

Abstract
Cortical interneurons activate GABA-A receptors to rapidly control electrical and biochemical signaling at pyramidal neurons. Different populations of interneurons are known to uniquely target the soma and dendrites of pyramidal neurons. However, the ability of these interneurons to inhibit Ca2+ signaling at spines and dendrites is largely unexplored. Here we use whole-cell recordings, two-photon microscopy, GABA uncaging and optogenetics to study dendritic inhibition at layer 5 (L5) pyramidal neurons in slices of mouse PFC. We first show that GABA-A receptors strongly inhibit action potential (AP)-evoked Ca2+ signals at both spines and dendrites. We find robust inhibition over tens of milliseconds that spreads along the dendritic branch. However, we observe no difference in the amount of inhibition at neighboring spines and dendrites. We then examine the influence of interneurons expressing parvalbumin (PV), somatostatin (SOM), or 5HT3a receptors. We determine that these populations of interneurons make unique contacts onto the apical and basal dendrites of L5 pyramidal neurons. We also show that SOM and 5HT3a but not PV interneurons potently inhibit AP Ca2+ signals via GABA-A receptors at both spines and dendrites. These findings reveal how multiple interneurons regulate local Ca2+ signaling in pyramidal neurons, with implications for cortical function and disease.
Keywords: calcium signaling, dendrite, inhibition, interneuron, prefrontal cortex, spine
Introduction
Rapid inhibition throughout the cerebral cortex is mediated by ionotropic GABA-A receptors (Isaacson and Scanziani, 2011). These receptors are found across the entire somatodendritic axis of pyramidal neurons (Fritschy and Mohler, 1995; Kubota et al., 2007). At the soma and axon, GABA-A receptor inhibition controls the timing of action potential (AP) firing (Freund and Katona, 2007). At the dendrites, inhibition regulates both electrical and biochemical signaling (Spruston, 2008; Palmer et al., 2012a). For example, inhibitory inputs onto the apical dendrites limit synaptic responses and local spiking (Kim et al., 1995; Larkum et al., 1999; Murayama et al., 2009). However, the impact of GABA-A receptors on local Ca2+ signaling is largely unexplored, particularly at the level of spines.
APs from the soma backpropagate into dendrites of pyramidal neurons to trigger Ca2+ signals that shape excitability and synaptic plasticity (Bloodgood and Sabatini, 2007). GABA-A receptor inhibition of AP Ca2+ signals strongly regulates the induction of synaptic plasticity (Hayama et al., 2013). Activation of these receptors is often thought to broadly inhibit a given dendritic branch (Liu, 2004; Gidon and Segev, 2012). Consistent with this idea, GABA-A receptors inhibit AP Ca2+ signals in the dendrites of hippocampal pyramidal neurons (Tsubokawa and Ross, 1996; Kanemoto et al., 2011). However, in layer 2/3 (L2/3) pyramidal neurons, equivalent inhibition of AP Ca2+ signals is highly restricted to only a subpopulation of spines and not their neighboring dendrites (Chiu et al., 2013). These findings raise the intriguing possibility that GABAergic interneurons may be able to precisely regulate local Ca2+ signaling within individual spines.
Multiple populations of interneurons are poised to activate GABA-A receptors and inhibit pyramidal neurons. These interneurons have distinct morphological, physiological, and molecular properties (Markram et al., 2004; Ascoli et al., 2008; DeFelipe et al., 2013), and make unique subcellular connections (Kawaguchi and Kubota, 1997; Somogyi et al., 1998; Di Cristo et al., 2004). For example, parvalbumin-expressing (PV) interneurons mainly contact the soma and axon initial segment. In contrast, many somatostatin-expressing (SOM) interneurons primarily innervate the distal dendrites. A third population of interneurons expresses 5HT3a receptors, but their subcellular targeting is unexamined (Rudy et al., 2011). While little is known about how these three types of interneurons regulate AP Ca2+ signals, recent results suggest that SOM interneurons selectively inhibit a subset of spines and not their nearby dendrites (Chiu et al., 2013).
Here we examine GABA-A receptor inhibition of AP Ca2+ signals at L5 pyramidal neurons in the mouse PFC. We first show that activation of GABA-A receptors strongly inhibits AP Ca2+ signals at both spines and dendrites. We find that inhibition occurs over tens of milliseconds and broadly extends within individual dendritic branches. We then show that PV, SOM, and 5HT3a interneurons make distinct functional connections onto the apical and basal dendrites. Finally, we show that different classes of interneurons have contrasting influences on AP Ca2+ signals. Together, these results indicate that multiple interneurons regulate local Ca2+ signaling at both spines and dendrites. Our findings have important implications for the role of rapid dendritic inhibition at cortical pyramidal neurons.
Materials and Methods
Preparation.
L5 pyramidal neurons and interneurons were studied in acute coronal slices of the prelimbic medial PFC from P21–P30 wild-type, heterozygous PV-Cre (Hippenmeyer et al., 2005), heterozygous SOM-Cre (Taniguchi et al., 2011), or heterozygous 5HT3a-Cre (Gong et al., 2007) male and female mice in a C57BL/6 or mixed background. Mice were anesthetized with a lethal dose of ketamine and xylazine and perfused intracardially with ice-cold external solution containing the following (in mm): 65 sucrose, 75 NaCl, 25 NaHCO3, 1.3 NaH2PO4, 25 glucose, 2.5 KCl, 1 CaCl2, 5 MgCl2, 0.4 Na-ascorbate, and 2 Na-pyruvate (295–305 mOsm), bubbled with 95% O2/5% CO2. Coronal slices (300 μm thick) were cut in ice-cold external solution and transferred to ACSF containing the following (in mm): 120 NaCl, 25 NaHCO3, 1.3 NaH2PO4, 20 glucose, 2.5 KCl, 2 CaCl2, 1 MgCl2, 0.4 Na-ascorbate, and 2 Na-pyruvate (295–305 mOsm), bubbled with 95% O2/5% CO2. After holding for 30 min at 35°C, slices were allowed to recover for 30 min at 24°C. All experiments were conducted at 30−32°C. For all experiments, 10 μm NBQX, 10 μm CPP, and 2 μm CGP-55845 were added to block AMPA, NMDA, and GABA-B receptors, respectively. For mapping experiments, 1 μm TTX was added to block AP propagation and 100 μm 4-AP was added to restore presynaptic release. For some experiments, 10 μm gabazine was added to block GABA-A receptors. All chemicals were from Sigma or Tocris Bioscience.
Stereotaxic injections.
P14–P17 mice were subcutaneously injected with 0.02 mg/kg atropine 10 min before surgery and then deeply anesthetized with 40 mg/kg ketamine and 5 mg/kg xylazine. PFC injection site coordinates were relative to bregma (dorsoventral axis, mediolateral axis, and rostrocaudal axis: −2.3, +0.4, and +2.0 mm, respectively). Borosilicate pipettes with 5–10 μm tip diameters were backfilled with 1 μl of AAV2/9-EF1a-dflox-hChR2(H134R)-mCherry (Penn Vector Core). Twenty to 25 boluses of virus (23 nl each) were pressure injected (Nanoject II; Drummond), with 30 s spacing between injections, for a total volume of 460–575 nl. After the final injection, the pipette was left in place for an additional 5 min to allow the virus to diffuse away from the pipette tip, and was then slowly removed from the brain. Animals were returned to their cages for 1–2 weeks before recordings.
Electrophysiology.
Whole-cell recordings were obtained from L5 pyramidal neurons located 450–550 μm from the pia and identified by infrared-differential interference contrast, as previously described (Chalifoux and Carter, 2011b). Interneurons expressing ChR2-mCherry were identified by their fluorescently labeled cell bodies. Borosilicate pipettes (2–5 MΩ) were filled with one of two internal solutions. Current-clamp recordings used the following (in mm): 135 K-gluconate, 7 KCl, 10 HEPES, 10 Na-phosphocreatine, 4 Mg2-ATP, and 0.4 Na-GTP, 290–295 mOsm, pH 7.35, with KOH. Voltage-clamp recordings used the following (in mm): 135 Cs-gluconate, 10 HEPES, 10 Na-phosphocreatine, 4 Mg2-ATP, and 0.4 Na-GTP, 10 EGTA, 10 TEA-chloride, and 2 QX314, 290–295 mOsm, pH 7.35, with CsOH. Both solutions contained 30 μm Alexa Fluor 594 to image morphology. Current-clamp recordings also included 200 μm Fluo-5F to monitor Ca2+ signals. This concentration of Fluo-5F was chosen to maximize detection of AP Ca2+ signals while staying in the linear range of the indicator (Yasuda et al., 2004). Dyes were allowed to diffuse throughout the dendrites for at least 20 min before imaging.
Electrophysiology recordings were made with a Multiclamp 700B amplifier, filtered at 6 kHz for current clamp and 2 kHz for voltage clamp, and sampled at 10 kHz. The series resistance was routinely measured in voltage clamp and maintained at <20 MΩ. All L5 pyramidal neurons were regular spiking and responded to hyperpolarizing current steps with a voltage sag. Somatic APs were triggered with a threshold current injection (2 ms duration), unless otherwise noted. For mapping and imaging experiments, basal dendrites were selected ∼150 μm from the soma and apical dendrites were selected ∼100 μm distal to the bifurcation point near the L1/L2 border.
Two-photon microscopy.
Two-photon imaging was performed on a custom microscope, as previously described (Chalifoux and Carter, 2010). A Ti:sapphire laser (Coherent) tuned to 810 nm was used to excite Alexa Fluor 594 and Fluo-5F to image morphology and monitor AP Ca2+ signals, respectively. Line scans were acquired at 500 Hz, divided by the total number of spines at each location (between 1 and 3, giving sampling rates between 167 and 500 Hz). Reference frame scans were routinely taken to correct for any spatial drifts over time at the imaging location. Ca2+ signals were quantified as the change in Fluo-5F fluorescence [green (G)] normalized to the Alexa Fluor 594 fluorescence [red (R)], giving units of ΔG/R. These signals were then normalized to the Gsat/R value measured with a saturating concentration of Ca2+ added to the internal solution in a thin-walled pipette, giving final measurements in units of ΔG/Gsat. Recordings were discarded if an increase in baseline fluorescence was detected, which could indicate photo damage. Recordings were also discarded if AP Ca2+ signals were <0.02 ΔG/Gsat, which prevented reliable analysis of inhibition. All imaging experiments were performed with a 60× 1.0 NA objective (Olympus).
GABA uncaging.
The back aperture of the objective was filled with collimated 473 nm light from a DPSS laser (OEM Laser) to form a spot at the sample. The duration and power of the uncaging laser light was controlled by a Pockels cell (Conoptics). A fast shutter (Uniblitz) was used to protect the photomultiplier tubes from the uncaging laser light. During two-photon imaging, a telescope of two plano-convex lenses was used to focus the collected fluorescence through the shutter aperture.
RuBi-GABA (20 μm) was prepared and bath applied in the dark. GABA uncaging was achieved using a single laser pulse of 2 ms duration and 4 mW power, unless otherwise noted. This duration and power was found to be sufficient to block somatic APs or dendritic AP Ca2+ signals without causing any photo damage. In all experiments, the laser was focused on either the soma or dendrites located within 40 μm of the slice surface. In most experiments, the uncaging spot was centered on the imaged spine–dendrite pair. In Figure 4, the uncaging spot was shifted perpendicular to the main apical dendrite, perpendicular to the imaged branch, or along the branch. In Figure 5, the uncaging spot was shifted to a nearby branch or to the main apical dendrite. Ca2+ imaging experiments consisted of at least four standard interleaved trials: 1 = no AP + no laser, 2 = AP + no laser, 3 = no AP + laser, and 4 = AP + laser. Trial 1 was used to detect photo-bleaching, Trial 2 was used to assess the control AP Ca2+ signal, Trial 3 was used to measure the laser artifact, and Trial 4 was used to assess the impact of inhibition on the AP Ca2+ signal. As needed, additional trials were used to vary uncaging power (Fig. 2), time delay (Fig. 3), or location (Figs. 4, 5). GABA uncaging preceded the somatic AP by 10 ms, coinciding with maximal inhibitory conductance, unless otherwise noted. Timing experiments (Fig. 3) consisted of the 10 ms time point and one to three other time points. Each trial was repeated up to 10 times and averaged for analysis.
Figure 4.

Spread of inhibition along a dendritic branch. A, Schematic of recording configuration, where blue spots indicate GABA uncaging at different distances from the main apical dendrite (Δx = 0, 10, 20, 30, and 40 μm). B, IPSCs evoked by GABA uncaging at different distances from the main apical dendrite, where the blue arrow indicates uncaging pulse. C, Left, Summary of IPSC amplitude versus uncaging distance, where dotted line indicates average exponential fit. Right, Space constants for individual cells (open circles) and mean ± SEM (closed circle). D, Similar to A with GABA uncaging at different distances (Δx = 0, 10, 20, and 30 μm) from the distal apical dendrite, where the dotted box indicates location of imaged spines and dendrites. E, Average Ca2+ signals evoked in apical spines by AP alone (black) or AP paired with GABA uncaging (red) at different distances from the imaging location, where the blue arrow indicates uncaging pulse and the black arrow indicates AP firing at 10 ms delay. F, Summary of impact of GABA uncaging at different distances on AP Ca2+ signals in spines (left) and dendrites (right). *p < 0.0125. G–I, Similar to D–F with GABA uncaging at centered, distal, or proximal locations along a dendritic branch; *p < 0.0167.
Figure 5.

Restricted inhibition across dendritic compartments. A, Schematic of recording configuration, where blue spots indicate GABA uncaging at centered or off-branch locations, and the dotted box indicates location of imaged spines and dendrites. B, Average Ca2+ signals evoked in apical spines by AP alone (black) or AP paired with GABA uncaging at the centered location (red) or off-branch location (blue), where the blue arrow indicates uncaging pulse and the black arrow indicates AP firing at 10 ms delay. C, Summary of impact of GABA uncaging at each location on AP Ca2+ signals in spines (left) and dendrites (right). *p < 0.025. D, Similar to A with GABA uncaging and imaging at the main apical dendrite. E, Magnified view of main apical dendrite with imaged spine (S), dendrite (D), and line-scan path (dashed line). F, Average Ca2+ signals evoked in main apical spines by AP alone (black) or AP paired with GABA uncaging (red). G, Summary of impact of GABA uncaging on AP Ca2+ signals in main apical spines (left) and dendrites (right). *p < 0.05. H–J, Similar to A–C with GABA uncaging at either the imaging location or the main apical dendrite. *p < 0.025.
Figure 2.

Similar inhibition at nearby spines and dendrites. A, Schematic of recording configuration, where the blue spot indicates GABA uncaging at the soma. B, IPSPs evoked by somatic GABA uncaging at different laser powers (red), and block with gabazine (GZ; dark blue), where the blue arrow indicates uncaging pulse. C, Summary of IPSP amplitude versus uncaging laser power. D, Similar to A with GABA uncaging at the apical dendrite, where the dotted box indicates location of imaged spines and dendrites. E, Average Ca2+ signals evoked in apical spines (left) and dendrites (right) by AP alone (black) or AP paired with GABA uncaging at different laser powers (red), where blue arrows indicate uncaging pulse and black arrows indicate AP firing at 10 ms delay. F, Summary of impact of GABA uncaging at different laser powers on AP Ca2+ signals in spines (left) and dendrites (right). *p < 0.0125. G, Summary of difference in inhibition at spines and dendrites, where values above zero indicate less inhibition at spines, and values below zero indicate more inhibition at spines, showing no difference at any stimulus intensity.
Figure 3.

Temporally precise inhibition of spines and dendrites. A, Schematic of recording configuration, where the blue spot indicates GABA uncaging at the soma. B, Influence of somatic GABA uncaging on AP firing, where the blue arrow and trace indicate uncaging pulse at t = 0 ms, and black arrows and traces indicate AP paired with uncaging at different delays. C, Summary of firing probability versus delay between AP and uncaging pulse. D, Similar to A with GABA uncaging at the apical dendrite, where the dotted box indicates location of imaged spines and dendrites. E, Average Ca2+ signals evoked in apical spines by AP alone (black) or AP paired with GABA uncaging (red), with 10 ms (left), 50 ms (middle), or 100 ms (right) delay, where blue arrows indicate uncaging pulse and black arrows indicate AP firing. F, Summary of impact of GABA uncaging on AP Ca2+ signals in spines (left) or dendrites (right) versus delay between AP and uncaging pulse. *p < 0.05. G, Summary of difference in inhibition at spines and dendrites, where values above zero indicate less inhibition at spines, and values below zero indicate more inhibition at spines, showing no difference at any delay.
Optogenetics.
Channelrhodopsin-2 (ChR2) was activated in PV, SOM, and 5HT3a interneurons to trigger inhibition of L5 pyramidal neurons. For wide-field illumination, ChR2 was activated using a 473 nm LED (Thorlabs) and 5× 0.9 NA objective (Olympus). The duration was varied and power was measured as 4.4 mW at the back aperture of the objective. For focused illumination, ChR2 was activated at axons using a similar method to GABA uncaging. The laser spot was focused on the dendrites of pyramidal neurons within 40 μm of the slice surface. The duration was fixed at 2 ms and the power was 1 mW at the back aperture of the objective. Ca2+ imaging experiments consisted of the same four interleaved trials: 1 = no AP + no laser, 2 = AP + no laser, 3 = no AP + laser, and 4 = AP + laser. ChR2 activation always preceded somatic AP firing by 5 ms. To overcome somatic inhibition, a suprathreshold current injection was used. We found that AP Ca2+ signals evoked by suprathreshold current injection were identical to those evoked by threshold current injection (data not shown).
Histology and fluorescence microscopy.
Mice were anesthetized with a lethal dose of ketamine and xylazine and perfused intracardially with ice-cold external solution, as described above. Brains were transferred into an ice-cold solution of 4% paraformaldehyde in 0.01 m PBS and fixed for 20–24 h at 4°C. Slices were prepared (Leica VT 1000S vibratome) at a thickness of 70 μm, and mounted under glass coverslips on gelatin-coated slides using ProLong Gold antifade reagent with DAPI (Invitrogen). Images were acquired using an automated microscope (Olympus VS120) with a 10× 0.25 NA objective. Excitation wavelengths were 555 and 359 nm for TRITC and DAPI, respectively.
Data analysis.
Electrophysiology and imaging data were acquired using National Instruments boards and MATLAB (MathWorks). Image processing was performed using ImageJ (NIH). Off-line analysis was performed using Igor Pro (WaveMetrics).
Input resistance was measured using the steady-state hyperpolarization after a small negative current injection. Membrane time constants were measured using exponential fits to these same voltage traces. AP width was measured as the time delay at the half-maximal values on the rising and falling phases. IPSC and IPSP amplitudes were measured as the average around 0.5 ms of the peak response. IPSC charge transfer was measured after baseline subtraction during a 200 ms period after the stimulus. IPSC decay times were measured using exponential fits after the peak response. In Ca2+ imaging experiments, laser artifacts were detected using Trial 3 (no AP + laser). Before any analysis, these artifacts were corrected by subtracting Trial 3 from Trial 4 (AP + laser). This correction was vital for analyzing AP Ca2+ signals, as the artifacts could otherwise obscure inhibition. AP Ca2+ signal amplitudes were measured over a 100 ms window after the end of the shutter period. The impact of inhibition was assessed by dividing the value of the corrected Trial 4 (AP + laser) by the value of Trial 2 (AP + no laser). In Figures 3–5, recordings were discarded if the control inhibition of AP Ca2+ signals in spines and dendrites was <50%, to enable accurate comparisons at different time delays or spatial locations.
Electrophysiology and imaging summary data are reported in the text and shown in figures as arithmetic mean ± SEM. For clarity, SEM is omitted from figures with three or more overlaid AP Ca2+ signals. In Figure 6, summary data are shown as box-plots of the median, interquartile range, and 10–90% range (whiskers). Comparisons between unpaired data were performed using Wilcoxon rank-sum tests. Comparisons between paired data were performed using paired Wilcoxon signed-rank tests. These nonparametric tests make no assumptions about the data distribution. Significance was defined as p < 0.05, adjusted for multiple comparisons using the Bonferroni correction, as indicated in figure legends.
Figure 6.
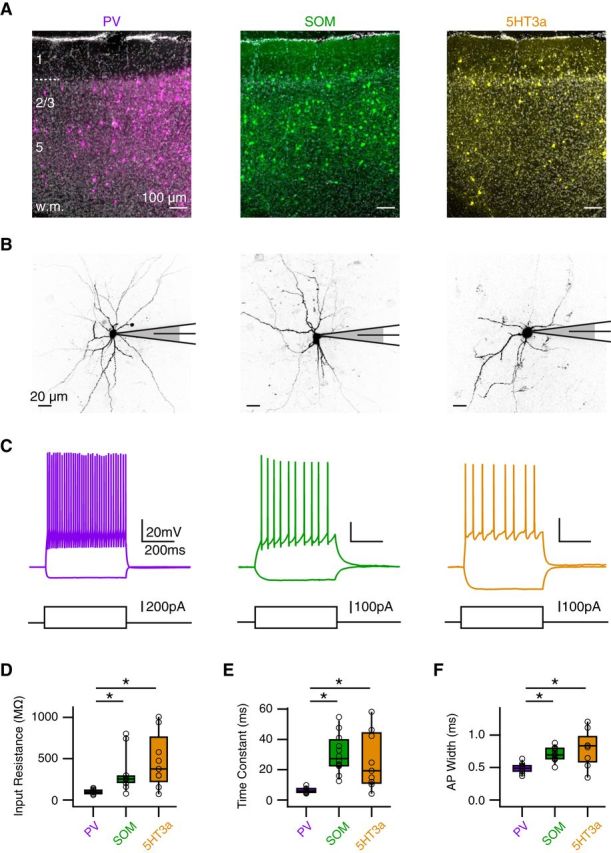
Three populations of GABAergic interneurons. A, Coronal sections of prelimbic PFC with PV (left), SOM (middle), or 5HT3 (right) interneurons expressing ChR2-mCherry (false color) and stained with DAPI (gray), where layers and white matter (w.m.) are indicated on the left. B, Two-photon images of PV (left), SOM (middle), and 5HT3a (right) interneurons during whole-cell recordings. C, Positive and negative current injections indicate distinct passive and active properties at different interneurons. D–F, Summary of input resistance (D), membrane time constant (E), and AP width (F) for the different interneurons. *p < 0.025.
Results
Inhibition of AP Ca2+ signals by GABA-A receptors
We examined dendritic inhibition at L5 pyramidal neurons in acute slices of mouse PFC. We used whole-cell recordings to fill neurons with the fluorescent dye Alexa Fluor 594 (30 μm) via the patch pipette. After allowing for dye diffusion, we used two-photon microscopy to image the soma and dendrites (Fig. 1A). We used a focused blue laser to uncage RuBi-GABA (20 μm) in the presence of AMPA, NMDA, and GABA-B receptor blockers (Rial Verde et al., 2008; Chalifoux and Carter, 2011b). In current-clamp recordings at resting membrane potentials, we found that GABA uncaging at the soma evoked robust IPSPs that blocked AP firing and were abolished by the GABA-A receptor antagonist gabazine (10 μm; n = 4; Fig. 1B). In contrast, GABA uncaging targeted to the apical dendrites generated smaller IPSPs that did not prevent somatic AP firing (n = 7; Fig. 1C).
Figure 1.
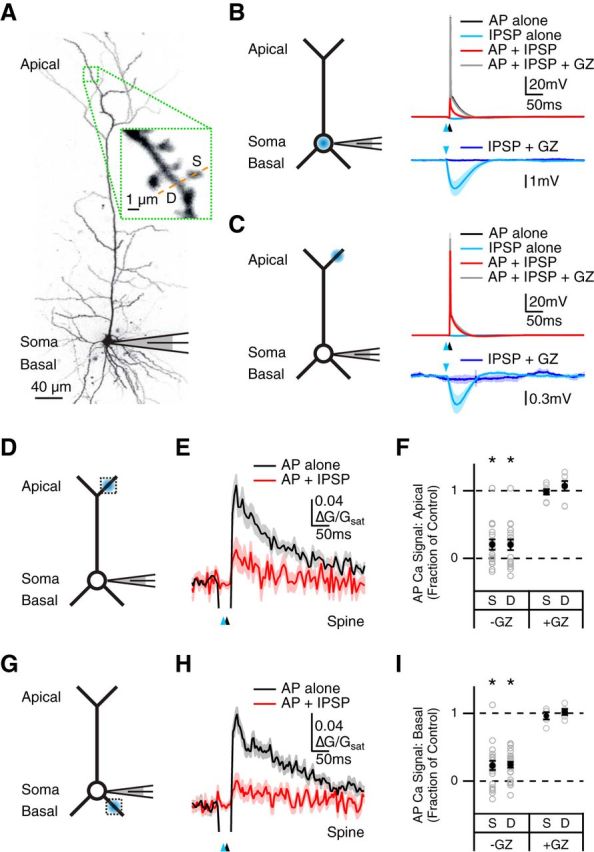
GABA-A receptor inhibition of AP Ca2+ signals. A, Two-photon image of a L5 pyramidal neuron, showing apical dendrites, soma, and basal dendrites, with magnification showing imaged spine (S), dendrite (D), and line-scan path (dashed line). B, Left, Schematic of recording configuration, where the blue spot indicates GABA uncaging at the soma. Right, Current-clamp recording of AP alone (black), GABA uncaging alone (light blue), AP paired with GABA uncaging (red), and paired response after addition of gabazine (GZ; gray), with expanded view of IPSP alone before and after the addition of GZ (dark blue). Blue arrows indicate uncaging pulse and black arrows indicate AP firing at 10 ms delay. C, Similar to B with GABA uncaging at the apical dendrite. D, Schematic of recording configuration, where the blue spot indicates GABA uncaging at the apical dendrite and the dotted box indicates location of imaged spines and dendrites. E, Average Ca2+ signals evoked in apical spines by AP alone (black) or AP paired with GABA uncaging (red). F, Summary of impact of GABA uncaging on AP Ca2+ signals in apical spines (S) and dendrites (D) in the absence and presence of gabazine. *p < 0.05. G–I, Similar to D–F with GABA uncaging and imaging at the basal dendrites.
We next determined the ability of GABA-A receptors to inhibit AP Ca2+ signals in spines and dendrites. To measure these signals, we also filled neurons with the fluorescent Ca2+ indicator Fluo-5F (200 μm), and used two-photon laser line scans through neighboring spines and dendrites (Fig. 1A). To accurately determine the impact of inhibition, we found that it was critical to first subtract the artifact generated by the uncaging laser (see Materials and Methods; Chalifoux and Carter, 2011a). After making this correction, we observed that GABA uncaging strongly reduced AP Ca2+ signals in both the apical dendrites (fraction of control: spines = 0.21 ± 0.08, p < 10−4; dendrites = 0.20 ± 0.08, p < 10−4; n = 17; Fig. 1D–F) and basal dendrites (spines = 0.22 ± 0.07, p < 10−6; dendrites = 0.24 ± 0.04, p < 10−6; n = 22; Fig. 1G–I). Importantly, this inhibition was also eliminated by bath wash-in of gabazine (10 μm; fraction of control: apical spines = 0.98 ± 0.04, p > 0.9; apical dendrites = 1.07 ± 0.07, p = 0.38; n = 8; basal spines = 0.97 ± 0.05, p > 0.9; basal dendrites = 1.02 ± 0.04, p = 0.63; n = 5; Fig. 1F,I). These results indicate that GABA-A receptor activation potently inhibits AP Ca2+ signals at spines and dendrites of L5 pyramidal neurons.
Equivalent inhibition at nearby spines and dendrites
We next sought to determine the sensitivity of AP Ca2+ signals in spines and dendrites to GABA-A receptor activation. Many previous studies indicate that GABA-A receptors are present on both spines and dendrites (Fritschy and Mohler, 1995; Kubota et al., 2007). However, recent findings at L2/3 pyramidal neurons suggest that spines can be more sensitive to inhibition than their parent dendrites (Chiu et al., 2013). To explore this possibility, we assessed the amount of inhibition generated by uncaging at different laser powers. We first measured IPSPs evoked by uncaging at the soma in current-clamp recordings (Fig. 2A). As expected, we found that IPSP amplitude progressively increased with stronger stimuli (0.5 mW = −0.31 ± 0.04 mV; 1 mW = −0.70 ± 0.13 mV; 2 mW = −1.34 ± 0.21 mV; 4 mW = −2.50 ± 0.43 mV; n = 4 cells; Fig. 2B,C), indicating that the amount of GABA-A receptor activation scales with laser power.
We then measured inhibition of AP Ca2+ signals in the apical dendrites while varying the laser power (Fig. 2D). We hypothesized that heightened sensitivity to inhibition at spines would be revealed at lower powers. We found that AP Ca2+ signals were progressively inhibited with stronger stimuli at both spines (fraction of control: 0.5 mW = 0.76 ± 0.06, p = 0.004; 1 mW = 0.70 ± 0.07, p = 0.006; 2 mW = 0.27 ± 0.11, p < 10−4; 4 mW = 0.18 ± 0.06, p < 10−4; n = 16) and their parent dendrites (fraction of control: 0.5 mW = 0.81 ± 0.05, p = 0.003; 1 mW = 0.62 ± 0.04, p < 10−4; 2 mW = 0.41 ± 0.07, p < 10−4; 4 mW = 0.14 ± 0.06, p < 10−4; Fig. 2E,F). However, the degree of inhibition in these compartments did not differ at any laser power (spine – dendrite: 0.5 mW = −0.05 ± 0.06, p = 0.67; 1 mW = 0.08 ± 0.07, p = 0.46; 2 mW = −0.14 ± 0.07, p = 0.10; 4 mW = 0.04 ± 0.06, p = 0.40; Fig. 2G). These results show that AP Ca2+ signals at spines and dendrites are similarly sensitive to GABA-A receptor activation at these neurons.
Time course of inhibition at spines and dendrites
The timing of GABA-A receptor inhibition is thought to play a key role in regulating Ca2+-dependent synaptic plasticity (Hayama et al., 2013). To investigate the temporal profile of dendritic inhibition, we first assessed the time window over which GABA uncaging blocks AP firing at the cell body (Fig. 3A). We varied the timing of the uncaging pulse relative to the threshold current injection (Δt = −5, 0, 5, 10, 20, 50, and 100 ms; Fig. 3B). Maximal inhibition occurred at 10 ms, with little inhibition by 100 ms (probability of firing: 10 ms = 0.03 ± 0.03; 100 ms = 0.69 ± 0.16; n = 7 cells; Fig. 3C). These results indicate that activation of GABA-A receptors inhibits AP firing over a brief time window.
We then examined inhibition of AP Ca2+ signals by varying the timing of the uncaging pulse in the apical dendrites relative to AP firing (Fig. 3D). We found that inhibition of AP Ca2+ signals also depended on timing at both spines and dendrites (Fig. 3E). Maximal inhibition occurred at 10 ms, with little inhibition by 100 ms at both spines (fraction of control: 10 ms = 0.22 ± 0.03, p < 10−10, n = 36; 100 ms = 1.01 ± 0.06, p = 0.90, n = 16) and dendrites (fraction of control: 10 ms = 0.19 ± 0.04, p < 10−10; 100 ms = 0.89 ± 0.05, p = 0.015; Fig. 3F). As a control, no inhibition was observed when the AP preceded the uncaging pulse by 5 ms (fraction of control: spines = 0.96 ± 0.06, p = 0.32; dendrites = 0.93 ± 0.08, p = 0.38; n = 11; Fig. 3F). We again found no significant difference in the amount of inhibition at spines and dendrites at any delay (spine – dendrite: −5 ms, p = 0.97, n = 11; 0 ms, p = 0.95, n = 8; 5 ms, p = 0.57, n = 9; 10 ms, p = 0.69, n = 36; 20 ms, p = 0.73, n = 9; 50 ms, p = 0.43, n = 9; 100 ms, p = 0.19, n = 16; Fig. 3G). These results indicate that GABA-A receptor inhibition of AP Ca2+ signals at spines and dendrites occurs in a window of tens of milliseconds.
Spatial extent of inhibition in the dendritic arbor
The impact of inhibitory inputs is thought to depend on their location within and across individual branches (Liu, 2004; Hao et al., 2009; Gidon and Segev, 2012; Jadi et al., 2012). We next sought to assess the spatial extent of GABA-A receptor inhibition of AP Ca2+ signals. It was first important to determine the spatial resolution of GABA uncaging in our brain slices. We recorded uncaging-evoked IPSCs at 0 mV while uncaging GABA in 10 μm steps moving away from the main apical dendrite (Fig. 4A). We found that IPSC amplitude decreased with distance from the dendrite (0 μm = 92 ± 12 pA; 10 μm = 45 ± 5.6 pA; 20 μm = 15 ± 4.5 pA; 30 μm = 6.5 ± 2.0 pA; 40 μm = 5.0 ± 1.0 pA; n = 3 cells; Fig. 4B,C). The average space constant was 11.9 ± 0.4 μm, indicating that GABA uncaging is highly restricted in our preparation.
In related experiments, we also assessed inhibition of AP Ca2+ signals while uncaging GABA in 10 μm steps moving away from the apical tuft (Fig. 4D). We found that this inhibition decreased with distance at both spines (fraction of control: 0 μm = 0.17 ± 0.06, p < 10−3; 10 μm = 0.37 ± 0.12, p < 10−3; 20 μm = 0.73 ± 0.06, p < 10−3; 30 μm = 1.09 ± 0.07, p = 0.36; n = 14) and dendrites (fraction of control: 0 μm = 0.19 ± 0.05, p < 10−3; 10 μm = 0.29 ± 0.09, p < 10−3; 20 μm = 0.91 ± 0.06, p = 0.15; 30 μm = 0.91 ± 0.06, p = 0.07; Fig. 4E,F). These results indicate that GABA uncaging directly activates GABA-A receptors within an ∼30 μm radius in our recordings. Thus, any effect of GABA uncaging on AP Ca2+ signals beyond this radius must reflect the spread of inhibition in the dendrites.
Having established the spatial resolution of our uncaging experiments, we asked whether inhibition could spread along a dendritic branch. We compared the impact of GABA uncaging at three locations along a single branch: centered on the imaged spine, 40 μm distal, or 40 μm proximal, using the same laser power at each location (Fig. 4G). We found that AP Ca2+ signals were significantly inhibited at all three locations at both spines (fraction of control: centered = 0.08 ± 0.11, p = 0.001; distal = 0.29 ± 0.13, p = 0.001; proximal = 0.29 ± 0.11, p = 0.002; n = 11) and dendrites (fraction of control: centered = 0.31 ± 0.06, p = 0.001; distal = 0.43 ± 0.15, p = 0.003; proximal = 0.18 ± 0.14, p = 0.001; Fig. 4H,I). Moreover, there was no significant difference in inhibition at the three locations (centered vs. distal: spines, p = 0.10; dendrites, p = 0.41; centered vs. proximal: spines, p = 0.12; dendrites, p = 0.65; distal vs. proximal: spines, p = 0.83; dendrites, p = 0.11). These results show that inhibition spreads along an individual dendritic branch, indicating that the reduction of AP Ca2+ signals can in principle also reflect the activation of GABA-A receptors at nearby locations.
Compartmentalization of inhibition in distal dendrites
Inhibition is often thought to be confined to a single dendritic branch, with adjacent branches functioning independently (Liu, 2004; Gidon and Segev, 2012). We next examined whether inhibition can spread between branches to influence AP Ca2+ signals at spines and dendrites. We compared the impact of GABA uncaging either centered on the imaged spine or targeted to another branch located 40–75 μm away from the imaging location (Fig. 5A). We found that AP Ca2+ signals were strongly inhibited by uncaging at the imaged spine (fraction of control: spines = −0.02 ± 0.07, p = 0.001; dendrites = −0.02 ± 0.08, p = 0.001; n = 11), but only weakly by uncaging at the other branch (fraction of control: spines = 0.74 ± 0.09, p = 0.019; dendrites = 0.75 ± 0.07; p = 0.007; Fig. 5B,C). Moreover, GABA-A receptor inhibition at these two locations was significantly different (centered vs. other branch: spines, p = 0.001; dendrites, p = 0.001). These results suggest that inhibition via GABA-A receptors is primarily confined to individual branches, and spreads across the tuft to a smaller degree.
We then assessed whether activation of GABA-A receptors on the main apical dendrite could also inhibit AP Ca2+ signals in the apical tuft. We first examined the impact of GABA uncaging at the main apical dendrite (Fig. 5D,E), and found a small impact on AP Ca2+ signals at both spines and dendrites (fraction of control: spines = 0.83 ± 0.04, p = 0.01; dendrites = 0.82 ± 0.02, p = 0.004; n = 9; Fig. 5F,G). We then measured AP Ca2+ signals in the apical tuft, comparing GABA uncaging at either the imaged spine or 50–85 μm away on the main apical dendrite (Fig. 5H). We found that AP Ca2+ signals were strongly inhibited by uncaging at the imaged spine (fraction of control: spines = 0.13 ± 0.05, p < 10−3; dendrites = 0.24 ± 0.06, p < 10−3; n = 12), but only modestly by uncaging at the main apical dendrite (fraction of control: spines = 0.57 ± 0.11, p = 0.001; dendrites = 0.70 ± 0.13, p = 0.009; Fig. 5I,J). Furthermore, GABA-A receptor inhibition at these two locations was significantly different (centered vs. main apical: spines, p = 0.007; dendrites, p = 0.002). These results suggest that GABA-A receptor activation at the main apical dendrite can also weakly reduce AP Ca2+ signals at distal locations. Our findings support the idea of rapid and localized inhibition of AP Ca2+ signals at both spines and dendrites.
Inhibition via three populations of cortical interneurons
Having established the properties of GABA-A receptor inhibition of AP Ca2+ signals, we next sought to determine which interneurons are responsible for this local effect in the apical and basal dendrites. Distinct interneuron populations are well known to make unique contacts onto cortical pyramidal neurons (Kawaguchi and Kubota, 1997; Somogyi et al., 1998; Di Cristo et al., 2004). We examined PV, SOM, and 5HT3a interneurons, which together comprise the vast majority of cortical interneurons (Rudy et al., 2011). To independently manipulate these populations, we used transgenic mice that express Cre recombinase under the regulatory elements of these molecular markers (Hippenmeyer et al., 2005; Gong et al., 2007; Taniguchi et al., 2011). We first injected AAV9-dfloxed-ChR2-mCherry into the PFC of these mice, waited 1–2 weeks for expression, and imaged red fluorescence in slices. We found all three types of interneurons were distributed across the deep and superficial layers of PFC (Fig. 6A). We then performed whole-cell recordings of labeled cells to characterize their morphology (Fig. 6B) and physiology (Fig. 6C). We found that PV interneurons (n = 11) had significantly lower input resistance, faster time constants, and shorter AP width than SOM (n = 12) and 5HT3a (n = 9) interneurons (Fig. 6D–F). These physiological properties are consistent with studies in other rodents (Kawaguchi, 1993), and suggest this is a viable approach for studying the functional roles of these different interneurons.
We then used optogenetics to assess the functional connections of PV, SOM, and 5HT3a interneurons onto L5 pyramidal neurons. We expressed ChR2 in interneurons by injecting AAV9-dfloxed-ChR2-mCherry into the PFC of Cre-expressing transgenic mice. After allowing 1–2 weeks for expression, we used wide-field illumination to activate presynaptic interneurons and evoke IPSCs at postsynaptic pyramidal neurons held at 0 mV in voltage clamp (Fig. 7A). We found that IPSCs increased in amplitude with stimulus duration and were completely blocked with gabazine (10 μm; Fig. 7B). Interestingly, we discovered that normalized IPSCs from each interneuron population displayed distinct kinetics (Fig. 7C). The time to peak was fastest at PV connections, and slowest at 5HT3a connections (PV = 8.6 ± 0.2 ms; SOM = 11.3 ± 0.8 ms; 5HT3a = 15.3 ± 1.0 ms; PV vs. SOM, p = 0.002; PV vs. 5HT3a, p = 0.002; SOM vs. 5HT3a, p = 0.015; n = 6; Fig. 7D). Similarly, the decay time was fastest at PV connections and slowest at 5HT3a connections (PV = 12.3 ± 1.2 ms; SOM = 24.9 ± 1.4 ms; 5HT3a = 42.1 ± 3.0 ms; PV vs. SOM, p = 0.002; PV vs. 5HT3a, p = 0.002; SOM vs. 5HT3a, p = 0.002; Fig. 7E). We hypothesized that these distinct kinetics could in part reflect differential targeting by these three populations of interneurons in the apical and basal dendrites.
Figure 7.

Distinct IPSCs evoked by multiple interneurons. A, Schematic of recording configuration, showing a pyramidal neuron (black) receiving putative connections from a nearby interneuron (gray), where the blue circle indicates wide-field illumination of ChR2-expressing interneurons. B, IPSCs evoked at pyramidal neurons by wide-field activation of PV (left), SOM (middle), or 5HT3a (right) interneurons at different pulse durations, and block by gabazine (GZ; dark blue), where the blue arrows indicate light pulse. C, Peak-scaled IPSCs evoked by each interneuron using the 4 ms light pulse. D, E, Summary of time to peak (D) and decay (E) of IPSCs evoked by different interneurons. *p < 0.025.
Dendritic targeting of inhibitory connections
We next used optogenetics to map the functional connections of PV, SOM, and 5HT3a interneurons onto the dendrites of L5 pyramidal neurons. We used a focused blue laser to release GABA from axons at the apical and basal dendrites, while measuring IPSCs in voltage-clamp recordings at 0 mV (Fig. 8A). To ensure local responses, we included TTX (1 μm) to block APs and 4-AP (100 μm) to restore presynaptic release (Petreanu et al., 2009; Little and Carter, 2012; MacAskill et al., 2012). We found that PV interneurons contacted the basal dendrites, but generated no response in the apical dendrites (Fig. 8B). In contrast, SOM and 5HT3a interneurons made contacts at both the apical and basal dendrites. Importantly, the ratio of charge transfer at the apical and basal dendrites was significantly different across the three populations (apical/basal ratio: PV = 0.007 ± 0.05; SOM = 1.4 ± 0.19; 5HT3a = 5.5 ± 2.0; PV vs. SOM, p < 10−3; PV vs. 5HT3a, p < 10−3; SOM vs. 5HT3a, p = 0.002; n = 7; Fig. 8C). These results indicate that PV, SOM, and 5HT3a interneurons make unique dendritic connections, suggesting they may have distinct effects on AP Ca2+ signals.
Figure 8.

Subcellular targeting of multiple interneurons. A, Schematic of recording configuration, where blue spots indicate local illumination of ChR2-expressing presynaptic terminals at the apical or basal dendrites in the presence of TTX and 4-AP. B, IPSCs evoked by local activation of PV (left), SOM (middle), and 5HT3a (right) inputs at the apical (dark) and basal (light) dendrites, where blue arrows indicate light pulse. C, Summary of the ratio of IPSC charge transfer at the apical and basal dendrites for each interneuron, where the dashed line equals unity. *p < 0.025. D, Peak-scaled IPSCs evoked by each interneuron at the apical (left) and basal (right) dendrites. E, F, Summary of time to peak (E) and decay (F) of IPSCs evoked by the different interneurons at the apical (left) and basal (right) dendrites. *p < 0.025.
Further analysis revealed that IPSC kinetics depended on both the type of interneuron and dendritic location (Fig. 8D). The time to peak was slower for connections onto the apical dendrites, but did not differ between interneurons (apical: SOM = 20.4 ± 1.1 ms; 5HT3a = 23.2 ± 1.8 ms; SOM vs. 5HT3a, p = 0.32; basal: PV = 12.3 ± 1.5 ms; SOM = 14.1 ± 0.5 ms; 5HT3a = 15.2 ± 1.5 ms; PV vs. SOM, p = 0.13; PV vs. 5HT3a, p = 0.10; SOM vs. 5HT3a, p = 0.62; Fig. 8E). The decay time was much slower for connections onto the apical dendrites, and also slower for SOM and 5HT3a interneurons at the basal dendrites (apical: SOM = 143 ± 21 ms; 5HT3a = 213 ± 36 ms; SOM vs. 5HT3a, p = 0.12; basal: PV = 19.0 ± 3.3 ms; SOM = 39.3 ± 4.4 ms; 5HT3a, 37.1 ± 3.9 ms; PV vs. SOM, p = 0.007; PV vs. 5HT3a, p = 0.007; SOM vs. 5HT3a, p = 0.87; Fig. 8F). These findings indicate that wide-field IPSC rise and decay times depend on both the type of presynaptic interneuron and dendritic targeting. These results indicate that PV, SOM, and 5HT3a interneurons make strong connections onto L5 pyramidal neurons, but have distinct targets and may serve unique functions.
Inhibition of AP Ca2+ signals by multiple interneurons
Having established the dendritic targeting of PV, SOM, and 5HT3a interneurons, we next tested their impact on AP Ca2+ signals in L5 pyramidal neurons (Fig. 9A). These current-clamp experiments were performed in the absence of TTX and 4-AP, which would otherwise disrupt AP firing. However, our measurements of AP Ca2+ signals continued to provide a measure of local inhibition. Our voltage-clamp results predicted that PV interneurons would have a minimal contribution to dendritic inhibition, whereas SOM and 5HT3a interneurons would have a large impact at the apical and basal dendrites. Consistent with this prediction, we found that PV interneurons did not inhibit AP Ca2+ signals in either the apical dendrites (fraction of control: spines = 1.03 ± 0.05, p = 0.58; dendrites = 0.98 ± 0.05, p = 0.93; n = 17) or basal dendrites (fraction of control: spines = 0.89 ± 0.05, p = 0.051; dendrites = 0.98 ± 0.06, p = 0.65; n = 19; Fig. 9B,E,F). In contrast, we found that SOM interneurons blocked AP Ca2+ signals in the apical dendrites (fraction of control: spines = 0.09 ± 0.04; p < 10−5; dendrites = −0.02 ± 0.05; p < 10−5; n = 19) and reduced them in the basal dendrites (fraction of control: spines = 0.48 ± 0.08, p < 10−5; dendrites = 0.45 ± 0.08, p < 10−4; n = 19; Fig. 9C,E,F). Last, we found that 5HT3a interneurons also blocked AP Ca2+ signals in the apical dendrites (fraction of control: spines = 0.01 ± 0.03, p < 10−4; dendrites = 0.00 ± 0.03, p < 10−4; n = 15), and inhibited them in the basal dendrites (fraction of control: spines = 0.59 ± 0.14, p = 0.007; dendrites = 0.63 ± 0.11, p < 10−3; n = 14; (Fig. 9D–F).
Figure 9.

Inhibition of AP Ca2+ signals by SOM and 5HT3a interneurons. A, Schematic of recording configuration, where blue spots indicate local illumination of ChR2-expressing interneurons at apical (top) or basal (bottom) dendrites, and dotted boxes indicate locations of imaged spines and dendrites. B–D, Average impact of PV (B), SOM (C), and 5HT3a (D) interneurons on AP Ca2+ signals in apical (top) or basal (bottom) spines, where blue arrows indicate light pulse and black arrows indicate AP firing at 5 ms delay. E, F, Summary of impact of different interneurons on AP Ca2+ signals in apical (Ap) and basal (Ba) spines (E) and dendrites (F). *p < 0.05. G, Summary of impact of PV (purple), SOM (green), and 5HT3a (yellow) interneurons on AP Ca2+ signals in spines versus dendrites. Light outlined markers indicate individual data points, dark solid markers indicate population averages with 2D SEM, solid line indicates unity, and dashed line and r2 value indicate linear regression of pooled data.
Finally, we examined whether any interneurons yielded differential inhibition at nearby spines and dendrites of L5 pyramidal neurons. We found that SOM and 5HT3a interneurons yielded stronger inhibition in both apical spines (apical vs. basal: SOM, p < 10−3; 5HT3a, p = 0.002) and their parent dendrites (apical vs. basal: SOM, p < 10−4; 5HT3a, p < 10−3; Fig. 9E,F). However, we found no difference in inhibition at spines and dendrites across all interneuron types and dendritic domains (spine vs. dendrite: PV apical, p > 0.9; PV basal, p = 0.18; SOM apical, p = 0.10; SOM basal, p = 0.77; 5HT3a apical, p = 0.85; 5HT3a basal, p = 0.54; pooled data, r2 = 0.71; Fig. 9G). Our findings establish how different populations of interneurons target the dendrites of L5 pyramidal neurons and robustly inhibit AP Ca2+ signals in both spines and dendrites.
Discussion
We have characterized GABA-A receptor inhibition of AP Ca2+ signals at L5 pyramidal neurons in the mouse PFC. Using GABA uncaging, we first showed similar inhibition via GABA-A receptors at spines and dendrites. We also found that inhibition is limited to tens of milliseconds and confined within a dendritic branch. Using optogenetics, we then showed that PV, SOM, and 5HT3a interneurons make distinct contacts onto the apical and basal dendrites. Finally, we found that SOM and 5HT3a interneurons strongly inhibit AP Ca2+ signals at both spines and dendrites. These findings provide new insights into rapid inhibition at spines and dendrites of pyramidal neurons, and underscore the importance of different populations of interneurons in regulating cortical function.
GABA-A receptors are found at both the dendrites and spines of cortical pyramidal neurons (Fritschy and Mohler, 1995; Kubota et al., 2007). Activation of GABA-A receptors is well known to inhibit synaptic responses and local spiking (Llinás et al., 1968; Kim et al., 1995; Miles et al., 1996; Larkum et al., 1999; Murayama et al., 2009; Lovett-Barron et al., 2012; Müller et al., 2012). However, much less is known about inhibition of Ca2+ signals in spines and dendrites, which help to regulate excitability and plasticity (Bloodgood and Sabatini, 2007). At hippocampal pyramidal neurons, GABA-A receptors can strongly inhibit AP Ca2+ signals in the apical dendrites (Tsubokawa and Ross, 1996; Kanemoto et al., 2011; Hayama et al., 2013). Here we observed widespread inhibition of AP Ca2+ signals in the apical and basal dendrites of L5 pyramidal neurons. Importantly, neighboring spines and dendrites were inhibited to the same degree across a range of GABA uncaging parameters. These results support the idea that GABA-A receptors control AP Ca2+ signals across the dendritic arbor.
The precise timing of GABA-A receptor inhibition is thought to regulate Ca2+-dependent synaptic plasticity (Hayama et al., 2013). We found that inhibition of AP Ca2+ signals peaked at 10 ms and had largely decayed by 100 ms. Moreover, the time course of inhibition was indistinguishable at neighboring spines and dendrites. Interestingly, this narrow time window is similar to GABA-A receptor inhibition of EPSPs (Koch et al., 1983). This time window also coincides with that of rapid disynaptic inhibition via GABAergic interneurons in cortical circuits (Isaacson and Scanziani, 2011). In contrast, we and others have shown that the slower metabotropic GABA-B receptor inhibition of Ca2+ signals peaks after hundreds of milliseconds (Pérez-Garci et al., 2006; Chalifoux and Carter, 2011b; Palmer et al., 2012b). These studies highlight two distinct epochs for GABAergic inhibition of electrical and biochemical signaling in the dendrites of cortical pyramidal neurons.
GABA-A receptor inhibition is often predicted to be confined to individual dendritic branches (Liu, 2004; Hao et al., 2009; Gidon and Segev, 2012; Jadi et al., 2012). Our uncaging experiments allowed us to test this prediction by focally activating GABA-A receptors at different locations in the dendrites. Because of scattering and diffusion, GABA can spread over tens of micrometers around a centered location. Inhibition within a 30 μm radius can reflect direct GABA-A receptor activation; inhibition outside this radius reflects spread along dendrites. We found that robust inhibition spreads proximally and distally within individual dendritic branches. We observed weaker influence from neighboring branches, suggesting that inhibition is spatially confined. We also found weaker influence from the main apical dendrite, which could reflect partial block of the backpropagating AP. Consistent with this idea, we found that AP Ca2+ signals in the main apical dendrite were inhibited but not blocked by local GABA uncaging. Notably, we observed broader inhibition than equivalent experiments in hippocampal pyramidal neurons (Kanemoto et al., 2011; Hayama et al., 2013), which could reflect differences in GABA uncaging parameters or brain slice preparations. These results show that GABA-A receptor inhibition of AP Ca2+ signals at spines and dendrites primarily occurs within a single branch.
Our initial uncaging experiments established GABA-A receptor inhibition of AP Ca2+ signals at both spines and dendrites. We then sought to determine the types of GABAergic interneurons that are responsible for this inhibition. PV, SOM, and 5HT3a receptors are nonoverlapping markers that label the vast majority of cortical interneurons (Rudy et al., 2011). Previous studies have largely focused on PV and SOM interneurons, which are thought to target either the axosomatic or dendritic regions, respectively (Kawaguchi and Kubota, 1997; Somogyi et al., 1998; Di Cristo et al., 2004). 5HT3a interneurons represent a distinct population, whose subcellular targeting has not been examined (Rudy et al., 2011). The combination of Cre transgenic mice and conditional viruses allowed us to study these three populations with whole-cell recordings and optical stimulation.
We first established how PV, SOM, and 5HT3a interneurons synapse onto L5 pyramidal neurons. Using wide-field illumination, we found that all three interneurons make functional connections, with the time course of IPSCs fastest for PV interneurons and slowest for 5HT3a interneurons. In principle, these kinetics could reflect dendritic targeting, with more distal inputs having slower IPSCs. Using subcellular mapping with focused illumination (Petreanu et al., 2009; Little and Carter, 2012; MacAskill et al., 2012), we found that PV inputs make limited connections with the basal dendrites, whereas SOM and 5HT3a inputs are more pronounced in the apical dendrites. Interestingly, IPSCs in the basal dendrites were much faster for PV interneurons, suggesting the possibility of differences in GABA-A receptor composition (Vicini, 1999). In contrast, IPSCs in the apical dendrites were not significantly different for SOM and 5HT3a interneurons. Because IPSCs evoked by wide-field illumination reflect weighted inputs from across the somatodendritic axis, our findings suggest that targeting of 5HT3a interneurons to apical dendrites contributed to their slower IPSCs.
Our mapping experiments predicted that SOM and 5HT3a interneurons could inhibit AP Ca2+ signals in the apical and basal dendrites. We found that both interneurons inhibited these signals in spines and dendrites, with no differences between these two compartments. Inhibition was more pronounced at the apical dendrites, consistent with larger IPSCs at this location. This inhibition may also reflect inhibitory connections along the main apical dendrite, which can reduce distal signaling. To our knowledge, this is the first demonstration of 5HT3a interneurons inhibiting dendritic Ca2+ signaling. In contrast, we found that PV interneurons had no influence on AP Ca2+ signals, consistent with weak IPSCs in the basal dendrites and selective targeting of the perisomatic regions (Kawaguchi and Kubota, 1997). These results highlight how different populations of interneurons uniquely inhibit the subcellular compartments of pyramidal neurons.
Several of our findings contrast with a recent study on GABA-A receptor inhibition at L2/3 pyramidal neurons in the PFC (Chiu et al., 2013). That study found that one-photon GABA uncaging and SOM interneuron activation selectively inhibited AP Ca2+ signals in a subset of spines. Using similar methods, we found robust inhibition at nearly all imaged spines of L5 pyramidal neurons, with no difference from parent dendrites. One possible explanation is that the influence of dendritic inhibition varies with the type of pyramidal neuron. For example, L2/3 pyramidal neurons have more hyperpolarized resting potentials, thereby reducing the driving force for GABA-A receptors. Consistent with this idea, the overall magnitude of inhibition appears smaller at L2/3 pyramidal neurons (data not shown; Chiu et al., 2013). Technical differences could also play a role, including the power and timing of the laser and presence of the laser artifact. For example, we observed that smaller uncaging powers or longer time intervals greatly reduce the amount of GABA-A receptor inhibition.
With these results in mind, it is interesting to compare subcellular responses to excitatory and inhibitory inputs at pyramidal neurons. Glutamate receptors are concentrated at spines, where single excitatory inputs evoke highly compartmentalized electrical and biochemical signals (Bloodgood and Sabatini, 2007). GABA-A receptors are also present at a subset of spines, which can receive inhibitory inputs (Kubota et al., 2007; Chiu et al., 2013). However, these receptors are more prominent at dendrites, where interneurons also make multiple contacts (Tamás et al., 1997; Silberberg and Markram, 2007). Consequently, inhibition is often thought to have a more extended influence along dendritic branches (Liu, 2004; Gidon and Segev, 2012). Our findings support this idea, showing that SOM and 5HT3a interneurons broadly inhibit AP Ca2+ signals. Our results indicate that GABA-A receptor inhibition can regulate bioelectrical signaling at both spines and dendrites.
Recent results highlight the roles of different GABAergic interneurons in cortical function (Kerlin et al., 2010; Letzkus et al., 2011; Adesnik et al., 2012; Gentet et al., 2012; Lee et al., 2012; Wilson et al., 2012; Kuhlman et al., 2013). In the PFC, these interneurons are thought to critically support cognitive behavior (Sohal et al., 2009; Yizhar et al., 2011; Kvitsiani et al., 2013; Courtin et al., 2014). Disruption of these interneurons is also thought to play a key role in neuropsychiatric disorders (Rubenstein and Merzenich, 2003; Lewis et al., 2005). Our findings provide new insights into the influence of SOM and 5HT3a interneurons at the cellular and subcellular levels. In the future, it will be interesting to examine specific subpopulations of PV, SOM, and 5HT3a interneurons found in different layers (Rudy et al., 2011). For example, 5HT3a receptor-expressing interneurons include both neurogliaform cells (Tamás et al., 2003; Oláh et al., 2009) and VIP-expressing interneurons (Lee et al., 2013; Pfeffer et al., 2013; Pi et al., 2013), which have distinct functions. Finally, it will also be important to explore how dendritic inhibition changes with patterned activity (Silberberg and Markram, 2007; Pouille et al., 2009) and neuromodulation (Couey et al., 2007; Brombas et al., 2014), which further regulate its influence on cortical pyramidal neurons.
Footnotes
This work was supported by the National Institutes of Health (R01-MH085974 to A.G.C.). We thank members of the Carter lab, Michael Long, and Bernardo Rudy for helpful discussions and comments on this manuscript. We thank Josh Huang for the SOM-Cre mice. We thank Nat Heintz and Gord Fishell for the 5HT3a-Cre mice.
The authors declare no competing financial interests.
References
- Adesnik H, Bruns W, Taniguchi H, Huang ZJ, Scanziani M. A neural circuit for spatial summation in visual cortex. Nature. 2012;490:226–231. doi: 10.1038/nature11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsáki G, Cauli B, Defelipe J, Fairén A, Feldmeyer D, Fishell G, Fregnac Y, Freund TF, Gardner D, Gardner EP, Goldberg JH, Helmstaedter M, Hestrin S, Karube F, et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci. 2008;9:557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2007;17:345–351. doi: 10.1016/j.conb.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Brombas A, Fletcher LN, Williams SR. Activity-dependent modulation of layer 1 inhibitory neocortical circuits by acetylcholine. J Neurosci. 2014;34:1932–1941. doi: 10.1523/JNEUROSCI.4470-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci. 2011a;31:4221–4232. doi: 10.1523/JNEUROSCI.4561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. Glutamate spillover promotes the generation of NMDA spikes. J Neurosci. 2011b;31:16435–16446. doi: 10.1523/JNEUROSCI.2777-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GC, Higley MJ. Compartmentalization of GABAergic inhibition by dendritic spines. Science. 2013;340:759–762. doi: 10.1126/science.1234274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Courtin J, Chaudun F, Rozeske RR, Karalis N, Gonzalez-Campo C, Wurtz H, Abdi A, Baufreton J, Bienvenu TC, Herry C. Prefrontal parvalbumin interneurons shape neuronal activity to drive fear expression. Nature. 2014;505:92–96. doi: 10.1038/nature12755. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, López-Cruz PL, Benavides-Piccione R, Bielza C, Larrañaga P, Anderson S, Burkhalter A, Cauli B, Fairén A, Feldmeyer D, Fishell G, Fitzpatrick D, Freund TF, González-Burgos G, Hestrin S, Hill S, Hof PR, Huang J, Jones EG, Kawaguchi Y, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci. 2013;14:202–216. doi: 10.1038/nrn3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristo G, Wu C, Chattopadhyaya B, Ango F, Knott G, Welker E, Svoboda K, Huang ZJ. Subcellular domain-restricted GABAergic innervation in primary visual cortex in the absence of sensory and thalamic inputs. Nat Neurosci. 2004;7:1184–1186. doi: 10.1038/nn1334. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Gentet LJ, Kremer Y, Taniguchi H, Huang ZJ, Staiger JF, Petersen CC. Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex. Nat Neurosci. 2012;15:607–612. doi: 10.1038/nn.3051. [DOI] [PubMed] [Google Scholar]
- Gidon A, Segev I. Principles governing the operation of synaptic inhibition in dendrites. Neuron. 2012;75:330–341. doi: 10.1016/j.neuron.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, Gerfen CR. Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci. 2007;27:9817–9823. doi: 10.1523/JNEUROSCI.2707-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Wang XD, Dan Y, Poo MM, Zhang XH. An arithmetic rule for spatial summation of excitatory and inhibitory inputs in pyramidal neurons. Proc Natl Acad Sci U S A. 2009;106:21906–21911. doi: 10.1073/pnas.0912022106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, Kasai H. GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat Neurosci. 2013;16:1409–1416. doi: 10.1038/nn.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–243. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadi M, Polsky A, Schiller J, Mel BW. Location-dependent effects of inhibition on local spiking in pyramidal neuron dendrites. PLoS Comput Biol. 2012;8:e1002550. doi: 10.1371/journal.pcbi.1002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemoto Y, Matsuzaki M, Morita S, Hayama T, Noguchi J, Senda N, Momotake A, Arai T, Kasai H. Spatial distributions of GABA receptors and local inhibition of Ca2+ transients studied with GABA uncaging in the dendrites of CA1 pyramidal neurons. PLoS One. 2011;6:e22652. doi: 10.1371/journal.pone.0022652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Groupings of nonpyramidal and pyramidal cells with specific physiological and morphological characteristics in rat frontal cortex. J Neurophysiol. 1993;69:416–431. doi: 10.1152/jn.1993.69.2.416. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- Kerlin AM, Andermann ML, Berezovskii VK, Reid RC. Broadly tuned response properties of diverse inhibitory neuron subtypes in mouse visual cortex. Neuron. 2010;67:858–871. doi: 10.1016/j.neuron.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Beierlein M, Connors BW. Inhibitory control of excitable dendrites in neocortex. J Neurophysiol. 1995;74:1810–1814. doi: 10.1152/jn.1995.74.4.1810. [DOI] [PubMed] [Google Scholar]
- Koch C, Poggio T, Torre V. Nonlinear interactions in a dendritic tree: localization, timing, and role in information processing. Proc Natl Acad Sci U S A. 1983;80:2799–2802. doi: 10.1073/pnas.80.9.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Hatada S, Kondo S, Karube F, Kawaguchi Y. Neocortical inhibitory terminals innervate dendritic spines targeted by thalamocortical afferents. J Neurosci. 2007;27:1139–1150. doi: 10.1523/JNEUROSCI.3846-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman SJ, Olivas ND, Tring E, Ikrar T, Xu X, Trachtenberg JT. A disinhibitory microcircuit initiates critical-period plasticity in the visual cortex. Nature. 2013;501:543–546. doi: 10.1038/nature12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitsiani D, Ranade S, Hangya B, Taniguchi H, Huang JZ, Kepecs A. Distinct behavioural and network correlates of two interneuron types in prefrontal cortex. Nature. 2013;498:363–366. doi: 10.1038/nature12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kwan AC, Zhang S, Phoumthipphavong V, Flannery JG, Masmanidis SC, Taniguchi H, Huang ZJ, Zhang F, Boyden ES, Deisseroth K, Dan Y. Activation of specific interneurons improves V1 feature selectivity and visual perception. Nature. 2012;488:379–383. doi: 10.1038/nature11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci. 2013;16:1662–1670. doi: 10.1038/nn.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SB, Meyer EM, Tovote P, Courtin J, Herry C, Lüthi A. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature. 2011;480:331–335. doi: 10.1038/nature10674. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Little JP, Carter AG. Subcellular synaptic connectivity of layer 2 pyramidal neurons in the medial prefrontal cortex. J Neurosci. 2012;32:12808–12819. doi: 10.1523/JNEUROSCI.1616-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat Neurosci. 2004;7:373–379. doi: 10.1038/nn1206. [DOI] [PubMed] [Google Scholar]
- Llinás R, Nicholson C, Freeman JA, Hillman DE. Dendritic spikes and their inhibition in alligator Purkinje cells. Science. 1968;160:1132–1135. doi: 10.1126/science.160.3832.1132. [DOI] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci. 2012;15:423–430. S1–3. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- MacAskill AF, Little JP, Cassel JM, Carter AG. Subcellular connectivity underlies pathway-specific signaling in the nucleus accumbens. Nat Neurosci. 2012;15:1624–1626. doi: 10.1038/nn.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Miles R, Tóth K, Gulyás AI, Hájos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/S0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Müller C, Beck H, Coulter D, Remy S. Inhibitory control of linear and supralinear dendritic excitation in CA1 pyramidal neurons. Neuron. 2012;75:851–864. doi: 10.1016/j.neuron.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457:1137–1141. doi: 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- Oláh S, Füle M, Komlósi G, Varga C, Báldi R, Barzó P, Tamás G. Regulation of cortical microcircuits by unitary GABA-mediated volume transmission. Nature. 2009;461:1278–1281. doi: 10.1038/nature08503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer L, Murayama M, Larkum M. Inhibitory regulation of dendritic activity in vivo. Front Neural Circuits. 2012a;6:26. doi: 10.3389/fncir.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M, Larkum ME. The cellular basis of GABA(B)-mediated interhemispheric inhibition. Science. 2012b;335:989–993. doi: 10.1126/science.1217276. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 2013;16:1068–1076. doi: 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, Kepecs A. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–524. doi: 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Marin-Burgin A, Adesnik H, Atallah BV, Scanziani M. Input normalization by global feedforward inhibition expands cortical dynamic range. Nat Neurosci. 2009;12:1577–1585. doi: 10.1038/nn.2441. [DOI] [PubMed] [Google Scholar]
- Rial Verde EM, Zayat L, Etchenique R, Yuste R. Photorelease of GABA with visible light using an inorganic caging group. Front Neural Circuits. 2008;2:2. doi: 10.3389/neuro.04.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183X.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg G, Markram H. Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron. 2007;53:735–746. doi: 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Rev. 1998;26:113–135. doi: 10.1016/S0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci. 2008;9:206–221. doi: 10.1038/nrn2286. [DOI] [PubMed] [Google Scholar]
- Tamás G, Lorincz A, Simon A, Szabadics J. Identified sources and targets of slow inhibition in the neocortex. Science. 2003;299:1902–1905. doi: 10.1126/science.1082053. [DOI] [PubMed] [Google Scholar]
- Tamás G, Buhl EH, Somogyi P. Fast IPSPs elicited via multiple synaptic release sites by different types of GABAergic neurone in the cat visual cortex. J Physiol. 1997;500:715–738. doi: 10.1113/jphysiol.1997.sp022054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubokawa H, Ross WN. IPSPs modulate spike backpropagation and associated [Ca2+]i changes in the dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1996;76:2896–2906. doi: 10.1152/jn.1996.76.5.2896. [DOI] [PubMed] [Google Scholar]
- Vicini S. New perspectives in the functional role of GABA(A) channel heterogeneity. Mol Neurobiol. 1999;19:97–110. doi: 10.1007/BF02743656. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Runyan CA, Wang FL, Sur M. Division and subtraction by distinct cortical inhibitory networks in vivo. Nature. 2012;488:343–348. doi: 10.1038/nature11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Science STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]