [DOI] [PubMed] [Google Scholar]
- 38.Kraegen E W, Chisholm D J, Young J D, Lazarus L. The gastrointestinal stimulus to insulin release. II. A dual action of secretin. J Clin Invest. 1970;49(3):524–529. doi: 10.1172/JCI106262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elrick H, Stimmler L, Hlad C J Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab. 1964;24:1076–1082. doi: 10.1210/jcem-24-10-1076. [DOI] [PubMed] [Google Scholar]
- 40.Krieger-Brauer H I, Kather H. Human fat cells possess a plasma membrane-bound H2O2-generating system that is activated by insulin via a mechanism bypassing the receptor kinase. J Clin Invest. 1992;89(3):1006–1013. doi: 10.1172/JCI115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee S P, Lane R H, Lynn W S. Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents. Biochem Pharmacol. 1978;27(22):2589–2594. doi: 10.1016/0006-2952(78)90332-5. [DOI] [PubMed] [Google Scholar]
- 42.May J M, de Haën C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem. 1979;254(7):2214–2220. [PubMed] [Google Scholar]
- 43.Goldstein B J, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7(7-8):1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Espinosa A, Campos C, Díaz-Vegas A. et al. Insulin-dependent H2O2 production is higher in muscle fibers of mice fed with a high-fat diet. Int J Mol Sci. 2013;14(8):15740–15754. doi: 10.3390/ijms140815740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stirpe F, Della Corte E, Bonetti E, Abbondanza A, Abbati A, De Stefano F. Fructose-induced hyperuricaemia. Lancet. 1970;2(7686):1310–1311. doi: 10.1016/s0140-6736(70)92269-5. [DOI] [PubMed] [Google Scholar]
- 46.Stavric B, Johnson W J, Clayman S, Gadd R E, Chartrand A. Effect of fructose administration on serum urate levels in the uricase inhibited rat. Experientia. 1976;32(3):373–374. doi: 10.1007/BF01940847. [DOI] [PubMed] [Google Scholar]
- 47.Muraoka S, Miura T. Inhibition by uric acid of free radicals that damage biological molecules. Pharmacol Toxicol. 2003;93(6):284–289. doi: 10.1111/j.1600-0773.2003.pto930606.x. [DOI] [PubMed] [Google Scholar]
- 48.Maples K R, Mason R P. Free radical metabolite of uric acid. J Biol Chem. 1988;263(4):1709–1712. [PubMed] [Google Scholar]
- 49.Vásquez-Vivar J, Santos A M, Junqueira V B, Augusto O. Peroxynitrite-mediated formation of free radicals in human plasma: EPR detection of ascorbyl, albumin-thiyl and uric acid-derived free radicals. Biochem J. 1996;314(Pt 3):869–876. doi: 10.1042/bj3140869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bagnati M, Perugini C, Cau C, Bordone R, Albano E, Bellomo G. When and why a water-soluble antioxidant becomes pro-oxidant during copper-induced low-density lipoprotein oxidation: a study using uric acid. Biochem J. 1999;340(Pt 1):143–152. [PMC free article] [PubMed] [Google Scholar]
- 51.Prasad K, Kalra J. Oxygen free radicals and hypercholesterolemic atherosclerosis: effect of vitamin E. Am Heart J. 1993;125(4):958–973. doi: 10.1016/0002-8703(93)90102-f. [DOI] [PubMed] [Google Scholar]
- 52.Prasad K. Reduction of serum cholesterol and hypercholesterolemic atherosclerosis in rabbits by secoisolariciresinol diglucoside isolated from flaxseed. Circulation. 1999;99(10):1355–1362. doi: 10.1161/01.cir.99.10.1355. [DOI] [PubMed] [Google Scholar]
- 53.Prasad K, Kalra J, Lee P. Oxygen free radicals as a mechanism of hypercholesterolemic atherosclerosis: effect of probucol. Int J Angiol. 1994;3:100–112. [Google Scholar]
- 54.Steinberg D. Antioxidants and atherosclerosis. A current assessment. Circulation. 1991;84(3):1420–1425. doi: 10.1161/01.cir.84.3.1420. [DOI] [PubMed] [Google Scholar]
- 55.Steinberg D. Antioxidant in the prevention of human atherosclerosis. Summary of the proceedings of a National Heart, Lung, and Blood Institute Workshop: September 5-6, 1991, Bethesda, Maryland. Circulation. 1992;85(6):2337–2344. doi: 10.1161/01.cir.85.6.2337. [DOI] [PubMed] [Google Scholar]
- 56.Prasad K. New York, NY: Springer Verlag; 2000. Pathophysiology of atherosclerosis; pp. 8–105. [Google Scholar]
- 57.Martin A, Foxall T, Blumberg J B, Meydani M. Vitamin E inhibits low-density lipoprotein-induced adhesion of monocytes to human aortic endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 1997;17(3):429–436. doi: 10.1161/01.atv.17.3.429. [DOI] [PubMed] [Google Scholar]
- 58.Faruqi R, de la Motte C, DiCorleto P E. Alpha-tocopherol inhibits agonist-induced monocytic cell adhesion to cultured human endothelial cells. J Clin Invest. 1994;94(2):592–600. doi: 10.1172/JCI117374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Devaraj S, Li D, Jialal I. The effects of alpha tocopherol supplementation on monocyte function. Decreased lipid oxidation, interleukin 1 β secretion, and monocyte adhesion to endothelium. J Clin Invest. 1996;98(3):756–763. doi: 10.1172/JCI118848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buñag R D, Tomita T, Sasaki S. Chronic sucrose ingestion induces mild hypertension and tachycardia in rats. Hypertension. 1983;5(2):218–225. doi: 10.1161/01.hyp.5.2.218. [DOI] [PubMed] [Google Scholar]
- 61.Martinez F J, Rizza R A, Romero J C. High-fructose feeding elicits insulin resistance, hyperinsulinism, and hypertension in normal mongrel dogs. Hypertension. 1994;23(4):456–463. doi: 10.1161/01.hyp.23.4.456. [DOI] [PubMed] [Google Scholar]
- 62.Tran L T, Yuen V G, McNeill J H. The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol Cell Biochem. 2009;332(1-2):145–159. doi: 10.1007/s11010-009-0184-4. [DOI] [PubMed] [Google Scholar]
- 63.Dhar I, Dhar A, Wu L, Desai K M. Increased methylglyoxal formation with upregulation of renin angiotensin system in fructose fed Sprague Dawley rats. PLoS ONE. 2013;8(9):e74212. doi: 10.1371/journal.pone.0074212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Preuss H G, Knapka J J. Sugar-induced hypertension in Fischer 344 and F1-hybrid rats (F344BN) at different ages. Geriatr Nephrol Urol. 1994;4(1):15–21. [Google Scholar]
- 65.Brown C M, Dulloo A G, Yepuri G, Montani J-P. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Regul Integr Comp Physiol. 2008;294(3):R730–R737. doi: 10.1152/ajpregu.00680.2007. [DOI] [PubMed] [Google Scholar]
- 66.Kitiyakara C, Wilcox C S. Antioxidants for hypertension. Curr Opin Nephrol Hypertens. 1998;7(5):531–538. doi: 10.1097/00041552-199809000-00008. [DOI] [PubMed] [Google Scholar]
- 67.Touyz R M, Schiffrin E L. Reactive oxygen species in vascular biology: implications in hypertension. Histochem Cell Biol. 2004;122(4):339–352. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- 68.Welch W J. Intrarenal oxygen and hypertension. Clin Exp Pharmacol Physiol. 2006;33(10):1002–1005. doi: 10.1111/j.1440-1681.2006.04478.x. [DOI] [PubMed] [Google Scholar]
- 69.Redón J, Oliva M R, Tormos C. et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41(5):1096–1101. doi: 10.1161/01.HYP.0000068370.21009.38. [DOI] [PubMed] [Google Scholar]
- 70.Zalba G, Beaumont F J, San José G. et al. Vascular NADH/NADPH oxidase is involved in enhanced superoxide production in spontaneously hypertensive rats. Hypertension. 2000;35(5):1055–1061. doi: 10.1161/01.hyp.35.5.1055. [DOI] [PubMed] [Google Scholar]
- 71.Elmarakby A A, Loomis E D, Pollock J S, Pollock D M. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET-1. Hypertension. 2005;45(2):283–287. doi: 10.1161/01.HYP.0000153051.56460.6a. [DOI] [PubMed] [Google Scholar]
- 72.Houston M C. Nutraceuticals, vitamins, antioxidants, and minerals in the prevention and treatment of hypertension. Prog Cardiovasc Dis. 2005;47(6):396–449. doi: 10.1016/j.pcad.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 73.Hong H J, Hsiao G, Cheng T H, Yen M H. Supplemention with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension. 2001;38(5):1044–1048. doi: 10.1161/hy1101.095331. [DOI] [PubMed] [Google Scholar]
- 74.Lacy F, Kailasam M T, O'Connor D T, Schmid-Schönbein G W, Parmer R J. Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension. 2000;36(5):878–884. doi: 10.1161/01.hyp.36.5.878. [DOI] [PubMed] [Google Scholar]
- 75.Sagar S, Kallo I J, Kaul N, Ganguly N K, Sharma B K. Oxygen free radicals in essential hypertension. Mol Cell Biochem. 1992;111(1-2):103–108. doi: 10.1007/BF00229580. [DOI] [PubMed] [Google Scholar]
- 76.Yasunari K, Maeda K, Nakamura M, Yoshikawa J. Oxidative stress in leukocytes is a possible link between blood pressure, blood glucose, and C-reacting protein. Hypertension. 2002;39(3):777–780. doi: 10.1161/hy0302.104670. [DOI] [PubMed] [Google Scholar]
- 77.Fortuño A, Oliván S, Beloqui O. et al. Association of increased phagocytic NADPH oxidase-dependent superoxide production with diminished nitric oxide generation in essential hypertension. J Hypertens. 2004;22(11):2169–2175. doi: 10.1097/00004872-200411000-00020. [DOI] [PubMed] [Google Scholar]
- 78.Higashi Y, Sasaki S, Nakagawa K, Matsuura H, Oshima T, Chayama K. Endothelial function and oxidative stress in renovascular hypertension. N Engl J Med. 2002;346(25):1954–1962. doi: 10.1056/NEJMoa013591. [DOI] [PubMed] [Google Scholar]
- 79.Lip G Y, Edmunds E, Nuttall S L, Landray M J, Blann A D, Beevers D G. Oxidative stress in malignant and non-malignant phase hypertension. J Hum Hypertens. 2002;16(5):333–336. doi: 10.1038/sj.jhh.1001386. [DOI] [PubMed] [Google Scholar]
- 80.Harrison D G, Gongora M C. Oxidative stress and hypertension. Med Clin North Am. 2009;93(3):621–635. doi: 10.1016/j.mcna.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 81.Vaziri N D. Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Curr Opin Nephrol Hypertens. 2004;13(1):93–99. doi: 10.1097/00041552-200401000-00013. [DOI] [PubMed] [Google Scholar]
- 82.Addabbo F, Montagnani M, Goligorsky M S. Mitochondria and reactive oxygen species. Hypertension. 2009;53(6):885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bharadwaj L A, Prasad K. Mechanism of superoxide anion-induced modulation of vascular tone. Int J Angiol. 2002;11:23–29. [Google Scholar]
- 84.Bharadwaj L A, Prasad K. Mediation of H2O2-induced vascular relaxation by endothelium-derived relaxing factor. Mol Cell Biochem. 1995;149-150:267–270. doi: 10.1007/BF01076587. [DOI] [PubMed] [Google Scholar]
- 85.Prasad K, Kalra J, Chan W P, Chaudhary A K. Effect of oxygen free radicals on cardiovascular function at organ and cellular levels. Am Heart J. 1989;117(6):1196–1202. doi: 10.1016/0002-8703(89)90396-7. [DOI] [PubMed] [Google Scholar]
- 86.Prasad K, Kalra J, Chaudhary A K, Debnath D. Effect of polymorphonuclear leukocyte-derived oxygen free radicals and hypochlorous acid on cardiac function and some biochemical parameters. Am Heart J. 1990;119(3 Pt 1):538–550. doi: 10.1016/s0002-8703(05)80276-5. [DOI] [PubMed] [Google Scholar]
- 87.Hughes A K, Stricklett P K, Padilla E, Kohan D E. Effect of reactive oxygen species on endothelin-1 production by human mesangial cells. Kidney Int. 1996;49(1):181–189. doi: 10.1038/ki.1996.25. [DOI] [PubMed] [Google Scholar]
- 88.Masumoto N, Tasaka K, Miyake A, Tanizawa O. Superoxide anion increases intracellular free calcium in human myometrial cells. J Biol Chem. 1990;265(36):22533–22536. [PubMed] [Google Scholar]
- 89.Dreher D, Junod A F. Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J Cell Physiol. 1995;162(1):147–153. doi: 10.1002/jcp.1041620118. [DOI] [PubMed] [Google Scholar]
- 90.Sun L, Yau H-Y, Lau O-C, Huang Y, Yao X. Effect of hydrogen peroxide and superoxide anions on cytosolic Ca2+: comparison of endothelail cells from large-sized and small-sized arteries. PLoS ONE. 2011;6(9):e25432. doi: 10.1371/journal.pone.0025432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Volk T, Hensel M, Kox W J. Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: role of hydrogen peroxide. Mol Cell Biochem. 1997;171(1-2):11–21. doi: 10.1023/a:1006886215193. [DOI] [PubMed] [Google Scholar]
- 92.Az-ma T, Saeki N, Yuge O. Cytosolic Ca2+ movements of endothelial cells exposed to reactive oxygen intermediates: role of hydroxyl radical-mediated redox alteration of cell-membrane Ca2+ channels. Br J Pharmacol. 1999;126(6):1462–1470. doi: 10.1038/sj.bjp.0702438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ignarro L J, Buga G M, Wood K S, Byrns R E, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kooy N W, Royall J A. Agonist-induced peroxynitrite production from endothelial cells. Arch Biochem Biophys. 1994;310(2):352–359. doi: 10.1006/abbi.1994.1178. [DOI] [PubMed] [Google Scholar]
- 95.Beckman J S, Beckman T W, Chen J, Marshall P A, Freeman B A. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee D S, McCallum E A, Olson D M. Effects of reactive oxygen species on prostacyclin production in perinatal rat lung cells. J Appl Physiol (1985) 1989;66(3):1321–1327. doi: 10.1152/jappl.1989.66.3.1321. [DOI] [PubMed] [Google Scholar]
- 97.Young J B, Landsberg L. Stimulation of the sympathetic nervous system during sucrose feeding. Nature. 1977;269(5629):615–617. doi: 10.1038/269615a0. [DOI] [PubMed] [Google Scholar]
- 98.Kopp W. Chronically increased activity of the sympathetic nervous system: our diet-related “evolutionary” inheritance. J Nutr Health Aging. 2009;13(1):27–29. doi: 10.1007/s12603-009-0005-1. [DOI] [PubMed] [Google Scholar]
- 99.Sarwar N, Gao P, Seshasai S R. et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375(9733):2215–2222. doi: 10.1016/S0140-6736(10)60484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Coutinho M, Gerstein H C, Wang Y, Yusuf S. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care. 1999;22(2):233–240. doi: 10.2337/diacare.22.2.233. [DOI] [PubMed] [Google Scholar]
- 101.Ray K K, Seshasai S R, Wijesuriya S. et al. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet. 2009;373(9677):1765–1772. doi: 10.1016/S0140-6736(09)60697-8. [DOI] [PubMed] [Google Scholar]
- 102.Selvin E, Coresh J, Golden S H, Brancati F L, Folsom A R, Steffes M W. Glycemic control and coronary heart disease risk in persons with and without diabetes: the atherosclerosis risk in communities study. Arch Intern Med. 2005;165(16):1910–1916. doi: 10.1001/archinte.165.16.1910. [DOI] [PubMed] [Google Scholar]
- 103.Liu S, Willett W C, Stampfer M J. et al. A prospective study of dietary glycemic load, carbohydrate intake, and risk of coronary heart disease in US women. Am J Clin Nutr. 2000;71(6):1455–1461. doi: 10.1093/ajcn/71.6.1455. [DOI] [PubMed] [Google Scholar]
- 104.Yudkin J. Sugar and ischaemic heart disease. Practitioner. 1967;198(187):680–683. [PubMed] [Google Scholar]
- 105.Lê K A, Tappy L. Metabolic effects of fructose. Curr Opin Clin Nutr Metab Care. 2006;9(4):469–475. doi: 10.1097/01.mco.0000232910.61612.4d. [DOI] [PubMed] [Google Scholar]
- 106.Fried S K, Rao S P. Sugars, hypertriglyceridemia, and cardiovascular disease. Am J Clin Nutr. 2003;78(4):873S–880S. doi: 10.1093/ajcn/78.4.873S. [DOI] [PubMed] [Google Scholar]
- 107.Ernst N, Fisher M, Smith W. et al. The association of plasma high-density lipoprotein cholesterol with dietary intake and alcohol consumption. The Lipid Research Clinics Prevalence Study. Circulation. 1980;62(4 Pt 2):IV41–IV52. [PubMed] [Google Scholar]
- 108.Liu S, Manson J E, Buring J E, Stampfer M J, Willett W C, Ridker P M. Relation between a diet with a high glycemic load and plasma concentrations of high-sensitivity C-reactive protein in middle-aged women. Am J Clin Nutr. 2002;75(3):492–498. doi: 10.1093/ajcn/75.3.492. [DOI] [PubMed] [Google Scholar]
- 109.Shanmugam N, Reddy M A, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes. 2003;52(5):1256–1264. doi: 10.2337/diabetes.52.5.1256. [DOI] [PubMed] [Google Scholar]
- 110.Castelli W P Cholesterol and lipids in the risk of coronary artery disease—the Framingham Heart Study Can J Cardiol 19884(Suppl A ):5A–10A. [PubMed] [Google Scholar]
- 111.Fuller J H, Shipley M J, Rose G, Jarrett R J, Keen H. Mortality from coronary heart disease and stroke in relation to degree of glycaemia: the Whitehall study. Br Med J (Clin Res Ed) 1983;287(6396):867–870. doi: 10.1136/bmj.287.6396.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vogel R A. Coronary risk factors, endothelial function, and atherosclerosis: a review. Clin Cardiol. 1997;20(5):426–432. doi: 10.1002/clc.4960200505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fielding J E. Smoking: health effects and control (1) N Engl J Med. 1985;313(8):491–498. doi: 10.1056/NEJM198508223130807. [DOI] [PubMed] [Google Scholar]
- 114.Kannel W B. Role of blood pressure in cardiovascular disease: the Framingham Study. Angiology. 1975;26(1 Pt. 1):1–14. doi: 10.1177/000331977502600101. [DOI] [PubMed] [Google Scholar]
- 115.Warnholtz A, Nickenig G, Schulz E. et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99(15):2027–2033. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- 116.Stokes K Y, Clanton E C, Russell J M, Ross C R, Granger D N. NAD(P)H oxidase-derived superoxide mediates hypercholesterolemia-induced leukocyte-endothelial cell adhesion. Circ Res. 2001;88(5):499–505. doi: 10.1161/01.res.88.5.499. [DOI] [PubMed] [Google Scholar]
- 117.Liu H-R, Tao L, Gao E. et al. Rosiglitazone inhibits hypercholesterolaemia-induced myeloperoxidase upregulation—a novel mechanism for the cardioprotective effects of PPAR agonists. Cardiovasc Res. 2009;81(2):344–352. doi: 10.1093/cvr/cvn308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prasad K. Hypocholesterolemic and antiatherosclerotic effect of flax lignan complex isolated from flaxseed. Atherosclerosis. 2005;179(2):269–275. doi: 10.1016/j.atherosclerosis.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 119.Prasad K, Mantha S V, Kalra J, Lee P. Prevention of hypercholesterolemic atherosclerosis by garlic, an antioxidant. J Cardiovasc Pharmacol Ther. 1997;2(4):309–320. doi: 10.1177/107424849700200409. [DOI] [PubMed] [Google Scholar]
- 120.Khan M A, Baseer A. Increased malondialdehyde levels in coronary heart disease. J Pak Med Assoc. 2000;50(8):261–264. [PubMed] [Google Scholar]
- 121.Dubois-Randé J L, Artigou J Y, Darmon J Y. et al. Oxidative stress in patients with unstable angina. Eur Heart J. 1994;15(2):179–183. doi: 10.1093/oxfordjournals.eurheartj.a060473. [DOI] [PubMed] [Google Scholar]
- 122.Kaur K, Bedi G, Kaur M, Vij A, Kaur I. Lipid peroxidation and the levels of antioxidant enzymes in coronary artery disease. Indian J Clin Biochem. 2008;23(1):33–37. doi: 10.1007/s12291-008-0008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vasanji Z, Cantor E J, Juric D, Moyen M, Netticadan T. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. Am J Physiol Cell Physiol. 2006;291(4):C772–C780. doi: 10.1152/ajpcell.00086.2005. [DOI] [PubMed] [Google Scholar]
- 124.Chang K C, Liang J T, Tseng C D. et al. Aminoguanidine prevents fructose-induced deterioration in left ventricular-arterial coupling in Wistar rats. Br J Pharmacol. 2007;151(3):341–346. doi: 10.1038/sj.bjp.0707223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davidoff A J, Mason M M, Davidson M B. et al. Sucrose-induced cardiomyocyte dysfunction is both preventable and reversible with clinically relevant treatments. Am J Physiol Endocrinol Metab. 2004;286(5):E718–E724. doi: 10.1152/ajpendo.00358.2003. [DOI] [PubMed] [Google Scholar]
- 126.Dutta K, Podolin D A, Davidson M B, Davidoff A J. Cardiomyocyte dysfunction in sucrose-fed rats is associated with insulin resistance. Diabetes. 2001;50(5):1186–1192. doi: 10.2337/diabetes.50.5.1186. [DOI] [PubMed] [Google Scholar]
- 127.Semeniuk L M, Kryski A J, Severson D L. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283(3):H976–H982. doi: 10.1152/ajpheart.00088.2002. [DOI] [PubMed] [Google Scholar]
- 128.Van den Bergh A, Flameng W, Herijgers P. Type II diabetic mice exhibit contractile dysfunction but maintain cardiac output by favourable loading conditions. Eur J Heart Fail. 2006;8(8):777–783. doi: 10.1016/j.ejheart.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 129.Fonarow G C Srikanthan P Diabetic cardiomyopathy Endocrinol Metab Clin North Am 2006353575–599., ix [DOI] [PubMed] [Google Scholar]
- 130.Ruddy T D, Shumak S L, Liu P P. et al. The relationship of cardiac diastolic dysfunction to concurrent hormonal and metabolic status in type I diabetes mellitus. J Clin Endocrinol Metab. 1988;66(1):113–118. doi: 10.1210/jcem-66-1-113. [DOI] [PubMed] [Google Scholar]
- 131.Severson D L. Diabetic cardiomyopathy: recent evidence from mouse models of type 1 and type 2 diabetes. Can J Physiol Pharmacol. 2004;82(10):813–823. doi: 10.1139/y04-065. [DOI] [PubMed] [Google Scholar]
- 132.Fein F S, Strobeck J E, Malhotra A, Scheuer J, Sonnenblick E H. Reversibility of diabetic cardiomyopathy with insulin in rats. Circ Res. 1981;49(6):1251–1261. doi: 10.1161/01.res.49.6.1251. [DOI] [PubMed] [Google Scholar]
- 133.Paolisso G, Giugliano D. Oxidative stress and insulin action: is there a relationship? Diabetologia. 1996;39(3):357–363. doi: 10.1007/BF00418354. [DOI] [PubMed] [Google Scholar]
- 134.Armstrong A M, Chestnutt J E, Gormley M J, Young I S. The effect of dietary treatment on lipid peroxidation and antioxidant status in newly diagnosed noninsulin dependent diabetes. Free Radic Biol Med. 1996;21(5):719–726. doi: 10.1016/0891-5849(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 135.Sundaram R K, Bhaskar A, Vijayalingam S, Viswanathan M, Mohan R, Shanmugasundaram K R. Antioxidant status and lipid peroxidation in type II diabetes mellitus with and without complications. Clin Sci (Lond) 1996;90(4):255–260. doi: 10.1042/cs0900255. [DOI] [PubMed] [Google Scholar]
- 136.Ceriello A, Giugliano D, Quatraro A, Dello Russo P, Lefèbvre P J. Metabolic control may influence the increased superoxide generation in diabetic serum. Diabet Med. 1991;8(6):540–542. doi: 10.1111/j.1464-5491.1991.tb01647.x. [DOI] [PubMed] [Google Scholar]
- 137.Prasad K, Mantha S V, Muir A D, Westcott N D. Protective effect of secoisolariciresinol diglucoside against streptozotocin-induced diabetes and its mechanism. Mol Cell Biochem. 2000;206(1-2):141–149. doi: 10.1023/a:1007018030524. [DOI] [PubMed] [Google Scholar]
- 138.Prasad K. Oxidative stress as a mechanism of diabetes in diabetic BB prone rats: effect of secoisolariciresinol diglucoside (SDG) Mol Cell Biochem. 2000;209(1-2):89–96. doi: 10.1023/a:1007079802459. [DOI] [PubMed] [Google Scholar]
- 139.Prasad K. Secoisolariciresinol diglucoside from flaxseed delays the development of type 2 diabetes in Zucker rat. J Lab Clin Med. 2001;138(1):32–39. doi: 10.1067/mlc.2001.115717. [DOI] [PubMed] [Google Scholar]
- 140.Prasad K, Kalra J, Bharadwaj L. Cardiac depressant effects of oxygen free radicals. Angiology. 1993;44(4):257–270. doi: 10.1177/000331979304400401. [DOI] [PubMed] [Google Scholar]
- 141.Nichols G A, Reinier K, Chugh S S. Independent contribution of diabetes to increased prevalence and incidence of atrial fibrillation. Diabetes Care. 2009;32(10):1851–1856. doi: 10.2337/dc09-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Johansen O E, Brustad E, Enger S, Tveit A. Prevalence of abnormal glucose metabolism in atrial fibrillation: a case control study in 75-year old subjects. Cardiovasc Diabetol. 2008;7:28. doi: 10.1186/1475-2840-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Movahed M R, Hashemzadeh M, Jamal M M. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int J Cardiol. 2005;105(3):315–318. doi: 10.1016/j.ijcard.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 144.Ostgren C J, Merlo J, Råstam L, Lindblad U. Atrial fibrillation and its association with type 2 diabetes and hypertension in a Swedish community. Diabetes Obes Metab. 2004;6(5):367–374. doi: 10.1111/j.1462-8902.2004.00358.x. [DOI] [PubMed] [Google Scholar]
- 145.Dublin S, Glazer N L, Smith N L. et al. Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J Gen Intern Med. 2010;25(8):853–858. doi: 10.1007/s11606-010-1340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Campbell A K, Wann K T, Matthews S B. Lactose causes heart arrhythmia in the water flea Daphnia pulex. Comp Biochem Physiol B Biochem Mol Biol. 2004;139(2):225–234. doi: 10.1016/j.cbpc.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 147.Binder G, Bosk A, Gass M, Ranke M B, Heidemann P H. Insulin tolerance test causes hypokalaemia and can provoke cardiac arrhythmias. Horm Res. 2004;62(2):84–87. doi: 10.1159/000079539. [DOI] [PubMed] [Google Scholar]
- 148.Oikawa S, Nomura H, Nishio M, Nagata R, Hata T. Doxapram hydrochloride aggravates adrenaline-induced arrhythmias accompanied by bidirectional ventricular tachycardia. ISRN Cardiol. 2014;2014:212045. doi: 10.1155/2014/212045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morita N, Sovari A A, Xie Y. et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297(5):H1594–H1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jeong E M, Liu M, Sturdy M. et al. Metabolic stress, reactive oxygen species, and arrhythmia. J Mol Cell Cardiol. 2012;52(2):454–463. doi: 10.1016/j.yjmcc.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sovari A A, Dudley S C Jr. Reactive oxygen species-targeted therapeutics interventions for atrial fibrillation. Front Physiol. 2012;3:311. doi: 10.3389/fphys.2012.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bonilla I M, Sridhar A, Györke S, Cardounel A J, Carnes C A. Nitric oxide synthases and atrial fibrillation. Front Physiol. 2012;3:105. doi: 10.3389/fphys.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shammas N W. Epidemiology, classification, and modifiable risk factors of peripheral arterial disease. Vasc Health Risk Manag. 2007;3(2):229–234. doi: 10.2147/vhrm.2007.3.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Murabito J M, D'Agostino R B, Silbershatz H, Wilson W F. Intermittent claudication. a risk profile from the Framingham heart study. Circulation. 1997;96(1):44–49. doi: 10.1161/01.cir.96.1.44. [DOI] [PubMed] [Google Scholar]
- 155.Yudkin J, Kakkar V V, Szanto S. Sugar intake, serum insulin and platelet adhesiveness in men with and without peripheral vascular disease. Postgrad Med J. 1969;45(527):608–611. doi: 10.1136/pgmj.45.527.608. [DOI] [PMC free article] [PubMed] [Google Scholar]