

Abstract
Platinum–acridine hybrid agents show low-nanomolar potency in chemoresistant non-small cell lung cancer (NSCLC), but high systemic toxicity in vivo. To reduce the promiscuous genotoxicity of these agents and improve their pharmacological properties, a modular build–click–screen approach was used to evaluate a small library of twenty hybrid agents containing truncated and extended chromophores of varying basicities. Selected derivatives were resynthesized and tested in five NSCLC cell lines representing large cell, squamous cell, and adenocarcinomas. 7-Aminobenz[c]acridine was identified as a promising scaffold in a hybrid agent (P1–B1) that maintained submicromolar activity in several of the DNA-repair proficient and p53-mutant cancer models, while showing improved tolerability in mice by 32-fold compared to the parent platinum–acridine (P1–A1). The distribution and DNA/RNA adduct levels produced by the acridine- and benz[c]acridine-based analogues in NCI-H460 cells (confocal microscopy, ICP-MS), and their ability to bind G-quadruplex forming DNA sequences (CD spectroscopy, HR-ESMS) were studied. P1–B1 emerges as a less genotoxic, more tolerable, and potentially more target-selective hybrid agent than P1–A1.
Keywords: anticancer drugs, cellular target, cytotoxicity, DNA adducts, intercalation
Introduction
Platinum-based drugs continue to be a mainstay in chemotherapy regimens for the treatment of a multitude of solid malignancies.[1] Despite improvements in their pharmacological properties and safety profile, many tumors are inherently insensitive to this form of therapy. In several intractable forms of cancer, such as non-small-cell lung cancer (NSCLC), current platinum chemotherapies provide only a moderate survival benefit both in the adjuvant and palliative setting.[2] To extend the spectrum of activity of the classical anticancer metal, platinum complexes have been introduced as reactive, electrophilic components into multifunctional DNA-targeted therapies, such as platinum–intercalator agents.[3,4]
The platinum–acridine agents represented by the general structure in Figure 1 form mixed monofunctional-intercalative DNA adducts distinct from the bifunctional cross-links induced by cisplatin[3] and structurally related monofunctional-noninter-calative DNA adducts.[5] The hybrid adducts form at a significantly higher rate and are a more severe form of DNA damage than the classical cross-links.[3] They effectively inhibit DNA synthesis in treated cells and lead to stalled replication forks and DNA double-strand breaks, which elicits responses from specialized DNA damage recognition and repair modules.[6] In addition, the hybrid adducts are significantly more potent inhibitors of RNA polymerase II (Pol II)-mediated transcription than the cross-links.[7] Fluorescent post-labeling in whole lung cancer cells recently revealed high levels of DNA adducts in all phases of the cell cycle, both in the loosely packaged and the condensed chromatin.[8] The cumulative effects of this damage result in cell cycle arrest in late G1/early S phase and efficiently trigger apoptosis in NSCLC models representing a diverse range of genetic backgrounds.[9] Hybrids derived from the general structure in Figure 1 effectively kill cancer cells at low-nanomolar concentrations, which places them among the most cytotoxic platinum-containing anticancer agents described in the literature to date.[6]
Figure 1.
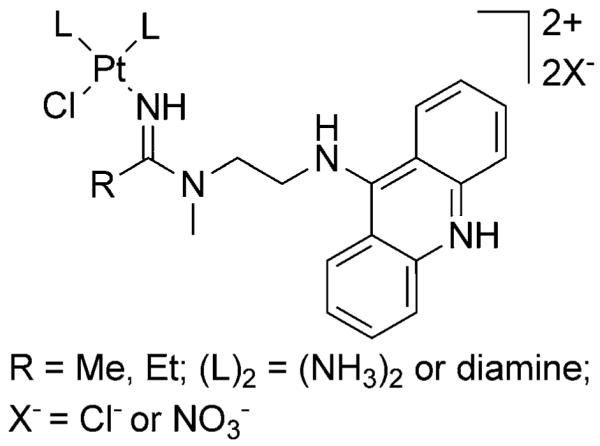
General structure of platinum–acridine hybrid agents.
Despite their promising performance in models of intractable cancers in vitro and in vivo, platinum–acridines show unfavorable ADME[10] (absorption, distribution, metabolism, and excretion) properties. They are highly hydrophilic, dicationic molecules, which insufficiently accumulate in tumor tissue and produce high levels of platinum in the kidneys and other normal tissues when administered to mice.[11] Like other cytotoxic agents, platinum–acridines exert their antitumor properties by indiscriminately damaging an essentially non-cancer-specific target, chromosomal DNA,[12] which likely also contributes to their low tolerability observed in test animals.[11] Thus, the remaining challenges with this type of anticancer agent revolve around improving its drug-like properties and reducing its dose-limiting toxicities, while retaining clinically useful anticancer activity.
In the current study we used a combination of computational and modular-synthetic library screening to devise a simple strategy by which the pharmacological and target binding properties of the platinum–acridines can be tuned. The approach involved attenuating the proton affinity and altering the size of the hybrid agent’s intercalator moiety. Benz[c]acridine was identified as a promising planar scaffold in a more tolerable hybrid that maintains submicromolar activity in a panel of NSCLC cell lines. Major differences between the acridine- and benz[c]acridine-based hybrids emerged with respect to their subcellular distribution and target binding levels.
Results
Design
In the new analogues the basic design of the hybrid agent (Figure 1), in particular the monofunctional platinum and the N-(2-aminoethyl)-N-alkylamidine linker connecting the platinum and intercalator moieties, was not changed. These previously optimized critical motifs allow strainless intercalation/p-stacking of the chromophore with DNA bases adjacent to the sites of platination.[3] Systematic changes to the chromophore were desired that reduce the electrostatically driven association and promiscuous reactivity of the platinum–acridines with nuclear DNA. The approach involved reducing the cationic charge of the DNA-interacting chromophore and modifying its shape and size. It consolidates two previously articulated concepts: i) the design of “less-than-ideal” intercalators (deprotonated/neutral, asymmetric chromophores featuring fewer or more than three fused aromatic rings), which reduces DNA affinity and enhances transcellular diffusion and tumor penetration,[13] and ii) the design of compounds that interact with the genome more selectively by targeting specific DNA structures and associated processes critical for cancer cell viability (e. g., G-quadruplex-forming sequences).[12]
The new generation of compounds modeled from the platinum–acridine hybrid agents therefore display a truncated or extended planar N-heterocyclic aromatic moiety and reduced basicity of the endocyclic nitrogen, resulting in an overall reduced cationic charge at neutral physiological pH. In addition to the desired effects on the target binding, reducing the basicity and hydrophilicity of the chromophore also has the potential to improve important pharmacokinetic parameters of the hybrids, such as uptake from circulation, membrane permeation, and tissue distribution.[14]
Computational study of DNA-targeted chromophores
We used self-consistent reaction field DFT-B3LYP level calculations[15] as a pre-screening platform to evaluate proton affinities for a set of extended or truncated aromatic moieties derived from the acridine scaffold. The virtual library included substituted and unsubstituted 7-aminobenz[c]acridine (3-3d), 7-aminodibenz[b,h][1,6]naphthyridine (4-4b), 7-aminobenz[b]-[1,10]phenanthroline (5-5b), and 7-aminodibenz[c,h]acridine (6) (Figure 2). Computations were also performed on the parent 9-aminoacridine (2) and the truncated chromophore, 4-aminoquinoline (1) (Figure 2). The final choice of modifications and substitution patterns to achieve the desired electronic and steric effects was based on chemical entities that are synthetically readily accessible. The proposed chromophores (except for 4-aminoquinoline, 1) can be assembled by using simple condensation and cyclization chemistries (see Scheme 1).
Figure 2.

Chromophores studied in computational screen.
Scheme 1.

Synthesis of benz[c]acridine chromophores. a) POCl3, 80 °C, b) (1) N-methylethylenediamine, dioxane, reflux, (2) 2m NH4OH; c) NaOH, phenol, 120 °C; d) (1) N-methylethylenediamine, dioxane, reflux, (2) 2m NH4OH; e) (1) tert-butyl(2-aminoethyl)methylcarbamate, triethylamine, dioxane, reflux, (2) HCl/AcOH, (3) 2m NaOH.
DFT calculations were performed for chromophores 1–6 in their protonated (at the endocyclic nitrogen atom highlighted in Figure 2) and unprotonated forms in a simulated dielectric. The differences between the free energies of solvated protonated and solvated unprotonated species (ΔGproton.) were used as a predictor of relative chromophore basicities.[16] On the basis of the calculated energies (Table 1), the nitrobenz[c]acridine derivatives 3d and 3c can be expected to be the least basic chromophores in the series and 5–5b the most basic, while the data suggests an intermediate basicity for the parent acridine (2). Only chromophores predictably less basic than 9-aminoacridine (pKa << 9.5[17]), which would effectively reduce the charge on the new hybrids at neutral physiological pH, were considered as candidates for the assembly of a synthetic library. To produce the desired structural and electronic diversity, the 7-aminobenz[c]acridine motifs 3, 3c, and 3d, along with 9-aminoacridine (2) and 4-aminoquinoline (1), were selected as components in the synthesized intercalator modules.
Table 1.
Relative free energies of protonation calculated for model chromophores.
| Chromophore | ΔGproton. (kcalmol−1)[a] |
|---|---|
| 3d | −238.01* |
| 3c | −238.10* |
| 4a | −240.09 |
| 4b | −240.89 |
| 4 | −243.35 |
| 3a | −243.58 |
| 3b | −244.51 |
| 1 | −244.89* |
| 6 | −245.65 |
| 3 | −246.99* |
| 2 | −249.22* |
| 5a | −254.33 |
| 5b | −255.27 |
| 5 | −257.60 |
Calculated for energy-optimized geometries in a self-consistent reaction field mimicking the dielectric constant of water using density functional theory (DFT). Proton affinities of structures increase from top to bottom. Chromophores synthesized in this study are highlighted by asterisks. Free energies were defined as ΔGproton.=ΔG(solv. BH+)−ΔG(solv. B). Because only relative energies between B and BH+ are relevant, ΔG(solv. H+) was not included in the calculations.
Synthesis of selected chromophores, library assembly, and pre-screening
Introducing the selected chromophores as components in platinum–intercalator hybrids required installation of 2-methylaminoethyl side chains at the 4-, 9-, and 7-amino positions of the aromatic systems to produce precursors Q1, A1, and B1–B3 (Figure 3A), respectively. Modules Q1 and A1 were synthesized from a selectively protected diamine linker and the appropriate phenoxy-substituted quinoline and acridine precursors using a previously reported scheme.[18] The synthesis of the benz[c]acridine derivatives, B1-B3 (Scheme 1), commenced with classical Ullman-type condensation of the appropriate substituted o-chlorobenzoic acid and 1-napthylamine, and subsequent cyclization of the condensation products with POCl3.[19] Because the 7-position in the nitrobenz[c]acridines was activated for nucleophilic aromatic substitution, both the 7-chloro and 7-phenoxy intermediates readily and selectively reacted with the primary amino group of N-methylethylenediamine to produce the desired linker-modified chromophores B2 and B3. By contrast, the unsubstituted benz[c]acridine intermediates B1.2 and B1.3 were significantly less reactive, and introduction of the (2-methylamino)ethyl linker to produce B1 required longer reaction times and protection of the secondary amino group.
Figure 3.

Chromophore modules (A), platinum modules (B), and micro-scale “click” reactions (C) used in library assembly.
Four platinum–nitrile precursor complexes, P1–P4, were synthesized as building blocks for assembling the library of hybrid molecules (Figure 3B). The diam(m)ine nonleaving groups were varied across the set of complexes as an additional means of tuning the DNA binding of the hybrids. Ammine (NH3), ethane-1,2-diamine (en), propane-1,3-diamine (pn), and N1,N1,N2,N2-tetramethylethane-1,2-diamine (tmeda) were introduced as substitution-inert ligands. Previous mechanistic studies have demonstrated that the monofunctional platinum moieties modified with pn (module P3) react with DNA most rapidly, whereas introduction of bulky tmeda (module P4) as a spectator ligand almost completely eliminates the formation of permanent platinum-DNA adducts.[3] We rationalized these observations for platinum–amidine model complexes by performing DFT calculations on transition states associated with both aquation and nucleobase binding. On the basis of the free energies of activation (ΔG≠) extracted from the modeled structures (see the Supporting Information), the predicted trend in reactivity is pn > NH3 ≈ en > tmeda, which is in agreement with previous experimental findings.[3]
Next, we used a combinatorial assay designed for platinum–acridines,[20] which takes advantage of metal-facilitated amine–nitrile addition chemistry, to assemble and pre-screen a small library of target molecules. In the process, an amidine donor group is formed, which establishes connectivity between the chromophore and the platinum moiety (Figure 3C). Using the five linker-modified chromophores and four platinum precursors, twenty micro-scale reactions were assembled to generate a library of twenty hybrid agents (Figure 3C). Reactions were monitored by in-line high performance liquid chromatography/electrospray mass spectrometry (LC-ESMS; see the Supporting Information) and subjected to cell viability pre-screening without isolation and purification of target compounds (see Experimental Section for details).
The entire library was tested in NCI-H460 (large cell carcinoma, wild-type p53) and NCI-H520 (squamous cell carcinoma, mutated p53) lung cancer cells. The chemosensitivity of the two cell lines was assessed for three concentrations of the newly generated platinum chromophores (P#-Q/A/B#) and platinum-free chromophores (Q1, A1, B1, B2, and B3; 0.1, 1.0, and 10 μm, 72 h incubations) using a colorimetric cell proliferation assay. (The precursor complexes P1, P2, P3, and P4, which are unstable in aqueous media and biologically inactive,[20] were not screened.)
The resulting cell viabilities for treated cells are plotted relative to controls in Figure 4. Analysis of the two data sets reveals the following trends: the hybrids and free chromophores generally perform better in NCI-H460 than in NCI-H520. In both cell lines, the acridine-based hybrids containing P1, P2, and P3 show the most pronounced cytotoxic response. They are the only derivatives that show a response at the lowest dose tested (0.1 μm). When grouped according to common chromophore, the order of cell kill potential across the entire five sets of compounds is A1 > B1 > B2 B3 > Q1 in NCI-H460, and A1 > B1 > B2 ≈ B3 > Q1 in NCI-H520. Within each set, hybrids containing P4 produce the lowest cytotoxicity, consistent with their inability to produce permanent DNA adducts.[3] Platinum–acridine hybrids generated from P1, P2, and P3 are cytotoxic at concentrations two orders of magnitude lower than those observed for unmodified A1. For the platinum–benz[c]acridine hybrids containing B1, a less pronounced enhancement is observed, and the opposite effect appears to exist for hybrids derived from the nitrobenz[c]acridines B2 and B3. On the basis of the preliminary structure–activity relationships established in this assay and the aforementioned design rationale, P1–A1, P1–B1, and P1–B2 were selected for scaled-up synthesis and testing in an extended panel of NSCLC cell lines.
Figure 4.

Biological activity profiles of 20 compounds based on viabilities of drug-treated cells compared to untreated controls as monitored by the MTS assay. Compounds were tested in a) NCI-H460 cells and b) NCI-H520 cells dosed for 72 h with 0.1 μm drug (white bars), 1 μm drug (shaded bars) and 10 μm drug (solid bars) concentrations. Compounds chosen for further evaluation, along with their precursors, are highlighted as red bars. Plotted data are the mean of three individual experiments ± the standard deviations.
Synthesis, physicochemical characterization, and biological activity of selected target compounds
The three hybrids, P1–A1, P1–B1, and P1–B2, were generated on a larger synthetic scale utilizing the same addition chemistry as shown in Figure 3. They contain an NH3-substituted platinum moiety of intermediate reactivity and the same amidine-modified linker group, but vary by the nature of the DNA-targeted chromophores (Figure 5). All hybrids were isolated in their monocationic form. The experimental pKa values for the fully protonated, dicationic forms of the hybrids (Table 2 and the Supporting Information) are in agreement with the trends predicted from computational data and support the overall design. Compound P1–A1 shows acid–base properties characteristic of the 9-aminoacridine chromophore in platinum–acridines (typical pKa range: 9–10[19]). By contrast, P1–B1, containing a significantly less basic, unsubstituted 7-aminobenz[c]acridine system, shows a decrease of two orders of magnitude in chromophore basicity. Modification of the benz[c]acridine moiety with an electron-withdrawing 2-nitro group in P1–B2 results in another decrease in pKa by 1.5 units. Thus, at close-to-neutral pH, P1–A1 will exist as a fully protonated dication, whereas P1–B1 and P1–B2 will form partially protonated and fully deprotonated monocationic species, respectively.
Figure 5.

Hybrid agents synthesized for further evaluation.
Table 2.
IC50 values (μm) for platinum–chromophore hybrids in lung cancer cell lines[a] and acid/base properties of their fully protonated, dicationic forms.[b]
| Compd | NCI-H460[c] | NCI-H520[d] | NCI-H522[d] | NCI-H1435[d] | A549[c] | pKa |
|---|---|---|---|---|---|---|
| P1–A1 | 0.0052±0.0001 (520)[e] | 0.043±0.004 (100) | 0.010±0.001 | 0.84±0.07 | 0.0065±0.0002 | 9.4±0.2 |
| P1–B1 | 0.24±0.01 (3.8) | 0.52±0.01 (3.5) | 0.12±0.02 | 1.48±0.01 | 0.32±0.06 | 7.6±0.3 |
| P1–B2 | 2.4±0.5 (0.9) | 2.2±0.1 (1.5) | 3.62±0.08 | 11.0±0.2 | 12.4±0.9 | 4.9±0.6 |
Concentrations of compound that reduce cell viability by 50%, extracted from dose-response curves for 72 h drug incubations using a colorimetric (MTS) cell proliferation assay. Values are means of two individual experiments performed in triplicate ±standard deviations.
Determined from Henderson-Hasselbalch plots of UV-visible absorbance data (averages for three titrations ±standard deviations; see the Supporting Information).
Wild-type p53.
Mutated p53.
Values in parentheses are enhancement factors relative to Pt-free ligands A1, B1, and B2 (see Supporting Information).
The benz[c]acridine hybrids are noticeably less soluble than P1–A1 in relevant aqueous buffers, most likely due to their reduced cationic charge and the increased lipophilicity of the chromophores, which show a tendency to self-aggregate in solution (for absorbance and fluorescence spectra, see the Supporting Information). Because of the high degree of aggregation, attempts to determine reliable partition coefficients for P1–B1 and P1–B2 in octanol/water were unsuccessful. Despite these features, the chloride salts of both hybrid agents remain sufficiently soluble (min. 200 μm in cell culture media when solubilized with DMF or dmso).
IC50 values were determined for the three hybrids in large cell carcinoma (NCI-H460), squamous cell carcinoma (NCI-H520), and adenocarcinoma (NCI-H522, NCI-H1435, A549) models of NSCLC (Table 2). For comparison, inhibitory concentrations were also determined for A1, B1, and B2 in selected cell lines (Supporting Information). The data acquired for the hybrids across the whole panel of cell lines confirms the general trend observed in the pre-screen, in which P1–A1 proved to be the most cytotoxic and P1–B2 the least cytotoxic of the three derivatives. P1–A1 shows the highest cytotoxicity in NCI-H460 (wild-type p53) and NCI-H522 (mutated p53), in agreement with previously reported data on chemically related hybrids.[3,9a] IC50 values in the low nanomolar range are also observed in A549. In NCI-H1435, a notoriously cisplatin-resistant adenocarcinoma cell line,[21] P1–A1 maintains submicromolar activity. By contrast, P1–B1 and P1–B2 are less effective and require higher submicromolar and micromolar doses, respectively, to achieve the same cell kill effect.
In NCI-H460 and NCI-H520 cells, P1–A1 shows greatly enhanced cytotoxicity compared to that of A1 by 520-fold and 100-fold, respectively. A less pronounced effect of approximately 4-fold is observed for P1–B1 when compared to B1, whereas no significant difference in cytotoxicities exists between P1–B2 and B2. The largest fold variation in sensitivity of a given cell line to the three hybrids is observed for A549 (2000-fold), while the smallest variation is observed for chemoresistant NCI-H1435 (14-fold). For a specific compound, variations in IC50 values of up to 10-fold are observed across the entire panel of cell lines, with the exception of compound P1-A1, which proves to be 160-fold more active in NCI-H460 than in NCI-H1435.
Distribution and nucleic acid binding in cancer cells
To gain insight into the intracellular distribution of the hybrids, we used confocal fluorescence microscopy by taking advantage of the intrinsic blue fluorescence of the 9-aminoacridine (in P1–A1) and 7-aminobenz[c]a-cridine (in P1–B1) chromophores. (The nitrobenzacridine derivative, B2, was not sufficiently fluorescent for this application.) A known drawback of this approach is that the fluorescence of the 9-aminoacridine chromophore is significantly quenched when intercalated between DNA base pairs,[17] which complicates the microscopic detection of platinum-acridine associated with chromatin in the cell’s nucleus. (A post-labeling strategy has recently been devised that facilitates the detection of adducts in the nuclear DNA and subnuclear structures in fixed cells.[8]) By contrast, the blue fluorescence of benz[c]acridine in P1–B1, which is relatively weak as a result of self-aggregation, is slightly enhanced in the presence of calf thymus DNA (see the Supporting Information).
To study the two hybrid molecules in intact cells, NCI-H460 cells were dosed with P1–A1 and P1–B1 at relatively high concentrations (5 mm), but only for a short period of time (max. 6 h). These conditions were necessary to produce a detectable fluorescence signal in target organelles without causing undesired cell death and the cytomorphological consequences associated with it (degradation of the nuclear envelope and fragmentation of the nuclear DNA[22]). Treated cells were then co-stained with suitable fluorescent dyes to study accumulation of the hybrids in several organelles, including lysosomes, mitochondria, and endoplasmic reticulum (ER). No nuclear staining was necessary to unequivocally map the nuclei in cells in interphase, which could be readily identified in the merged and bright field images.
In all analyzed samples, cells incubated with P1–B1 for 1, 3, or 6 h showed higher fluorescence levels in the blue channel than cells treated with P1–A1, consistent with a higher uptake of the less basic and more lipophilic benz[c]acridine derivative. P1–A1 and P1–B1 show a high degree of colocalization with LysoTracker, indicating accumulation of the hybrids in this organelle (Figure 6A), but do not localize to the mitochondria (see Supporting Information). The most intense blue fluorescence at each time point is detected for both hybrids in the lysosomes. Lysosomal accumulation of P1–B1 is accompanied by a pronounced decrease of LysoTracker-related red fluorescence relative to levels in cells treated with P1–A1 (Figure 6A, lower panel, and Supporting Information). This observation may indicate that P1–B1, a protonable species at neutral cytosolic pH (pKa=7.6), but not P1–A1 (pKa=9.4), increases the lysosomal pH, resulting in reduced accumulation of the acidotropic Lyso-Tracker dye (pKa=7.5[23]).
Figure 6.

Subcellular distribution of compounds P1–A1 and P1–B1 in NCI-H460 cells. Confocal fluorescence microscopy images of cells co-stained with LysoTracker Red (A) and ER Tracker Red (B). Panel (C) shows a comparison of colocalization images captured for cells treated with P1–A1 and P1–B1 for 6 h and co-stained with LysoTracker Red. The arrows indicate blue-fluorescent intranuclear vesicular structures and regions of intense blue fluorescence. Scale bars represent a distance of 10 μm.
Cells incubated with P1–A1 for 1 h and subsequently treated with red-fluorescent ER Tracker to stain the endoplasmic reticulum of the cytoplasm show acridine-related blue fluorescence confined to distinct areas identified as lysosomes (Figure 6B, upper panel). By contrast, P1–B1 produces significant blue fluorescence in the area of the rough and smooth endoplasmic reticulum across the entire cytoplasm, in the perinuclear region, and in the nucleus (Figure 6B, lower panel). Confocal images captured of cells treated with P1–A1 and P1–B1 for 6 h and stained with LysoTracker show a high degree of perinuclear clustering of lysosomes (Figure 6C). In these areas a large number of blue-fluorescent vesicles are observed, which are not stained by LysoTracker. From these vesicles, which appear to localize to, and penetrate, the nuclear membrane, emanate areas of intense blue fluorescence in the nuclear region, possibly indicating release of fluorescent hybrid agent (see highlighted features in Figure 6C). While this causes high levels of blue fluorescence across the entire nuclear region in the case of P1–B1, relatively weaker P1–A1-related fluorescence is observed only in contrast-enhanced images captured after 6 h of continuous dosing in subnuclear structures previously identified as nucleoli[8] (Supporting Information).
Analysis of the microscopic images confirms that both P1–A1 and P1–B1 rapidly enter lung cancer cells and accumulate at high levels in the lysosomes. The fluorescence intensity observed for P1–B1 is significantly higher than for P1–A1 in the nucleus and in the cytoplasm, suggesting major differences in subcellular levels of the two hybrids. The results also suggest that lysosome-like vesicles may be involved in the cellular trafficking of the hybrid agents and their delivery to the nucleus.
Because fluorescence intensities cannot be directly correlated with the levels of irreversible damage in chromatin (Pt adducts) produced by P1–A1 and P1–B1, we extracted and purified the nuclear DNA from treated NCI-H460 cells and quantified the platinum content by inductively-coupled plasma mass spectrometry (ICP-MS). For comparison, the fraction of cellular RNA was also recovered and analyzed. Cells were dosed with P1–A1 and P1–B1 at the same time points applied in the imaging study, but at a lower concentration (1 μm for 3 and 6 h). The amounts of DNA extracted from cells treated with the hybrids, in particular for the 6 h time point, do not vary significantly from the controls (Figure 7a). This observation confirms the absence of apoptotic cell death in the population of treated cells, which would have resulted in major fragmentation and loss of DNA during the extraction procedure. By contrast, a more dramatic time-dependent reduction in extractable RNA of approximately 70% relative to control cells at the 6 h time point is observed for both P1–A1 and P1–B1 (Figure 7b), corroborating the notion that both hybrids interfere with the transcription machinery or cause post-transcriptional degradation of RNA in treated cells.
Figure 7.

Amounts of DNA (a) and RNA (b) extracted from untreated NCI-H460 cells and cells treated with 1 mm P1––A1 or P1–B1 for 3 h (black) and 6 h (white). These data were used to determine the Pt/nucleic acid ratio (c) in DNA after 3 h (black) and 6 h (shaded black), and in RNA after 3 h (gray) and 6 h (shaded gray) of continuous dosing. Data with one asterisk (*) displays no statistical difference from control at 95% confidence (α = 0.05). Data with two asterisks (**) displays no statistical difference at 95% confidence (α = 0.05), but displays a statistical difference at 90 % confidence (α = 0.10) when using a two-tailed t-test assuming unequal variances.
In DNA samples analyzed by ICP-MS, a rapid and time-dependent accumulation of platinum is observed for P1–A1 (Figure 7c) at levels consistent with data reported previously for platinum–acridines.[24] Quite unexpectedly, accumulation of platinum at similar high levels was also observed in the extracted RNA for P1–A1. A different scenario is observed for P1–B1, for which concentrations of DNA-associated platinum in cells treated for 3 and 6 h were not statistically different from control levels. Although P1–B1 does not seem to accumulate in chromosomal DNA, it is possible that this agent selectively targets noncanonical DNA motifs in the genome at levels undetectable by ICP-MS. Likewise, only slightly higher levels of platinum than in controls were detected for P1–B1 in RNA.
Interactions with G-quadruplex DNA
The cumulative data from the experiments performed in cancer cells suggest that chromosomal dsDNA may not be the primary cellular target of the benz[c]acridine containing pharmacophore and that it may trigger cell death by alternate mechanisms (see also the Discussion). One potential target of the benz[c]acridine derivatives is G-quadruplex (G4)-forming DNA, which is ubiquitous in cancer-related genomic sequences and considered a preclinically validated anticancer target.[25] It is possible to shift the π–π stacking potential of DNA intercalative molecules from classical Watson–Crick to these non-canonical DNA structures. Favorable stacking with the G4 motifs is typically observed for molecules containing extended, asymmetric aromatic moieties, such as quindoline[26] and ellipticine,[27] which are structurally similar to the benz[c]acridine scaffold, or with chromophores bearing side chains that disfavor insertion into the DNA duplex (e. g., in 3,6-disubstituted acridines[28]).
We have used a combination of circular dichroism (CD) spectroscopy and high-resolution electrospray mass spectrometry (HR-ESMS) to study the interactions of P1–A1, P1–B1, and P1–B2, as well as the platinum-free precursors A1, B1, and B2 with selected G4-forming model sequences. They include a 24-mer representing the human telomeric repeat ([T2AG3]n),[29] as well as three of the guanine-rich sequences found in the genes encoding 45S pre-ribosomal RNA (rDNA),[30] which are transcribed by RNA polymerase I (Pol I) in the cell’s nucleolus (Figure 8a).[31] The ability of A1, B1, and B2 to induce the G4 structure in each of the sequences was first tested in an unbiased fashion in a Na+- and K+-free buffer. In titrations with the three chromophores, the random-coil form of TEL24 is converted into a quadruplex structure. While A1 produces a mixture of parallel and antiparallel topologies, as indicated by the positive CD bands at ~260 nm and ~290 nm, respectively,[32] B1 and B2 stabilize antiparallel forms of the sequence (Figure 8b–d). CD spectra recorded for the titrations of the three rDNA sequences show characteristic features of parallel (major component) and antiparallel (minor component, except for B2 in NUC23A) G4 DNA (see the Supporting Information), suggesting that multiple topologies, or mixed topologies with parallel and antiparallel strand polarities, exist in solution. Among the 12 titrations, addition of B1 into a solution of TEL24 showed the most distinct change in ellipticities and gave the cleanest conversion from unfolded single-strand into a single G4 structure, based on distinct isoelliptic points. The CD features of TEL24 in complex with B1 are unique among all combinations tested, particularly the strong negative bands at ~260 and ~310 nm. While the former band is a characteristic feature of the antiparallel basket conformation of the human telomeric sequence observed in Na+ solution,[32] the band at longer wavelength most likely arises from induced circular dichroism (ICD) or exciton-coupled benz[c]acridine chromophores,[33] which absorb in that region (see the Supporting Information).
Figure 8.

Interaction of chromophores and the corresponding hybrid agents with G-quadruplex forming sequences (a) (10 μm), and new chromophore and platinum-chromophore hybrid agents. Circular dichroism (CD) spectra (b–d) of in the interaction between unfolded TEL24 (black dashed line) and increasing equivalents of a) A1, b) B1, and c) B2 to a final ratio of 5 equiv drug to one quadruplex (thick black line) (10 mm tetrabutylammonium phosphate/1 mm EDTA buffer, pH 6.75). CD spectra were also acquired of P1–A1 (red), P1–B1 (blue), and P1–B2 (green) incubated with pre-folded G-quadruplex-forming sequences (same buffer with 100 mm KCl added) for e) TEL24, f) NUC23A, g) NUC23P, and h) NUC25M. i) Induced circular dichroism (ICD) spectra for the chromophore region of P1–A1 (red), P1–B1 (blue), and P1–B2 (green) in complex with pre-folded TEL24. For representative structures of G4 topologies, see the Supporting Information.
Hybrids P1–A1, P1–B1, and P1–B2 were also incubated with the four sequences in physiologically relevant K+ buffer, in which TEL24 and NUC23A form a parallel/antiparallel hybrid structure[32] and a mixture of the two forms, respectively (Figure 8e, f). Under these conditions, NUC23P and NUC25M exist predominantly in a parallel conformation. P1–A1 causes a noticeable perturbation in the CD bands of TEL24 and NUC23A in favor of the parallel structural component, whereas P1–B1 and P1–B2 effect relatively minor structural changes (Figure 8e–h). To demonstrate differences in the binding modes in TEL24, ICD spectra were recorded in the ligand region where the π–π transitions of the aromatic chromophores are observed (325–500 nm; Figure 8i). The acridine chromophore in P1–A1 does not show significant ICD features in complex with the telomeric G-quadruplex, consistent with multiple non-specific binding modes giving rise to inefficient coupling between the DNA’s and ligand’s transition dipoles. An entirely different situation is observed for P1–B1 and P1–B2, which produce strong positive ICD bands for the transition centered at ~400 nm and ~450 nm (short-axis π–π transition), respectively, and a weaker negative band around 350 nm (long-axis π–π transition). This CD signature has been used as a diagnostic of coplanar orientation of the intercalator and adjacent nucleobases in covalent-intercalative DNA adducts,[34] which strongly supports efficient π–π stacking interactions between the G4 tetrad and benz[c]acridine chromophores.
To confirm adduct formation in the quadruplex-forming sequences, samples of the pre-folded G4 structures incubated with hybrid agent (Figure 8e–i) were also analyzed by HR-ESMS. The spectra recorded in negative-ion mode provide evidence of substitution of the chloro ligand on platinum by nucleobase nitrogen in all of the sequences for which formation of 1:1 adducts was predicted from CD/ICD data. (For mass spectra and assignments of molecular ions, see the Supporting Information.) On the basis of peak intensities and assuming in-source dissociation of the resulting platinum–DNA coordinative bonds is negligible or occurs to a similar extent for the three hybrids, P1–A1 and P1–B1 generally produce the highest level of binding, whereas P1–B2 forms adducts at a much lower yield or proves to be completely unreactive (see the Supporting Information). Thus, the combined CD and HR-MS data confirm that the hybrids form coordinative adducts with the G-quadruplex structures in which the planar chromophores are π-stacked with the guanine tetrads.
Equilibrium dialysis experiments were also conducted to establish the selectivity of the chromophores (and their platinum derivatives) for G4 versus double-stranded DNA. Unfortunately, these experiments were unsuccessful due to irreversible aggregation and deposition of the benz[c]acridine derivatives on all components of the microdialysis apparatus and membranes.
In vivo toxicity
The acridine-based hybrid agents have previously shown relatively low toxicity in non-transformed cells,[35] but were quite toxic when administered to mice.[11] The target tissue and cell-type specificity of the systemic toxicity caused by platinum-acridines are unknown. To determine if the design of P1–B1 indeed translates into improved in vivo tolerability, a dose escalation study was performed in mice. No signs of toxicity and no weight loss were observed at a dose of 3.2 mgkg−1 when administered intraperitoneally (i.p.) on five consecutive days (see the Supporting Information). By contrast, the maximum tolerated dose (MTD) for P1–A1 was found to be 0.1 mgkg−1 using the same dosing scheme.[11] These results confirm that the simple structural modification of extending the aromatic chromophore is able to reduce the acute toxicity of this type of hybrid molecule by 32-fold.
Discussion
The 7-aminobenz[c]acridine chromophore was identified as a promising new building block in platinum–intercalator hybrid agents, which may help overcome several of the drawbacks of the platinum–acridines. The goal was to generate a less genotoxic derivative of the parent platinum–acridine pharmacophore. As an additional benefit, the desired molecule would also act by an altered mechanism at the cellular and molecular level, thereby potentially overcoming resistance in repair-proficient cancers. Unsubstituted benz[c]acridine (B1) in combination with a monofunctional DNA platinating moiety was able to produce submicromolar cell kill in chemoresistant forms of lung cancer. Hybrids containing substituted nitrobenz[c]acridine B3 and quinoline Q1 were not pursued further because they showed poor activity, low solubility, and/or no significant advantages over the platinum-free chromophores.
NSCLC cell lines characterized by wild-type p53 protein were exquisitely sensitive to the acridine-based hybrid, P1–A1. P1–A1 was approximately 50-fold more potent than P1–B1 in NCI-H460 and A549 cells (Table 2). This observation suggests that a p53-mediated mechanism[36] is an important contributor to the cell kill effected by this highly DNA damaging agent. Characteristically, the cytotoxic advantage of P1–A1 is less pronounced in DNA repair-proficient cells harboring a p53 mutation where relatively higher IC50 values are observed (Table 2). The cell kill potential of P1–B1 appears to be affected by these parameters to a lesser extent, and the new derivative proved to be even more cytotoxic in NCI-H522 than in NCI-H460 despite the p53 status and the higher DNA repair activity observed in the former cell line.[21] P1–B1, while significantly less cytotoxic than P1–A1, clearly outperforms cisplatin in cell lines representing this form of cancer.[9,37]
Lung adenocarcinomas are often highly resistant to common cytotoxics,[2b,38] such as cisplatin (e. g., NCI-H1435), and show high expression levels of antiapoptotic proteins.[36,39] On the other hand, the treatment of squamous cell lung cancers (e. g., NCI-H520), which show genomic alterations similar to basal (triple-negative) breast cancer and squamous head and neck cancer, continues to rely on traditional systemic therapies.[40] Unlike the more common adenocarcinomas, they are notoriously insensitive to common molecularly targeted therapies, such as inhibitors targeted at epidermal growth factor receptor tyrosine kinase.[41] P1–B1 may be a suitable candidate for tackling these aggressive phenotypes.
The acridine- and benz[c]acridine-based agents share some common features but also show some critical differences at the cellular level. Our imaging data demonstrate that the lysosomes are a major target organelle of both agents. Excessive sequestration of drugs into this acidic organelle has been linked to multidrug resistance, but may also lead to lysosomal injury, resulting in cell death.[42] P1–A1 and P1–B1 contain weakly basic chromophores, making them prime candidates for lysosomal accumulation. Contributions of lysosome disruption to the cell death, in particular for the presumably less genotoxic benz[c]acridine derivative, cannot be ruled out.[43] On the basis of confocal images, lysosome-type vesicles may also be involved in the subcellular trafficking of the hybrid molecules and their delivery across the nuclear membrane.[44]
Previous studies on the subcellular distribution of the platinum-acridines have shown that while these agents produce approximately 60-fold higher intracellular levels than cisplatin they result in 5-fold higher DNA adduct levels than the clinical drug.[24] This observation suggests that a major fraction of the hybrid agents must be present in the cytosol or be associated with cytoplasmic organelles, which is consistent with their lysosomal accumulation observed in the current study. P1–A1 in the nucleus of NCI-H460 cells is only very weakly blue fluorescent as a consequence of the quenched fluorescence of DNA-associated 9-aminoacridine, and ICP-MS detection was required to demonstrate its accumulation in this target organelle. P1–B1 produced significantly higher fluorescence levels in cytoplasmic structures and membranes and also significantly more intense blue staining of the cells’ nuclei than compound P1–A1, but ICP-MS was unable to detect platinum in extracted nuclear DNA. A plausible explanation for this observation would be that P1–B1 enters the nucleus but does not accumulate in chromatin because DNA adduct formation is disfavored. Alternatively, it may be reasoned that the blue fluorescence and platinum do not colocalize due to decomposition of the hybrid agent, resulting in cleavage of the platinum–benz[c]acridine linkage prior to entering the nucleus. No such fragmentation, however, has been observed for relevant conditions in analytical assays mimicking the cell incubations performed with P1–B1, and this possibility can be firmly ruled out.
The reduced reactivity of P1--B1 relative to P1–A1 with nucleic acids is also reflected in the RNA binding levels. Despite the difference in nucleic acid affinity, both analogues show the same effect on the total RNA content of treated cells, which was unexpected. RNA itself is generally not considered a primary cancer target of platinum-based therapies, although intracellular platinum levels in this biomolecule are usually high.[45] A recent mechanistic study of platinum–acridines using a yeast-based chemical genomics platform indicated that severe DNA damage is the major cause of cell kill produced by these agents.[6] It also demonstrated that cytotoxic action on non-DNA targets involved in RNA metabolism as well as on components of the translational machinery (ribosomal subunits) compromise cell viability.[6] The distinct nucleic acid damage profile of P1–B1 in cancer cells warrants further experimentation to determine if RNA-mediated mechanisms may be involved in its cell kill.
Finally, there has been a longstanding interest in targeting anticancer agents to G-quadruplex DNA with the goal of triggering apoptotic signals in a more cancer-cell specific way.[25] The discovery that platinum-acridines produce adducts in the human telomeric G-quadruplex at a faster rate than in dsDNA[46] prompted us here to also explore the newly designed chromophores as G4-directed ligands. We demonstrated that benz[c]acridine alone is able to induce the G-quadruplex structure in various target sequences and, when introduced into hybrid agents, promotes platination of the DNA secondary structure without disrupting the G4 tetrad. It is unclear if this reactivity is sufficiently selective to produce significant damage in therapeutically useful G-quadruplex-forming sequences.
In conclusion, we have investigated structure–activity relationships in novel derivatives of platinum–acridines containing truncated and extended forms of the DNA-targeted 9-aminoacridine intercalating moiety. Replacement of acridine with a benz[c]acridine moiety profoundly alters the target binding properties, cellular pharmacology, and in vivo tolerability of this type of agent. If the improved safety offsets the relative loss in cytotoxic potential observed for P1–B1 and results in a more favorable therapeutic window, this derivative should have an advantage over P1–A1 in vivo. In addition to redesigning the chromophore, introduction of cleavable, lipophilic “protecting” groups may turn the benz[c]acridine-based agent into an even more tolerable prodrug (as demonstrated for the analogous acridine-based agents).[50] Finally, potential non-DNA-mediated mechanisms and mechanisms involving non-canonical nucleic acid structures remain to be investigated.
Experimental Section
Reagents and general procedures
All reagents were used as obtained from commercial sources without further purification unless indicated otherwise. Compounds A1,[18] P1,[11] and tert-butyl(2-aminoethyl)methylcarbamate[18] were synthesized according to reported procedures. Synthetic (desalted) oligodeoxyribonucleotides were purchased from IDT, Inc. (Coralville, IA). 1H NMR and proton-decoupled 13C spectra of the target compounds and intermediates were recorded on Bruker Advance 300 or Bruker DRX-500 instruments. Chemical shifts (d) are reported in parts per million (ppm) relative to the internal standard tetramethylsilane (TMS). In-line LC-ESMS analyses were performed on an Agilent 1100 LC/MSD ion trap mass spectrometer equipped with an electrospray source. HR-ESMS was performed on a Thermo Scientific LTQ Orbitrap XL equipped with an electrospray source. All NMR and LC-ESMS spectra were processed by using the MestReNova Suite (version 8.1.2) equipped with MS plugins. HR-ESMS data was processed with Xcalibur 2.1 (Thermo Scientific). The analytical purity of target compounds was determined by reverse-phase HPLC in conjunction with product analysis by ESMS and was found to be greater than 95% in all cases (see the Supporting Information). For the preparation of biological buffers, biochemical grade chemicals (Fisher/Acros) were used. HPLC grade solvents were used for all HPLC and mass spectrometry experiments. All reagents were used as obtained from commercial sources without further purification unless indicated otherwise. Stock solutions of platinum complexes in DMF (DMSO for P1–B2) were stored at −20°C and brought to room temperature immediately before use.
Computational studies
Chromophore models were built using the Gaussview5 program.[47] Optimizations and single-point energy calculations were performed with the Gaussian 09 (G09) software package.[48] All geometries were fully optimized in the gas phase at the gradient-corrected DFT level using the spin-unrestricted B3LYP functional and the 6–311G** basis set.[15] Vibrational frequencies were used to confirm that the optimized structures had converged to their local minima and the equilibrium structures contained no imaginary frequencies. Single-point energy calculations were performed using the self-consistent reaction field (SCRF) approach,[49] which simulates a constant dielectric constant of ε=78.3553 for water. All models of platinum complexes and transition states were built, optimized, and calculated according to a previously published procedure.[7]
Synthesis of N1-methyl-N2-(quinolin-4-yl)ethane-1,2-diamine (Q1)
A mixture of 4-phenoxyquinoline (Q1.1) (504 mg, 2.28 mmol), tert-butyl(2-aminoethyl)methylcarbamate (773 mg, 4.43 mmol), and freshly distilled triethylamine (1 mL) in anhydrous dioxane (1 mL) was heated at reflux for 72 h. The solvent was removed, and the residue was stirred overnight in 4m HCl. The reaction mixture was partitioned between a 10% NaOH and chloroform. The chloroform layer was washed with saturated aqueous NaCl and 2m NaOH, treated with activated carbon, dried over sodium sulfate, and filtered through a Celite pad. Evaporation of the solvent afforded Q1 as a light yellow oil, which was dried in a vacuum. Yield: 273 mg, 60%. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (300 MHz, [D6]DMSO): d=8.38 (d, J=5.3 Hz, 1H), 8.29–8.11 (m, 1H), 7.90–7.71 (m, 1H), 7.59 (ddd, J=8.3, 6.7, 1.4 Hz, 1H), 7.50–7.32 (m, 1H), 7.04 (s, 1H), 6.47 (d, J=5.4 Hz, 1H), 3.35 (t, J=6.5 Hz, 2H), 2.79 (t, J= 6.4 Hz, 2H), 2.34 ppm (s, 3H); 13C NMR (75 MHz, [D6]DMSO): δ= 150.58, 149.85, 148.20, 128.92, 128.55, 123.64, 121.51, 118.74, 98.10, 49.36, 41.95, 35.75 ppm; UV/Vis (BPES buffer, pH 7.2, 1% SDS): λmax =339 nm, ε=13008m−1cm−1.
Synthesis of N1-(benz[c]acridin-7-yl)-N2-methylethane-1,2-diamine (B1)
A mixture of 7-phenoxybenz[c]acridine (B1.3) (918 mg, 2.86 mmol), tert-butyl(2-aminoethyl)methylcarbamate (778 mg, 4.46 mmol), anhydrous triethylamine (400 μL, 2.87 mmol) in anhydrous dioxane (2 mL) was stirred at reflux for five days. The solvent was removed by rotary evaporation and the remaining residue stirred overnight in 4 mL of a 3:1 mixture of concentrated glacial acetic acid and concentrated hydrochloric acid. The acid was removed by rotary evaporation and the residue was neutralized with 2m NaOH. The reaction mixture was partitioned between the 2m NaOH solution and chloroform. The chloroform layer was washed with saturated NaCl and 2m NaOH, treated with activated carbon, dried over sodium sulfate, and filtered through a Celite pad. Rotary evaporation of the solvent afforded B1 as a brown oil, which was dried in a vacuum. Yield: 778 mg, 90%. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (300 MHz, CDCl3): δ=9.54–9.41 (m, 1H), 8.32–8.18 (m, 2H), 8.01 (d, J=9.4 Hz, 1H), 7.87–7.53 (m, 5H), 7.47 (ddd, J= 8.3, 6.7, 1.3 Hz, 1H), 6.05 (s, 1H), 3.84–3.73 (m, 2H), 2.94–2.84 (m, 2H), 2.53 ppm (s, 3H); 13C NMR (75 MHz, [D6]DMSO): δ=151.23, 147.50, 147.23, 133.38, 131.27, 129.47, 129.40, 128.55, 127.36, 126.41, 124.94, 123.31, 123.27, 123.08, 121.67, 118.09, 112.67, 51.42, 49.02, 35.66 ppm; UV/Vis (BPES buffer, pH 7.2, 1% SDS): λmax=427 nm, ε=10071m−1cm−1.
Synthesis of N1-methyl-N2-(9-nitrobenz[c]acridin-7-yl)ethane-1,2-diamine (B2)
A mixture of 9-nitro-7-chlorobenz[c]acridine (B2.3) (298 mg, 0.81 mmol) and N-methylethylenediamine (150 μL, 1.72 mmol) was stirred at reflux in anhydrous dioxane (10 mL) for 24 h. After the mixture had cooled to room temperature, the solvent was removed by rotary evaporation and the residue was suspended in 2m NaOH. After 2 h of stirring, a dark red solid was collected by vacuum filtration and washed with 2m NH4OH to afford B2 as a red solid, which was dried in a vacuum. Yield: 280 mg, 99%. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (300 MHz, [D6]DMSO): δ=9.47 (d, J=2.5 Hz, 1H), 9.30 (d, J=7.7 Hz, 1H), 8.36 (dd, J=9.4, 2.4 Hz, 1H), 8.19 (d, J=9.4 Hz, 1H), 8.10 (d, J=9.4 Hz, 1H), 8.01–7.92 (m, 1H), 7.85–7.66 (m, 3H), 3.96 (t, J=6.3 Hz, 2H), 2.93 (t, J= 6.2 Hz, 2H), 2.33 ppm (s, 3H); 13C NMR (75 MHz, [D6]DMSO): δ= 153.42, 149.57, 149.19, 141.41, 133.98, 130.76, 130.65, 129.36, 127.49, 126.74, 125.39, 123.87, 122.56, 122.35, 121.10, 114.94, 111.93, 50.96, 48.58, 35.62 ppm; UV/Vis (BPES buffer, pH 7.2, 1% SDS): λmax=441 nm, ε=4851m−1cm−1.
Synthesis of N1-methyl-N2-(10-nitrobenz[c]acridin-7-yl)ethane-1,2-diamine (B3)
A mixture of 10-nitro-7-chlorobenz[c]acridine (B3.2) (251 mg, 0.81 mmol) and N-methylethylenediamine (141 μL, 1.62 mmol) was stirred at reflux in anhydrous dioxane (4 mL) for 24 h. After the mixture had cooled to room temperature, the dioxane was removed by rotary evaporation and the residue was suspended in 2m NaOH. After 2 h of stirring, the dark red solid was collected by vacuum filtration and washed with 2m NH4OH. The crude product was partitioned between chloroform and saturated NaCl. The chloroform layer was washed with 2m NH4OH, treated with activated carbon, dried over sodium sulfate, and filtered through a Celite pad. Rotary evaporation of the solvent afforded B3 as a red solid, which was dried in a vacuum. Yield: 229 mg, 81%. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (300 MHz, [D6]DMSO): δ= 9.39–9.23 (m, 1H), 8.82 (d, J=2.5 Hz, 1H), 8.65 (d, J=9.4 Hz, 1H), 8.31–8.02 (m, 2H), 8.04–7.87 (m, 1H), 7.88–7.68 (m, 3H), 3.87 (t, J= 6.2 Hz, 2H), 2.86 (t, J=6.2 Hz, 2H), 2.30 ppm (s, 3H);13C NMR (75 MHz, [D6]DMSO): δ=151.31, 148.87, 147.48, 146.22, 133.57, 130.75, 129.25, 127.54, 126.91, 126.21, 125.14, 125.06, 124.70, 121.23, 120.30, 115.16, 113.34, 51.22, 48.80, 35.64 ppm; UV/Vis BPES Buffer, pH 7.2 with 1% SDS): λmax=403 nm, ε=6922m−1cm−1.
Synthesis of [PtCl(NH3)2(N-(2-(acridin-9-ylamino)ethyl)-N-methylpropionimidamide)]Cl (P1-A1)
(An improved procedure for this previously synthesized[11] derivative is described.) A mixture of [PtCl(EtCN)(NH3)2]Cl (P1) (107 mg, 0.30 mmol) and N1-(acridin-9-yl)-N2-methylethane-1,2-diamine (A1) (81 mg, 0.32 mmol) in anhydrous DMF (2 mL) was stored at −20°C for 72 h. Activated carbon was added to the DMF solution, which was then filtered through a 0.2 μm membrane into vigorously stirred anhydrous diethyl ether. The yellow precipitate was recovered by vacuum filtration, washed excessively with diethyl ether, and dried in a vacuum to afford P1–A1 as a yellow solid. Yield: 162 mg, 88%. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (500 MHz, [D4]MeOH): δ=8.25 (d, J=8.6 Hz, 2H), 7.92–7.55 (m, 5H), 7.35 (dd, J=9.6, 7.5 Hz, 2H), 4.06 (t, J=6.4 Hz, 2H), 3.74 (t, J= 6.3 Hz, 2H), 3.19–2.86 (m, 5H), 1.27 ppm (t, J=7.5 Hz, 4H); 13C NMR (75 MHz, [D4]MeOH): δ=171.62, 155.25, 132.36, 129.24, 126.62, 125.96, 123.66, 29.08, 11.53 ppm; HR-ESMS (positive-ion mode): m/z for C19H28ClN6Pt [M]+, calcd: 570.1706; found: 570.1073 (tolerance: 0.549 ppm).
Synthesis of [PtCl(NH3)2(N-(2-(benz[c]acridin-7-ylamino)ethyl)-N-ethylpropionimidamide)]Cl (P1-B1)
A mixture of [PtCl(EtCN)(NH3)2]Cl (P1) (251 mg, 0.70 mmol) and N1-(benz[c]acridin-7-yl)-N2-methylethane-1,2-diamine (B1) (325 mg, 1.02 mmol) in anhydrous DMF (1.5 mL) was stirred at 4°C for four days and then slowly warmed to room temperature and allowed to stir for an additional 24 h. The solvent was removed by vacuum distillation at 35°C, and the resulting brown residue was redissolved in a minimal amount of anhydrous MeOH. The solution was treated with a small amount of activated carbon, filtered through a 0.2 μm membrane into vigorously stirred anhydrous diethyl ether, and allowed to stir for 24 h. A yellow precipitate formed, which was recovered by vacuum filtration, washed with diethyl ether, and dried in a vacuum to afford P1–B1 as a yellow solid. Yield: 385 mg, 83%. Analytical purity: ≥ 90% by LC-ESMS. Prior to biological testing P1–B1 was subjected to additional purification by dissolving the product in anhydrous CH2Cl2, passing the solution through a celite pad, and recovering batches of precipitated product after reducing the solvent volume by rotary evaporation. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (500 MHz, [D4]MeOH): δ=9.24 (d, J=5.3 Hz, 1H), 8.35–8.22 (m, 1H), 8.23–8.12 (m, 1H), 8.00 (d, J=9.3 Hz, 1H), 7.92–7.79 (m, 1H), 7.79–7.43 (m, 5H), 3.87 (t, J=6.6 Hz, 2H), 3.66 (t, J=6.6 Hz, 2H), 2.92 (d, J = 35.3 Hz, 7H), 1.22 ppm (t, J=7.6 Hz, 3H); 13C NMR (126 MHz, [D4]MeOH): δ=169.94, 151.27, 147.66, 134.07, 129.71, 128.69, 128.36, 127.48, 126.56, 124.82, 124.52, 124.07, 122.59, 120.54, 118.78, 113.90, 10.19 ppm; HR-ESMS (positive-ion mode): m/z for C19H28ClN6Pt [M]+, calcd: 620.1863; found: 620.1862 (tolerance: 0.134 ppm).
Synthesis of [PtCl(NH3)2(N-methyl-N-(2-((9-nitrobenz[c]acridin-7-yl)amino)ethyl)propionimidamide)]Cl 0.33 DMF (P1–B2·0.33DMF)
This derivative was generated by the same procedure as P1–B1. Starting from P1 (203 mg, 0.57 mmol) and N1-methyl-N2-(9-nitrobenz[c]acridin-7-yl)ethane-1,2-diamine (B2) (297 mg, 0.86 mmol) in anhydrous DMF (5 mL), 166 mg (41%) of the DMF solvate of P1–B2 were recovered. Analytical purity: ≥ 95% by LC-ESMS. 1H NMR (500 MHz, [D6]DMSO): δ=9.49–9.38 (m, 1H), 9.31 (d, J=8.0 Hz, 1H), 8.40 (dd, J=9.4, 2.4 Hz, 1H), 8.20 (dd, J=32.7, 9.5 Hz, 2H), 8.07–7.67 (m, 4H), 6.13 (s, 1H), 4.29 (s, 3H), 3.98 (d, J=24.8 Hz, 5H), 3.81 (t, J=7.1 Hz, 2H), 3.02 (s, 5H), 1.24 ppm (s, 3H); 13C NMR (126 MHz, [D6]DMSO): δ=168.81, 153.25, 149.54, 149.42, 141.89, 134.07, 130.95, 130.85, 129.58, 127.69, 126.98, 125.46, 124.46, 122.55, 122.27, 121.18, 115.40, 112.64 ppm; HR-ESMS (positive-ion mode): m/z for C23H29O2ClN7Pt [M]+, calcd: 665.1719; found: 665.1701 (tolerance: 2.766 ppm).
For synthetic details of new precursors and intermediates, see the Supporting Information.
Combinatorial library
Stock solutions of the chromophores (Q1, A1, B1, B2, B3) and the platinum complexes (P1, P2, P3, P4) were prepared in dry DMF at a concentration of 300 mm. To 0.6 mL micro-centrifuge tubes were added 20 μL of each chromophore, and the solutions were cooled to −20°C. Aliquots of the platinum stock solutions/suspensions were homogenized by pipetting and added directly to the cooled chromophores. Mixtures were incubated at 4°C for seven days in a benchtop multi-tube vortexer. Small aliquots were removed from each reaction and diluted with methanol containing 0.1% (v/v) formic acid to generate 15 μm samples for in-line LC-ESMS analysis of precursor conversion and product identity. Chromatographic separations were performed with a 2.1× 30 mm Rapid Resolution ZORBAX StableBond C-18 (3.5 μm) column, which was maintained at 40°C. The LC-ESMS analysis was performed on an Agilent 1100 LC/MSD ion trap mass spectrometer with an autosampler thermostatted at 4°C using optimized separation and ionization conditions for each sample (see the Supporting Information). Prior to dosing cancer cells, micro-scale reactions were first diluted to 10 mm in DMF and then serially diluted with media to a final (non-toxic) DMF content of less than 1%. The cytotoxicity of the 20 combinations was assessed at chromophore/platinum concentrations of 0.1 μm, 1.0 μm, and 10 μm in NCI-H460 and NCI-H520 cells using a previously reported colorimetric cell proliferation assay.[20]
Cell culture
The human NSCLC cell lines, NCI-H460 (large cell), NCI-H520 (squamous cell), NCI-H522, NCI-H1435, and A549 (adenocarcinomas) were obtained from the American Type Culture Collection (Rockville, MD, USA). NCI-H460, NCI-H520, and NCI-H522 cells were cultured in RPMI-1640 media (HyClone) supplemented with 10% fetal bovine serum (FBS), 10% penstrep (P&S), 10% l-glutamine, and 1.5 gL−1 NaHCO3. A549 cells were cultured in HAM’s F12 K media (Gibco) with the same additives as above. NCI-H1435 cells were cultured in serum-free 1:1 DMEM/F12 media (Gibco) containing 2.436 gL−1 NaHCO3, 0.02 mg mL−1 insulin, 0.01 mg mL−1 transferrin, 25 nm sodium selenite, 50 nm hydrocortisone, 1 ng mL−1 epidermal growth factor, 0.01 mm ethanolamine, 0.01 mm phosphorylethanolamine, 100 pM triiodothyronine, 0.5% (w/v) bovine serum albumin (BSA), 10 mm HEPES, 0.5 mm sodium pyruvate, and an extra 2 mm L-glutamine (final concentration 4.5 mm). Cells were incubated at a constant temperature at 37°C in a humidified atmosphere containing 5% CO2 and were subcultured every 2–3 days to maintain cells in logarithmic growth. The slowly dividing NCI-H1435 cells were subcultured every 7 days.
Cytotoxicity assay
The cytotoxicity studies were carried out according to a standard protocol using the Celltiter 96 aqueous nonradioactive cell proliferation assay kit (Promega, Madison, WI). Stock solutions (10 mm) of the test compounds were prepared in DMF, except for P1–B2 which was prepared in DMSO, and serially diluted with media prior to incubation with cancer cells. In all cases, cells were incubated with drug for 72 h before quantifying cell viability using the MTS assay. Relative cell viability was determined from the viability of treated and untreated (control) cells. IC50 values were calculated from sigmoidal curve fits using the appropriate dose-response equation in GraphPad Prism (GraphPad Software, La Jolla, CA).
Confocal microscopy
NCI-H460 cells were seeded into poly-d-lysine coated glass-bottom Petri dishes (MatTeck Corporation, Ashland, MD, USA) with 105 cells mL−1 suspended in 2 mL of medium per dish. Cells were incubated overnight and then treated with P1–A1, P1–B1 (5 μm), or medium for controls for 1, 3, and 6 h. After treatment, medium was removed and the cells were stained with 75 nm of LysoTracker Red DND-99 (Invitrogen), 1 μm of ER-Tracker™ Red (BODIPY® TR Glibenclamide, Invitrogen), and 100 nm MitoTracker Deep Red TM (Invitrogen) in 2 mL pre-warmed Hank’s Balanced Salt Solution (HBSS) at 37°C for 30 min. After staining with each organelle marker, solutions were replaced with fresh medium. Images were collected using a Zeiss LSM 710 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY) using a 63× (PLAN APO, 1.2 NA) objective lens. All images were acquired in multi-track configuration mode to minimize excitation cross talk and emission bleed-through. The fluorescence of P1–A1 (blue), P1–B1 (blue), Lyso–Tracker Red DND-99, ER-Tracker Red, and MitoTracker Deep Red, was excited/collected at 405/477, 440/470, 561/591, 561/636, or 633/661 nm, respectively. ZEN software (blue edition, Carl Zeiss, 2011) was used for image processing. Confocal image planes for each channel were not contrast-adjusted (unless indicated) or otherwise changed. Panels were assembled and annotated without any additional manipulation of images in Adobe Photoshop CS2.
Extraction of cellular nucleic acids
The nucleic acid content was extracted from cells and purified using the Qiagen AllPrep DNA/RNA Mini Kit (Qiagen, Alameda, CA, USA). 1.5 million exponentially-growing NCI-H460 cells were seeded into 60 mm cell culture dishes with 2 mL of media and were allowed to attach for 24 h. Cells were dosed with 1 μm P1–A1 or P1–B1 at 37°C for 3 and 6 h. Experiments were performed in triplicate for each derivative and time point. Cells were washed three times in cold PBS, trypsinized, and removed from the dishes using PBS washes. Samples were centrifuged at 1500 rpm for 5 min at 4°C to generate cell pellets, which were resuspended in 5 mL of cold PBS and again centrifuged at 1500 rpm for 5 min at 4°C. The supernatant was removed and the DNA/RNA was extracted from the pellets according to the manufacturer’s protocol. The concentration of recovered nucleic acids and the purity of each sample were determined from absorbances at 260 and 280 nm (triplicate readings).
ICP-MS analysis
The concentration of Pt was determined by quadrupole inductively coupled mass spectrometry (ICP-MS). Prior to analysis, samples were subjected to a rigorous digestion procedure in the presence of high-purity, Ultrex-grade nitric (HNO3) and hydrochloric (HCl) acids (J.T. Baker, Phillipsburg, NJ). Nominal 0.200 mL aliquots of each acid were added to the samples directly in 15 mL centrifuge tubes (NUNC, Rochester, NY), along with 0.600 mL of approximately 18-MW quality deionized water (DI H2O), obtained from a Pure Water Solutions (Hillsborough, NC) system. Samples were then capped and placed in a controlled temperature water bath (VWR, Model 750HT) maintained at 90°C. After four hours, samples were removed from the bath and were allowed to cool to room temperature before adding of DI H2O to a total volume of 4 mL. An aliquot (3 mL) of each digested sample was transferred to an ICP-MS acid-washed autosampler tube and was fortified to contain a nominal 10 ng mL−1 concentration of bismuth (Bi) internal standard. Prior to the sample analysis, the Thermo X-Series II ICP-MS (Waltham, MA, USA) was calibrated with Pt-containing acid matrix matched standards ranging from 0.001 to 5.00 ng PtmL−1. Single element Pt and Bi stock solutions traceable to the National Institute of Standards and Technology (NIST) were obtained from a commercial vendor (High Purity Standards, Charleston, SC) and were used to prepare all solutions. After calculating pg Pt/μg DNA values, few outliers were identified using Grubb’s outlier test at 90% confidence and removed from the data set. For additional details of the sample analysis and quality control, see the Supporting Information.
CD spectroscopy
CD spectra were recorded in quartz cuvettes at 25°C on an AVIV Model 215 Circular Dichroism Spectrometer equipped with a thermoelectrically controlled cell holder in the 200–350 nm range with 1 nm resolution and an averaging time of 2 s. Each spectrum was baseline corrected using a blank spectrum and smoothed using a moving average of 10–15 periods. DNA sequences were dissolved in buffer (10 mm tetrabutylammonium phosphate-monobasic, 1 mm EDTA, pH 6.75) and diluted to a final concentration of 10 mm (strand). DNA samples for incubations with the hybrids were prepared in the same buffer supplemented with 100 mm KCl. Stock solutions of A1, B1, B2, P1–A1, and P1–B1 were prepared in anhydrous DMF to a final concentration of 1 mm. P1–B2 was dissolved in DMSO. Samples were mixed for 30 s and allowed to equilibrate for an additional 30 s before spectra were acquired. To allow proper folding of the sequences into G-quadruplexes, samples were heated to 95°C for 10 min and slowly cooled overnight in a dry bath heat block. Pre-folded DNA sequences were incubated with 1.05 equivalents of hybrid agents for 48 h at 37°C. CD spectra were recorded of each untreated DNA sample and each treated sample using the acquisition parameters described above. CD spectra in the ligand (ICD) region of each hybrid agent in complex with TEL24 were recorded in the 325–500 nm range at a concentration of 65 μm (strand).
HR-ESMS analysis of platinum-modified G-quadruplex sequences
All HR-ESMS spectra were recorded on a Thermo Scientific LTQ Orbitrap XL. Unmodified and platinum-modified G-quadruplex sequences in annealing buffer were desalted using Amicon Ultra-0.5 mL centrifugal filters with a 3 kDa molecular weight cut-off (Millipore Corporation). Samples were diluted to a final concentration of 1 μm in 50% 5 mm ammonium acetate in water (pH 7.2)/50% 5 mm ammonium acetate in methanol and introduced into the electrospray ionization source by direct injection utilizing the same solvent system at a flow rate of 100 μL min−1. Spectra were recorded in negative-ion mode. Blank injections were run between samples to avoid carryover.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (Grant CA101880) and Wake Forest Innovations. HR-ESMS spectra were acquired on a Thermo Scientific LTQ Orbitrap instrument (NSF grant 947028). Computations were performed on the Wake Forest University DEAC Cluster, a centrally managed resource with support provided in part by the University. We thank Dr. Marcus W. Wright and Tiffany K. West for technical assistance.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201404845.
References
- [1].Kelland L. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- [2].a) Burris HA., III Oncogene. 2009;28(Suppl 1):S4–13. doi: 10.1038/onc.2009.196. [DOI] [PubMed] [Google Scholar]; b) Seve P, Dumontet C. Curr. Appl. Phys. Curr. Med. Chem. Anticancer Agents. 2005;5:73–88. doi: 10.2174/1568011053352604. [DOI] [PubMed] [Google Scholar]; c) Archer VR, Billingham LJ, Cullen MH. Oncologist. 1999;4:470–477. [PubMed] [Google Scholar]; d) Gloeckler Ries LA, Eisner MP. Fast Facts: An Interactive Tool for Access to SEER Cancer Statistics. National Cancer Institute; Bethesda: 2011. pp. 73–80. http://seer.cancer.gov/faststats. [Google Scholar]
- [3].Suryadi J, Bierbach U. Chem. Eur. J. 2012;18:12926–12934. doi: 10.1002/chem.201202050. [DOI] [PubMed] [Google Scholar]
- [4].Momekov G, Bakalova A, Karaivanova M. Curr. Med. Chem. 2005;12:2177–2191. doi: 10.2174/0929867054864877. [DOI] [PubMed] [Google Scholar]
- [5].a) Park GY, Wilson JJ, Song Y, Lippard SJ. Proc. Natl. Acad. Sci. USA. 2012;109:11987–11992. doi: 10.1073/pnas.1207670109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Margiotta N, Savino S, Gandin V, Marzano C, Natile G. ChemMedChem. 2014;9:1161–1168. doi: 10.1002/cmdc.201402028. [DOI] [PubMed] [Google Scholar]
- [6].Cheung-Ong K, Song KT, Ma Z, Shabtai D, Lee AY, Gallo D, Heisler LE, Brown GW, Bierbach U, Giaever G, Nislow C. ACS Chem. Biol. 2012;7:1892–1901. doi: 10.1021/cb300320d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kostrhunova H, Malina J, Pickard AJ, Stepankova J, Vojtiskova M, Kasparkova J, Muchova T, Rohlfing ML, Bierbach U, Brabec V. Mol. Pharm. 2011;8:1941–1954. doi: 10.1021/mp200309x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ding S, Qiao X, Suryadi J, Marrs GS, Kucera GL, Bierbach U. Angew. Chem. Int. Ed. 2013;52:3350–3354. doi: 10.1002/anie.201210079. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013;125:3434–3438. [Google Scholar]
- [9].a) Smyre CL, Saluta G, Kute TE, Kucera GL, Bierbach U. ACS Med. Chem. Lett. 2011;2:870–874. doi: 10.1021/ml2001888. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guddneppanavar R, Bierbach U. Anticancer Agents Med. Chem. 2007;7:125–138. doi: 10.2174/187152007779313991. [DOI] [PubMed] [Google Scholar]
- [10].Huttunen KM, Raunio H, Rautio J. Pharmacol. Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- [11].Ma Z, Choudhury JR, Wright MW, Day CS, Saluta G, Kucera GL, Bierbach U. J. Med. Chem. 2008;51:7574–7580. doi: 10.1021/jm800900g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hurley LH. Nat. Rev. Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- [13].Denny WA. In: DNA and RNA Binders. Demeunynck M, Bailly C, Wilson WD, editors. Vol. 2. VCH-Wiley; Weinheim: 2003. pp. 482–502. [Google Scholar]
- [14].Lullmann H, Mohr K, Ziegler A, Bieger D. Color Atlas of Pharmacology. 2ednd ed Thieme Medical Publishers; Stuttgart: 2000. p. 386. [Google Scholar]
- [15].a) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]; b) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- [16].Álvarez-Diduk R, Ramirez-Silva MT, Galano A, Merkoci A. J. Phys. Chem. B. 2013;117:12347–12359. doi: 10.1021/jp4049617. [DOI] [PubMed] [Google Scholar]
- [17].Baruah H, Rector CL, Monnier SM, Bierbach U. Biochem. Pharmacol. 2002;64:191–200. doi: 10.1016/s0006-2952(02)01107-3. [DOI] [PubMed] [Google Scholar]
- [18].Martins ET, Baruah H, Kramarczyk J, Saluta G, Day CS, Kucera GL, Bierbach U. J. Med. Chem. 2001;44:4492–4496. doi: 10.1021/jm010293m. [DOI] [PubMed] [Google Scholar]
- [19].Acheson RM. In: The Chemistry of Heterocyclic Compounds. Weissberger A, editor. Interscience Publishers; New York: 1956. [Google Scholar]
- [20].Ding S, Qiao X, Kucera GL, Bierbach U. J. Med. Chem. 2012;55:10198–10203. doi: 10.1021/jm301278c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Weaver DA, Crawford EL, Warner KA, Elkhairi F, Khuder SA, Willey JC. Mol. Cancer. 2005;4:18. doi: 10.1186/1476-4598-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Saraste A, Pulkki K. Cardiovasc. Res. 2000;45:528–537. doi: 10.1016/s0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- [23].Johnson I, Spence MTZ. A Guide to Fluorescent Probes and Labeling Technologies. 11edth ed Life Technologies Corporation; Carlsbad: 2010. [Google Scholar]
- [24].Qiao X, Zeitany AE, Wright MW, Essader AS, Levine KE, Kucera GL, Bierbach U. Metallomics. 2012;4:645–652. doi: 10.1039/c2mt20031g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Balasubramanian S, Hurley LH, Neidle S. Nat. Rev. Drug Discovery. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dai J, Carver M, Hurley LH, Yang D. J. Am. Chem. Soc. 2011;133:17673–17680. doi: 10.1021/ja205646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ghosh S, Kar A, Chowdhury S, Dasgupta D. Biochemistry. 2013;52:4127–4137. doi: 10.1021/bi400080t. [DOI] [PubMed] [Google Scholar]
- [28].Moore MJB, Schultes CM, Cuesta J, Cuenca F, Gunaratnam M, Tanious FA, Wilson WD, Neidle S. J. Med. Chem. 2006;49:582–599. doi: 10.1021/jm050555a. [DOI] [PubMed] [Google Scholar]
- [29].Blackburn EH. Biochemistry. 1997;62:1196–1201. [PubMed] [Google Scholar]
- [30].Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP, Lim JK, Von Hoff D, Anderes K, Rice WG. Cancer Res. 2009;69:7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- [31].Pickard AJ, Bierbach U. ChemMedChem. 2013;8:1441–1449. doi: 10.1002/cmdc.201300262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gray RD, Li J, Chaires JB. J. Phys. Chem. B. 2009;113:2676–2683. doi: 10.1021/jp809578f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fornasiero D, Kurucsev T. Biophys. Chem. 1985;23:31–37. doi: 10.1016/0301-4622(85)80061-2. [DOI] [PubMed] [Google Scholar]
- [34].Baruah H, Wright MW, Bierbach U. Biochemistry. 2005;44:6059–6070. doi: 10.1021/bi050021b. [DOI] [PubMed] [Google Scholar]
- [35].Hess SM, Anderson JG, Bierbach U. Bioorg. Med. Chem. Lett. 2005;15:443–446. doi: 10.1016/j.bmcl.2004.10.049. [DOI] [PubMed] [Google Scholar]
- [36].Siddik ZH. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- [37].a) Graham LA, Suryadi J, West TK, Kucera GL, Bierbach U. J. Med. Chem. 2012;55:7817–7827. doi: 10.1021/jm300879k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Olaussen KA, Adam J, Vanhecke E, Vielh P, Pirker R, Friboulet L, Popper H, Robin A, Commo F, Thomale J, Kayitalire L, Filipits M, Le Chevalier T, Andre F, Brambilla E, Soria JC. Lung Cancer. 2013;80:216–222. doi: 10.1016/j.lungcan.2013.01.014. [DOI] [PubMed] [Google Scholar]; c) Chen J, Emara N, Solomides C, Parekh H, Simpkins H. Cancer Chemother. Pharmacol. 2010;66:1103–1111. doi: 10.1007/s00280-010-1268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rabik CA, Fishel ML, Holleran JL, Kasza K, Kelley MR, Egorin MJ, Dolan ME. J. Pharmacol. Exp. Ther. 2008;327:442–452. doi: 10.1124/jpet.108.141291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].a) Fujii T, Toyooka S, Ichimura K, Fujiwara Y, Hotta K, Soh J, Suehisa H, Kobayashi N, Aoe M, Yoshino T, Kiura K, Date H. Lung Cancer. 2008;59:377–384. doi: 10.1016/j.lungcan.2007.08.025. [DOI] [PubMed] [Google Scholar]; b) Wakelee H, Dubey S, Gandara D. Oncologist. 2007;12:331–337. doi: 10.1634/theoncologist.12-3-331. [DOI] [PubMed] [Google Scholar]
- [39].Stewart DJ. Crit. Rev. Oncol. Hematol. 2010;75:173–234. doi: 10.1016/j.critrevonc.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].The Cancer Genome Atlas Research Network Nature. 2012;489:519–525. [Google Scholar]
- [41].Dempke WC, Suto T, Reck M. Lung Cancer. 2010;67:257–274. doi: 10.1016/j.lungcan.2009.10.012. [DOI] [PubMed] [Google Scholar]
- [42].Kirkegaard T, Jaattela M. Biochim. Biophys. Acta Mol. Cell Res. 2009;1793:746–754. doi: 10.1016/j.bbamcr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- [43].Sancho-Martinez SM, Prieto-Garcia L, Prieto M, Lopez-Novoa JM, Lopez-Hernandez FJ. Pharmacol. Ther. 2012;136:35–55. doi: 10.1016/j.pharmthera.2012.07.003. [DOI] [PubMed] [Google Scholar]
- [44].Sakhrani NM, Padh H. Drug Des. Dev. Ther. 2013;7:585–599. doi: 10.2147/DDDT.S45614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a) Akaboshi M, Kawai K, Maki H, Akuta K, Ujeno Y, Miyahara T. Jap. J. Cancer Res. 1992;83:522–526. doi: 10.1111/j.1349-7006.1992.tb01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hostetter AA, Osborn MF, DeRose VJ. ACS Chem. Biol. 2012;7:218–225. doi: 10.1021/cb200279p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rao L, Bierbach U. J. Am. Chem. Soc. 2007;129:15764–15765. doi: 10.1021/ja077390a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dennington R, Keith T, Millam J. GaussView, Version 5. Semichem Inc., Shawnee Mission; 2009. [Google Scholar]
- [48].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Laham A, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 09, Revision D.01. Gaussian Inc.; Wallingford: 2009. [Google Scholar]
- [49].Miertus S, Scrocco E, Tomasi J. Chem. Phys. 1981;55:117–129. [Google Scholar]
- [50].Ding S, Pickard AJ, Kucera GL, Bierbach U. Chem. Eur. J. 2014 doi: 10.1002/chem.201404675. DOI: 10.1002/chem.201404675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.