

Abstract
Mutations in the presenilin (PSEN1 and PSEN2) genes are linked to familial Alzheimer's disease (AD) and cause loss of its essential function. Complete inactivation of presenilins in excitatory neurons of the adult mouse cerebral cortex results in progressive memory impairment and age-dependent neurodegeneration, recapitulating key features of AD. In this study, we examine the effects of varying presenilin dosage on cortical neuron survival by generating presenilin-1 conditional knock-out (PS1 cKO) mice carrying two, one, or zero copies of the PS2 gene. We found that PS1 cKO;PS2+/− mice at 16 months exhibit marked neurodegeneration in the cerebral cortex with ∼17% reduction of cortical volume and neuron number, as well as astrogliosis and microgliosis compared with ∼50% reduction of cortical volume and neuron number in PS1 cKO;PS2−/− mice. Moreover, there are more apoptotic neurons labeled by activated caspase-3 immunoreactivity and TUNEL assay in PS1 cKO;PS2+/− mice at 16 months, whereas apoptotic neurons are increased in the PS1 cKO;PS2−/− cerebral cortex at 4 months. The accumulation of the C-terminal fragments of the amyloid precursor protein is inversely correlated with PS dosage. Interestingly, levels of PS2 are higher in the cerebral cortex of PS1 cKO mice, suggesting a compensatory upregulation that may provide protection against neurodegeneration in these mice. Together, our findings show that partial to complete loss of presenilin activity causes progressively more severe neurodegeneration in the mouse cerebral cortex during aging, suggesting that impaired presenilin function by PSEN mutations may lead to neurodegeneration and dementia in AD.
Keywords: Alzheimers disease, conditional knockout, dementia, gamma-secretase, neurodegeneration, neuronal survival
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder, and mutations in the presenilin (PSEN) genes account for ∼90% of the identified familial AD (FAD) mutations. Presenilins are expressed broadly throughout life and serve as the integral catalytic subunit of the γ-secretase complex (Li et al., 2000). PS1 and PS2 have overlapping functions and are required for development and adult brain functions (Wines-Samuelson and Shen, 2005; Ho and Shen, 2011). The presenilin-1 (PS1) knock-out (KO) mice show perinatal lethality and impaired neural development (Shen et al., 1997; Handler et al., 2000; Wines-Samuelson et al., 2005), whereas PS2 KO mice have little detectable phenotypes (Steiner et al., 1999), but PS1/2 double KO mice exhibit more severe phenotypes and die at embryonic day 9 (Donoviel et al., 1999).
To circumvent the perinatal lethality of PS1 KO mice, we previously generated conditional KO (cKO) mice lacking PS1 in excitatory neurons of the adult cerebral cortex and found memory impairment in these mice (Yu et al., 2001). Furthermore, loss of both presenilins in conditional double KO (cDKO) mice results in striking age-dependent neurodegeneration, as well as synaptic and memory impairment (Beglopoulos et al., 2004; Feng et al., 2004; Saura et al., 2004; Zhang et al., 2009, 2010; Wines-Samuelson et al., 2010; Wu et al., 2013). Interestingly, presenilins promote memory and neuronal survival and maintain synaptic function in a γ-secretase-dependent manner, because conditional inactivation of another component of the γ-secretase complex, nicastrin, results in similar memory impairment, synaptic dysfunction, and age-related neurodegeneration (Tabuchi et al., 2009; Lee et al., 2014). Thus, complete loss of PS/γ-secretase function in the adult mouse cerebral cortex recapitulates key features of AD, including age-dependent neurodegeneration, progressive memory impairment, tau hyperphosphorylation, and gliosis.
PSEN mutations confer partial loss of its presenilin function, and some mutations result in complete loss of its activity (Levitan et al., 1996; Song et al., 1999; Moehlmann et al., 2002; Seidner et al., 2006; Heilig et al., 2010). These findings together with striking age-dependent neurodegeneration observed in PS cDKO mice suggested that PSEN mutations might lead to AD as a result of partial loss of PS essential functions (Shen and Kelleher, 2007). However, it has not been tested whether partial loss of presenilin would lead to neurodegeneration in the mouse brain. In this study, we address this question by generating three groups of mutant mice bearing different copies of the Psen genes. We found that partial to complete loss of presenilin leads to increasingly more severe neurodegeneration, as indicated by ∼17% and ∼50% reduction of cortical volume and neuron number, respectively, in the cerebral cortex of PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 16 months of age. The neurodegeneration observed in these mice are accompanied by astrogliosis and microgliosis, as well as increased apoptosis. Furthermore, inactivation of PS1 leads to compensatory upregulation of PS2, which may provide protection against neurodegeneration in PS1 cKO mice. Together, these findings provide direct experimental evidence showing that partial loss of PS activity indeed causes age-dependent neurodegeneration in the mouse cerebral cortex.
Materials and Methods
Mice.
Generation of PS1 cKO (PS1floxed/floxed;PS2+/+;Cre), PS cDKO (PS1floxed/floxed;PS2−/−;Cre), and αCaMKII–Cre transgenic mice was described previously (Yu et al., 2001; Saura et al., 2004). To generate postnatal forebrain-specific PS1 cKO mice carrying different dosages of the PS2 gene, female PS cDKO mice were bred with male PS1floxed/floxed;PS2+/+ mice to obtain PS1floxed/floxed;PS2+/−;Cre and PS1floxed/floxed;PS2+/− mice. Next, male mice from PS1floxed/floxed;PS2+/+, PS1floxed/floxed;PS2+/−, or PS1floxed/floxed;PS2−/− mice were crossed to female PS1floxed/floxed;PS2+/−;Cre mice to obtain control (PS1floxed/floxed;PS2+/+; for histological analysis, PS1floxed/floxed;PS2+/− mice were also included as control), PS1 cKO;PS2+/+ (PS1floxed/floxed;PS2+/+;Cre, also known as PS1 cKO), PS1 cKO;PS2+/− (PS1floxed/floxed;PS2+/−;Cre), and PS1 cKO;PS2−/− (PS1floxed/floxed;PS2−/−;Cre, also known as PS cDKO) mice. The genetic background of all the mice used in this study was C57BL/6J 129 hybrid. For all histological analysis and stereological quantification, the experimenter was blind to the genotype of the mice. All procedures relating to animal care and treatment conformed to institutional and National Institutes of Health guidelines.
Western blot analysis.
Dissected cortices (2–3, 9, or 16 months of age) were homogenized in RIPA buffer [50 mm Tris-Cl, pH 7.6, 150 mm NaCl, 0.5 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, protease inhibitor mixture (Sigma), and 1 mm PMSF]. Equal amounts (10–40 μg/lane) of proteins were separated in NuPAGE gels (Invitrogen) and transferred to nitrocellulose membranes. The membranes were blocked in 5% nonfat milk/TBS for 1 h and incubated with specific primary antibodies as shown below: rabbit anti-PS1 (catalog #529591 and #529592; Calbiochem), rabbit anti-PS2 (ab51249; Abcam), rabbit anti-nicastrin (N1660; Sigma), rabbit anti-Aph-1 (PA1-2010; Pierce), rabbit anti-Pen-2 (catalog #36-7100; Zymed), rabbit anti-amyloid precursor protein (APP; C8; kind gift from Dr. Dennis Selkoe, Center for Neurologic Diseases, Brigham and Women's Hospital and Harvard Medical School, Boston, MA), rabbit anti-amyloid precursor-like protein-1 (APLP1; catalog #171615; Calbiochem), mouse anti-glial fibrillary acidic protein (GFAP; G6171; Sigma), and mouse anti-actin (A1978; Sigma). The membrane was then incubated with IRDye 800CW or IRDye 680-labeled secondary antibodies (LI-COR Bioscience). Signals were developed and quantified with an Odyssey Infrared Imaging System (LI-COR Bioscience).
Nissl staining.
Mice were anesthetized and perfused with PBS including heparin and procaine. Brains were dissected, and hemispheres were immersed in 4% paraformaldehyde at 4°C for 3 h and then processed for paraffin embedding. Serial sagittal sections were collected by microtome at 10 μm thickness. Every 40th sections were deparaffinized, dehydrated, and stained with 0.5% cresyl violet (Sigma).
Immunohistochemistry.
Paraffin-embedded brain sections were deparaffinized, alcohol dehydrated, and blocked in 5% normal goat serum/TBS for 1 h. Then sections were reacted with primary antibodies against NeuN (1:400; Millipore Bioscience Research Reagents), cleaved caspase-3 (1:250, Cell Signaling Technology), ionized calcium-binding adapter molecule 1 (Iba1; 1:300; Wako), or GFAP (1:500; Sigma) at 4°C overnight. These slides were then incubated with biotinylated secondary antibody (Vector Laboratories) at room temperature for 1 h. Specific signals were developed by Vectastain Elite ABC kit and DAB peroxidase substrate (Vector Laboratories) and analyzed by BX40 microscope system (Olympus).
Stereological quantification.
NeuN-stained sections (seven to eight sections per brain, spaced 0.4 mm apart) were analyzed using an unbiased fractionator and optical dissector method, and the images were analyzed using the BioQuant image analysis software that was connected to the Olympus BX51 microscope with a CCD camera. Approximately 40 optical dissectors were used for the entire neocortex area. Each optical dissector was a 100 × 100 μm sampling box, and NeuN-positive (NeuN+) neurons were counted through the 40× objective lens. The coefficient of error was <0.10. The volume of the neocortex was determined using Nissl-stained series sections and the BioQuant image analysis software (Yamaguchi and Shen, 2013). Values were presented as means ± SEM.
GFAP-stained sections (five sections per brain, spaced 0.5 mm apart) were analyzed using the Olympus BX51 microscope with a CCD camera. The GFAP+ area and the total area of the neocortex and the hippocampus were measured under 4× objective lens using the BioQuant image analysis software. The GFAP+ area was determined using the set threshold value of immunostained color that covers all the GFAP signals. The percentage of GFAP+ area was calculated as GFAP+ area/total area (n = 3–6 mice per genotype). Iba1-stained sections (five sections per brain, spaced 0.5 mm apart) were analyzed, and the number of Iba1+ cells was counted using the unbiased fractionator and optical dissector method and the BioQuant image analysis software. The Iba1+ signal was calculated as the Iba1+ cell number/total area (n = 3–6 per genotype). All values were presented as means ± SEMs.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining.
Every 30th sections (seven to nine sections per brain) were blocked using 5% of goat serum, 1% BSA for 1 h, followed by the protocol of the manufacturer of the In Situ Cell Death Detection kit (Roche). The slides were then washed using PBS for three times. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining was analyzed using a fluorescent microscope. TUNEL+ signals in the neocortical area were counted, and the number of positive cells per section was averaged.
Data analysis.
Two-tailed Student's t test was conducted for all the pairwise comparisons in the biochemical results, unless stated otherwise. In Figure 6, statistical analysis was performed using one-way ANOVA, followed by post hoc analysis (Tukey's test) to measure the genotypic effect. Three to 10 mice were used per genotype group. A value of p < 0.05 is considered statistically significant. All the data were reported as mean ± SEM, except Figure 1C (mean ± SD).
Figure 6.

Upregulation of PS2 in the cerebral cortex of PS1 cKO;PS2+/+ and PS1 cKO;PS2+/− mice. A, Western blot analysis of PS1 NTF, PS1 CTF, PS2 CTF, nicastrin, Aph-1a, Pen-2, and β-actin. Representative blots are shown for each protein. B, Quantitative analysis of control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mouse brains at 2 months of age (n = 4 per genotype) shows ∼60% decreases of NTFs and CTFs of PS1 in PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− at 2 months of age (n = 4 per genotype). PS2 CTFs are increased by ∼30% in cortical lysates of PS1 cKO;PS2+/+ and PS1 cKO;PS2+/− mice compared to the expected values, suggesting compensatory upregulation in the absence of PS1. Levels of nicastrin and Pen-2 are decreased by ∼40% and ∼60% in the mutant mice compared with controls, respectively, whereas levels of another γ-secretase component, Aph-1a, is present at comparable levels between the genotypic groups. NS, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001. All data are expressed as mean ± SEM.
Figure 1.
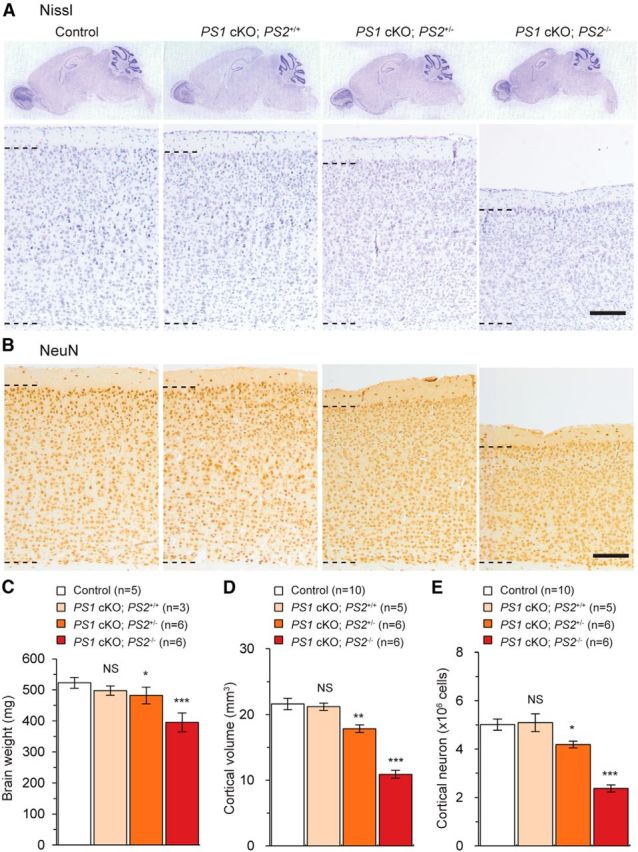
Neurodegeneration in the cerebral cortex of PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 16 months of age. A, Representative images of the Nissl-stained sagittal sections of the whole brain (top) and the neocortex (bottom) of control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 16 months of age are shown. Black dashed bars delineate cortical layers II–VI. Whereas striking decreases in neocortical thickness are observed in PS1 cKO;PS2−/− mice, milder cortical atrophy is seen in PS1 cKO;PS2+/− mice. Scale bar, 200 μm. B, Representative images of the neocortex stained with NeuN immunoreactivity from control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 16 months of age are shown. Scale bar, 200 μm. C–E, Stereological measurement of whole-brain weight (C) and the volume (D) and the neuron number (E) of the neocortex from control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 16 months. NS, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001; n = 5–10 mice per genotype. Data are presented as the mean ± SEM, except C (mean ± SD). Small decreases of brain weight are present in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice compared with controls. The cortical volume is significantly reduced (∼17%) in PS1 cKO;PS2+/− mice and is further reduced in PS1 cKO;PS2−/− mice (∼50%). Values are presented as per hemisphere. Neuron numbers are also significantly decreased (∼16%) in PS1 cKO;PS2+/− mice and are further decreased in PS1 cKO;PS2−/− mice (∼52%).
Results
Neurodegeneration in the cerebral cortex of aged PS1 cKO;PS2+/− mice
Complete loss of γ-secretase activity in excitatory neurons of the cerebral cortex in presenilin cDKO and nicastrin (Nct) cKO mice exhibit severe cerebral atrophy and dramatic loss of cortical volume and neurons by 6–9 months (Saura et al., 2004; Tabuchi et al., 2009; Wines-Samuelson et al., 2010). To address whether partial loss of γ-secretase activity also causes neurodegeneration in vivo, we generated PS1 cKO mice carrying varying doses of the PS2 gene, in which the floxed PS1 gene is inactivated in excitatory neurons of the postnatal forebrain under control of the αCaMKII–Cre transgene (Yu et al., 2001; Saura et al., 2004). These mice were born at the expected ratio, and their gross appearances were normal at birth or at young adult ages.
We first performed histological analysis of postnatal forebrain-specific PS1 cKO mice carrying different dosage of the PS2 gene (PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/−) and littermate control mice at 16–18 months of age. A striking cortical atrophy was observed in the cerebral cortex of PS1 cKO;PS2−/− mice compared with littermate controls (Fig. 1A). In addition, comparable Nissl-stained (Fig. 1A) and NeuN-immunostained (Fig. 1B) brain sections revealed a subtle atrophy of the neocortex in the brain of PS1 cKO;PS2+/− mice compared with littermate controls, as well as small reduction of whole-brain weight (Fig. 1C). To examine the subtle cortical atrophy of PS1 cKO;PS2+/− mice thoroughly, we next performed unbiased quantitative stereological analysis using Nissl-stained series brain sections. Compared with littermate controls, a marked reduction of neocortical volume was observed in PS1 cKO;PS2+/− (∼17%; p = 0.005) and PS1 cKO;PS2−/− (∼50%; p = 2.423 × 10−7) mice, respectively (Fig. 1D; control, 21.65 ± 0.86 mm3; PS1 cKO;PS2+/−, 17.85 ± 0.58 mm3; PS1 cKO;PS2−/−, 10.91 ± 0.60 mm3), whereas no obvious change was seen in PS1 cKO;PS2+/+ mice (21.21 ± 0.55 mm3, p = 0.740). Likewise, as measured by counting NeuN+ cells, the number of neocortical neurons was significantly decreased by ∼16.4% (p = 0.019) and ∼52.4% (p = 6.554 × 10−7) in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice, respectively (Fig. 1E; control, 5.02 × 106 cells; PS1 cKO;PS2+/−, 4.19 × 106 cells; PS1 cKO;PS2−/−, 2.37 × 106 cells), whereas no obvious change was seen in PS1 cKO;PS2+/+ mice (5.09 × 106 cells, p = 0.854). These results show that partial loss of PS dosage also results in cortical atrophy and cortical neuronal loss in PS1 cKO;PS2+/− mice, although the extent of neurodegeneration is less severe compared with PS1 cKO;PS2−/− mice.
We further performed similar stereological analysis at 4 months of age and found no reduction of cortical volume in PS1 cKO;PS2+/− mice (Fig. 2A; control, 21.78 ± 0.62 mm3; PS1 cKO;PS2+/−, 20.97 ± 1.62 mm3, p = 0.604). Using NeuN-stained series sections, we also did not see alteration of cortical neuron number in PS1 cKO;PS2+/− mice at this age (Fig. 2B,C; control, 4.45 × 106; PS1 cKO;PS2+/−, 4.48 × 106, p = 0.853), whereas ∼11% reduction of cortical neuron number was observed in PS1 cKO;PS2−/− mice at this age, as we reported previously (Wines-Samuelson et al., 2010). Together, these results show the later age of onset for neurodegeneration in PS1 cKO;PS2+/− mice relative to PS1 cKO;PS2−/− mice.
Figure 2.

No significant reduction of cortical neuron number in the neocortex of PS1 cKO;PS2+/− mice at 4 months of age. A, Representative images of the Nissl-stained sagittal sections of control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 4 months of age are shown. B, Representative images of the neocortex stained with NeuN immunoreactivity from control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 4 months of age are shown. Black dashed bars delineate cortical layers II–VI. Cortical atrophy is seen at this age in PS1 cKO;PS2−/− mice. Scale bar, 200 μm. C, Stereological measurement of neuron number in the neocortex from control, PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 4 months shows significant reduction (∼11%) of neuron number in PS1 cKO;PS2−/− mice relative to controls, whereas no cortical neuron loss is detected in PS1 cKO;PS2+/− mice. NS, Not significant. *p < 0.05; n = 4–6 mice per genotype. Data are presented as the mean ± SEM.
Increased apoptosis in PS1 cKO;PS2+/− mice
Previous studies showed increases of neuronal apoptosis as early as 2 months of age in the cerebral cortex of PS1 cKO;PS2−/− mice (Wines-Samuelson et al., 2010). To determine whether apoptosis is increased in the cerebral cortex of PS1 cKO;PS2+/− mice, we performed immunostaining using antibodies specific for active (cleaved) forms of caspase-3, which is an excellent marker for apoptosis (Earnshaw et al., 1999). Similar to the previous report (Wines-Samuelson et al., 2010), significant increases of cells positive for active caspase-3 were found in the neocortex of PS1 cKO;PS2−/− mice at 4 months of age relative to controls (Fig. 3A; control, 3.35 ± 0.48; PS1 cKO;PS2−/−, 9.71 ± 1.62 cells per section, p = 0.020), whereas at 16 months, the increase of active caspase-3+ cells in the PS1 cKO;PS2−/− neocortex is not significant compared with the control (control, 2.95 ± 0.44; PS1 cKO;PS2−/−, 4.21 ± 1.30 cells per section, p = 0.274; n = 6–11). Interestingly, the number of cells positive for active caspase-3 was increased in PS1 cKO;PS2+/− mice compared with littermate controls at 16 months of age (Fig. 3A; 7.08 ± 1.09 cells per section, p = 7.872 × 10−4) but not at 4 months (4.21 ± 0.27 cells per section, p = 0.191). In contrast, PS1 cKO;PS2+/+ mice did not exhibit a significant change in the number of cells positive for active caspase-3 (4 months, 3.04 ± 0.42 cells per section, p = 0.658; 16 months, 3.73 ± 1.23 cells per section, p = 0.465). These results indicate that neuronal cell death occurs in the cerebral cortex of PS1 cKO;PS2+/− mice, but the age of onset of apoptosis is delayed in the cerebral cortex of PS1 cKO;PS2+/− mice compared with PS1 cKO;PS2−/− mice.
Figure 3.
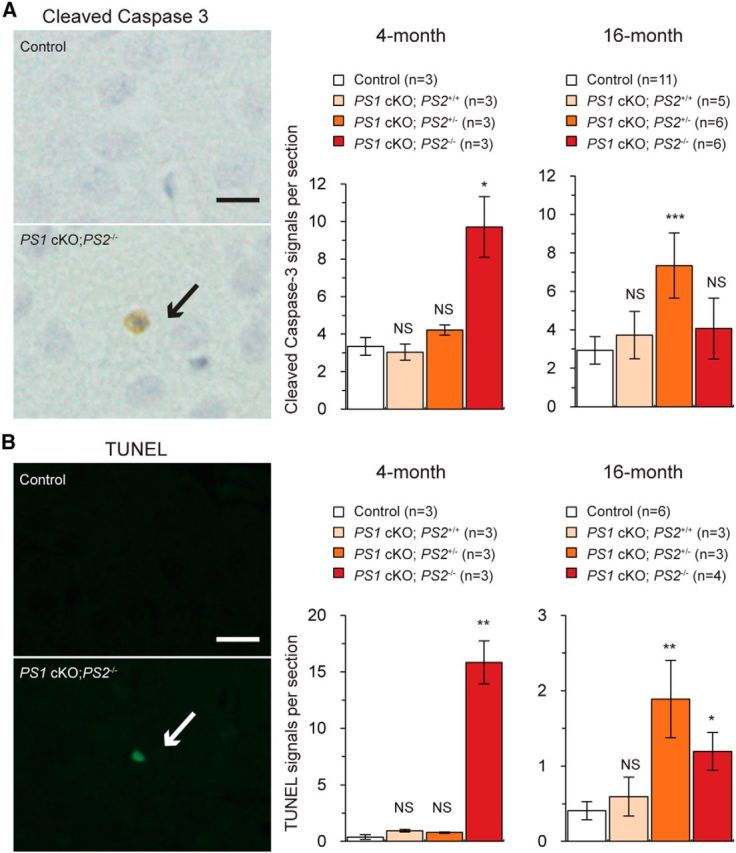
Delayed onset of apoptosis in the cerebral cortex of PS1 cKO;PS2+/− mice relative to PS1 cKO;PS2−/− mice. A, Quantification of cells positive for activated caspase-3. Left, Representative optical micrographs are indicated from control (top) and PS1 cKO;PS2−/− (bottom) mice. Right, The number of active caspase-3+ cells is significantly increased in the neocortex of PS1 cKO;PS2+/− mice at 16 months of age but not at 4 months (n = 3–11 mice per genotype per age; 8–9 sections analyzed per mouse). Data are presented as the mean ± SEM. B, Left, Fluorescent microscopic images of TUNEL+ cells in the neocortex of control (top) and PS1 cKO;PS2−/− (bottom) mice. Right, Quantification of TUNEL+ cells shows considerable increases in the PS1 cKO;PS2+/− neocortex at 16 months compared with controls (n = 3–6 mice per genotype; 9 sections analyzed per mouse) but not at 4 months. Scale bar, 20 μm. NS, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001. Data are presented as the mean ± SEM.
To confirm further the occurrence of apoptosis in the cortex of PS1 cKO;PS2+/− mice, we also performed the TUNEL assay using mice at 16 months of age, in which PS1 cKO;PS2+/− mice exhibit significant increases of active caspase-3 signals. Similarly, we found a significant increase in the number of TUNEL+ cells in the neocortex of PS1 cKO;PS2+/− mice at 16 months (p = 0.005) compared with control mice (Fig. 3B; TUNEL+ cells per section: control, 0.41 ± 0.12; PS1 cKO;PS2+/+, 0.59 ± 0.26;PS1 cKO;PS2+/−, 1.89 ± 0.51; n = 3–6) but not at 4 months (control, 0.39 ± 0.20; PS1 cKO;PS2+/−, 0.78 ± 0.06 per section, p = 0.135). TUNEL+ cells are also increased in the cerebral cortex of PS1 cKO;PS2−/− mice at 16 months of age (1.19 ± 0.25 cells per section, p = 0.011), although the increase of apoptosis is greater at 4 months of age (15.83 ± 1.90 cells per section, p = 1.260 × 10−3). Together, these results suggest that apoptotic cell death occurs in the neocortex of both PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice, although the age of onset is later in PS1 cKO;PS2+/− mice with increases of apoptosis occurring at 16 months in PS1 cKO;PS2+/− mice and at 4 months in PS1 cKO;PS2−/− mice.
Progressive gliosis in PS1 cKO;PS2+/− mice
Because resident astrocytes are activated with ongoing neurodegeneration (Beglopoulos et al., 2004; Saura et al., 2004; Lobsiger and Cleveland, 2007; Heneka et al., 2010), we next looked for the presence of astrogliosis, as performed by immunohistochemical analysis on GFAP, a marker of astrogliosis, using brain sections of PS1 cKO;PS2+/− mice and littermate controls. As expected, we did not detect increases of GFAP immunoreactivity in the hippocampus and the neocortex in PS1 cKO;PS2+/+ mice at both 4 and 16 months of age, whereas we could easily see robust increases of GFAP immunoreactivity in PS1 cKO;PS2−/− mice even at 4 months (Fig. 4A,B), when prominent apoptosis had taken place in the cerebral cortex (Fig. 3). Interestingly, moderate increases of GFAP signals were detected in PS1 cKO;PS2+/− mice at 16 months of age but not 4 months of age (Fig. 4A,B). To determine the extent of astrogliosis further, we performed unbiased stereological quantification of GFAP signals in the neocortex and hippocampus (percentage GFAP+ area/tissue area). GFAP signals are unaltered in the neocortex (control, 1.01 ± 0.31%; PS1 cKO;PS2+/−, 2.43 ± 0.80%; p = 0.096) and the hippocampus (control, 3.77 ± 1.14%; PS1 cKO;PS2+/−, 4.67 ± 1.33%; p = 0.634) of PS1 cKO;PS2+/− mice at 4 months (Fig. 4C), but GFAP signals are significantly increased in the neocortex of PS1 cKO;PS2+/− mice at 16 months of age (Fig. 4D, left; control, 3.68 ± 0.82%; PS1 cKO;PS2+/−, 10.94 ± 3.42%; p = 0.0247). However, we did not see significant increases of GFAP in the hippocampus of PS1 cKO;PS2+/− mice at 16 months (Fig. 4D, right; control, 18.72 ± 6.13%; PS1 cKO;PS2+/−, 29.98 ± 11.20%; p = 0.3636), perhaps because of the higher and more variable basal GFAP signals in the hippocampus of control mice at this age (Fig. 4B,D). PS1 cKO;PS2+/+ mice did not exhibit any alteration at 4 months (neocortex, 1.00 ± 0.52%, p = 0.987; hippocampus, 3.79 ± 1.24%, p = 0.991) or 16 months (neocortex, 6.86 ± 2.48%, p = 0.160; hippocampus, 14.84 ± 4.91%, p = 0.696). These results show an incidence of mild astrogliosis accompanying neurodegeneration in PS1 cKO;PS2+/− mice.
Figure 4.

Astrogliosis in the cerebral cortex of PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 16 months. A, B, Immunostaining of GFAP in the neocortex and hippocampus from each genotype at 4 (A) and 16 (B) months of age. There is a progressive astrogliosis in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice. Scale bar, 100 μm. C, Stereological measurement of GFAP+ areas from the neocortex (left) and the hippocampus (right) at 4 months of age (n = 3–5 mice per genotype; 5 sections analyzed per mouse) shows a significant increase of GFAP signals in the neocortex and hippocampus of PS1 cKO;PS2−/− mice but not in PS1 cKO;PS2+/− mice. D, Stereological measurement of GFAP+ areas from the neocortex (left) and the hippocampus (right) at 16 months of age (n = 3–6 mice per genotype; 5 sections analyzed per mouse) shows a significant increase of GFAP signals in the neocortex of PS1 cKO;PS2+/− mice, whereas there is greater variability in GFAP signals in the hippocampus. NS, Not significant. *p < 0.05; ***p < 0.001. Data are presented as the mean ± SEM.
We next performed immunohistochemical analysis on Iba1, which is specifically expressed in microglia in the brain. We found that Iba1 immunoreactivity was increased in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 16 months of age but not in PS1 cKO;PS2+/+ mice (Fig. 5A). Unbiased stereological quantification uncovered significant increases of Iba1 signals (Iba1+ cell number/tissue area) in the neocortex (control, 33.45 ± 1.79 cells/mm2; PS1 cKO;PS2+/−, 64.96 ± 3.99 cells/mm2; p = 6.107 × 10−5) and the hippocampus (control, 38.24 ± 3.26 cells/mm2; PS1 cKO;PS2+/−, 66.36 ± 10.57 cells/mm2; p = 0.012) of PS1 cKO;PS2+/− mice compared with littermate controls at 16 months of age (Fig. 5B), whereas robust increases were observed in PS1 cKO;PS2−/− mice (neocortex, 139.39 ± 12.93 cells/mm2, p = 7.726 × 10−6; hippocampus, 126.07 ± 13.31 cells/mm2, p = 5.326 × 10−5). In contrast, PS1 cKO;PS2+/+ mice did not exhibit any alteration (neocortex, 36.35 ± 8.81 cells/mm2, p = 0.662; hippocampus, 52.16 ± 12.49 cells/mm2, p = 0.185) relative to controls. Together, PS1 cKO;PS2+/− mice exhibit modest gliosis in both astrocytes and microglia accompanied by neuronal cell death, but these phenotypes are less severe than that in cerebral cortex of PS1 cKO;PS2−/− mice.
Figure 5.

Microgliosis in the cerebral cortex of PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 16 months. A, Iba1 immunoreactivity appears higher in the neocortex and hippocampus of PS1 cKO;PS2+/− mice and further increased in PS1 cKO;PS2−/− mice. The insets in the pictures show higher-magnification views of the Iba1+ cells. Scale bars, 100 μm; insets 10 μm. B, Stereological quantification of Iba+ cells from the neocortex (left) and the hippocampus (right) at 16 months of age (n = 3–6 mice per genotype; 4–5 sections analyzed per mouse) shows a significant increase of Iba1+ cells in the neocortex and hippocampus of PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice. NS, Not significant. *p < 0.05; ***p < 0.001. Data are presented as the mean ± SEM.
Upregulation of PS2 in the cerebral cortex of PS1 cKO;PS2+/+ and PS1 cKO;PS2+/− mice
To determine whether levels of PS1, PS2, and other components of the γ-secretase complex were altered, we performed Western blot analysis using cell lysates isolated from the cerebral cortex of PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, PS1 cKO;PS2−/−, and littermate control mice at 2 months of age. We found similar reductions (∼60%) of PS1 N-terminal fragments (NTFs) and C-terminal fragments (CTFs) in the cerebral cortex of PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice compared with littermate controls (Fig. 6). An overall ANOVA showed significant genotypic effect between controls and other genotypes (NTFs, F(3,12) = 222.9, p < 0.0001; CTFs, F(3,12) = 90.84, p < 0.0001), but there was no difference by post hoc test among PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice. Interestingly, PS2 CTFs were increased by ∼30% in the cerebral cortex of PS1 cKO;PS2+/+ compared with littermate controls, suggesting compensatory upregulation attributable to the lack of PS1 protein, consistent with other reports (Dewachter et al., 2002; Lai et al., 2003). The compensatory upregulation of PS2 was also observed at a similar extent at 16 months of age (data not shown), suggesting that this increase is consistent from 2 to 16 months of age. Compared with PS1 cKO;PS2+/+ mice, levels of PS2 CTFs were decreased by 50% in the cerebral cortex of PS1 cKO;PS2+/− mice and completely eliminated in PS1 cKO;PS2−/− mice. Given the fact that PS1 is inactivated selectively in excitatory neurons but not in interneurons and glia, which is consistent with the ∼40% PS1 protein remaining in the cortical lysates of PS1 cKO mice, the increase of PS2 levels in excitatory neurons is likely to be greater than the 30% increase detected in total cortical lysates of PS1 cKO mice (Fig. 6). Thus, the compensatory upregulation of PS2 in PS1 cKO;PS2+/+ and PS1 cKO;PS2+/− mice likely protect the excitatory neurons during aging.
We further quantified other components of the γ-secretase complex. For quantification of nicastrin, we first treated peptide-N-glycosidase F to remove saccharide groups from the mature glycosylated form of nicastrin proteins and then performed Western blot analysis. Levels of nicastrin and Pen-2 proteins were significantly reduced in the cortical lysates from PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 2 months of age compared with littermate controls (Fig. 6; nicastrin, F(3,12) = 68.74, p < 0.0001; Pen-2, F(3,12) = 54.92, p < 0.0001). Conversely, Aph-1a protein was unchanged among all four genotypes (F(3,12) = 0.931, p = 0.456), consistent with our previous findings in nicastrin cKO mice (Tabuchi et al., 2009). These data show that loss of PS1 disrupts the γ-secretase complex, resulting in reduction of nicastrin and Pen-2 in the cortex of PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice.
Decreased γ-secretase activity in the cerebral cortex of PS1 cKO;PS2+/− mice
We next evaluated γ-secretase activity by measuring levels of the APP CTFs, because accumulation of APP CTFs is a sensitive marker for impairment of γ-secretase activity in mouse brains (Yu et al., 2001; Dewachter et al., 2002; Saura et al., 2004; Saura et al., 2005). We performed Western blot analysis to measure levels of APP CTFs in the cortex of PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice, as well as the littermate controls. We found dramatic accumulation (∼30-fold) of APP CTFs in the cortex of PS1 cKO;PS2+/+ mice (Fig. 7A). Interestingly, the extent of the accumulation is inversely correlated with the dosage of the PS2 gene in PS1 cKO mice (Fig. 7A). Compared with PS1 cKO;PS2+/+ mice, levels of APP CTFs are increased significantly by ∼1.2-fold (p = 2.225 × 10−2) and ∼2-fold (p = 9.352 × 10−6) in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 2 months, respectively. Likewise, its accumulation is increased significantly by ∼1.5-fold (p = 1.927 × 10−3) and ∼2.2-fold (p = 6.555 × 10−4) in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice at 9 months of age, respectively. These results suggest that γ-secretase activity is further impaired in the cortex of PS1 cKO;PS2+/− mice during aging.
Figure 7.

Accumulation of the CTFs of APP and APLP1 in the cerebral cortex of PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice. A, B, Unchanged levels of full-length APP and increased levels of the CTFs of APP (A) and APLP1 (B) in the cerebral cortex of PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice at 2 and 9 months of age. Immunoblotting (top) was performed using antibodies specific for the APP C terminus (A) or the APLP1 C terminus (B). Varying amounts of cortical lysates were loaded in each lane. Lanes 1 and 2 were loaded with 3× and 1× amounts of cortical lysates from control mice, and the rest of the lanes were loaded with 1× (lanes 3–5), 0.25× (A, lanes 6–8), or 0.5× (B, lanes 6–8) of cortical lysates from PS1 cKO;PS2+/+, PS1 cKO;PS2+/−, and PS1 cKO;PS2−/− mice. Levels of the CTFs were quantified and normalized to the full-length proteins (bottom). Quantification analysis shows a massive increase of the CTFs in the cerebral cortex of PS1 cKO;PS2+/+ mice and additional increases in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice. The value of the CTFs in PS1 cKO;PS2+/+ mice is set as 100%. NS, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
We further examined the C-terminal stub of another γ-secretase substrate, APLP1 (Naruse et al., 1998). Similar to APP, APLP1 CTFs accumulate by ∼10-fold in the cortex of PS1 cKO;PS2+/+ mice, and the extent of the accumulation is inversely correlated with the dosage of the wild-type PS2 gene (Fig. 7B). For example, at 2 months of age, levels of APLP1 CTFs are increased at ∼1.2-fold (p = 0.156) and ∼1.6-fold (p = 2.700 × 10−3) in PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice, respectively (Fig. 7B). Furthermore, at 9 months of age, levels of APLP1 CTFs are increased by ∼1.4-fold (p = 2.493 × 10−3) and ∼1.8-fold (p = 8.411 × 10−4) in the cerebral cortex of PS1 cKO;PS2+/− and PS1 cKO;PS2−/− mice, respectively. These results demonstrated that γ-secretase activities are modulated by the presence of both PS1 and PS2 proteins and that, in the absence of PS1, γ-secretase activity is dependent on the dosage of the PS2 gene.
Discussion
Complete inactivation of presenilin or nicastrin in excitatory neurons of the adult cerebral cortex results in striking age-dependent, progressive neurodegeneration, as well as synaptic and memory impairment (Beglopoulos et al., 2004; Saura et al., 2004; Tabuchi et al., 2009; Zhang et al., 2009, 2010; Wines-Samuelson et al., 2010; Wu et al., 2013; Lee et al., 2014). These findings demonstrate the essential role of presenilin/γ-secretase in neuronal survival, synaptic function, and memory in the adult brain. The fact that complete loss of presenilin or γ-secretase recapitulates key features of AD, including age-dependent loss of synapses and neurons, increases of apoptotic neuronal death, inflammatory responses, tau hyperphosphorylation, and progressive impairment of memory and other behavior, suggested that loss of essential PS function by PSEN mutations may underlie neurodegeneration and dementia in AD (Shen and Kelleher, 2007). This view is further supported by findings in Caenorhabditis elegans, Drosophila, and mammalian cultured cells showing that PSEN mutations result in partial to complete loss of its function or γ-secretase activity (Levitan et al., 1996; Song et al., 1999; Seidner et al., 2006; Heilig et al., 2010). However, whether partial loss of PS indeed results in neurodegeneration has not been tested experimentally. The current study was designed to address this question through the generation of PS1 cKO mice carrying varying doses of the PS2 gene. We found that PS1 cKO;PS2+/− mice exhibit significant neurodegeneration in the cerebral cortex, with ∼17% loss of cortical volume and neuron number at 16 months of age compared with the more severe neurodegeneration in PS1 cKO;PS2−/− mice, with ∼50% reduction of cortical volume and neuron number (Fig. 1). Neurodegeneration in PS1 cKO;PS2+/− mice is also accompanied by increases of apoptosis (Fig. 3), as well as astrogliosis (Fig. 4) and microgliosis (Fig. 5). At 4 months of age, significant loss of cortical volume and neuron number was observed in PS1 cKO;PS2−/− mice but not in PS1 cKO;PS2+/− mice (Fig. 2). Consistent with these findings, increased apoptosis was seen in PS1 cKO;PS2+/− mice at 16 months but not at 4 months compared with dramatic increases of apoptosis in PS1 cKO;PS2−/− mice at 4 months, suggesting a later onset of apoptosis in PS1 cKO;PS2+/− mice (Fig. 3). Thus, loss of PS activity indeed leads to neurodegeneration and increases of apoptotic cell death in an age- and PS dose-dependent manner.
PS1 and PS2 share ∼67% sequence homology and are functional homologs (Levy-Lahad et al., 1995; Rogaev et al., 1995). PS1 appears to be more important than PS2 in performing presenilin functions, because PS1 germ-line KO mice die at birth and PS2 KO mice have little detectable phenotypes. However, in the absence of PS1, PS2 does play essential roles. For example, mice lacking both PS1 and PS2 die much earlier than PS1 KO mice at embryonic day 9 (Shen et al., 1997; Donoviel et al., 1999), and the neurogenesis defects caused by loss of both presenilins in neural progenitor cells are much more severe than those observed in neural progenitor cell-restricted PS1 cKO mice (Shen et al., 1997; Handler et al., 2000; Wines-Samuelson et al., 2005; Kim and Shen, 2008). Similarly, in the adult brain, PS cDKO mice lacking both presenilins in excitatory neurons of the adult cerebral cortex develop striking neurodegeneration and severe memory impairment during aging, whereas PS1 cKO mice exhibit mild memory impairment but no neurodegeneration (Yu et al., 2001; Saura et al., 2004; Wines-Samuelson et al., 2010; Fig. 1). Consistent with these in vivo findings, in vitro γ-secretase assay showed that membrane fractions from PS1+/−;PS2−/− and PS1−/−;PS2+/+ cells have 56 and 11% of γ-secretase activity, respectively, compared with those derived from PS1+/+;PS2+/+ cells, suggesting that PS1 is ∼10-fold more active than PS2 (Lai et al., 2003). Furthermore, ablation of the PS2 gene alone in mice did not affect both accumulation of APP CTFs and β-amyloid (Aβ) production (Steiner et al., 1999), whereas conditional inactivation of PS1 selectively in excitatory neurons of the adult cerebral cortex leads to 30-fold accumulation of APP CTFs and reduction of Aβ production at 6 months of age (Yu et al., 2001). In this study, we showed that accumulation of the CTFs of APP and APLP1 in the mouse brain correlates with PS dosage and that loss of one or two copies of the PS2 gene in PS1 cKO mice results in increasingly greater accumulation of the CTFs (Fig. 7). The accumulation of APP CTFs in the cerebral cortex of PS1 cKO;PS2−/− mice is approximately twice as much as that in PS1 cKO mice, which accumulate APP CTFs >10-fold compared with control mice (Fig. 7). Thus, both in vivo and in vitro analysis showed that PS1/γ-secretase is much more potent than PS2/γ-secretase, although a recent study using a yeast reconstitution system showed that the amount of active PS1 containing γ-secretase complex is much greater than PS2 containing γ-secretase and that the activity of PS1 or PS2 in single γ-secretase complex is similar (Yonemura et al., 2011).
The failure to detect significant neuronal degeneration in PS1 cKO mice at 16 months of age is somewhat surprising, because most, if not all, cortical pyramidal neurons lack PS1 in the cerebral cortex of PS1 cKO mice beginning at ∼1 month of age (Yu et al., 2001). Interestingly, PS2 is upregulated in these mice, as indicated by ∼30% increase of PS2 CTFs in the cerebral cortex of PS1 cKO;PS2+/+ mice at 2 months of age (Fig. 6). Given the fact that the PS1 cKO cerebral cortex is mosaic with PS1 inactivated only in excitatory neurons but not in interneurons or glia, the increase of PS2 in individual excitatory neurons is likely to be greater than the 30% increase detected by Western blotting using cortical lysates. Thus, the upregulation of PS2 may provide additional protection against neurodegeneration in PS1 cKO mice at this age (Fig. 1). The upregulation of PS2 protein in PS1-deficient cells might be attributable to enhanced formation of the PS2/γ-secretase complex, which stabilizes PS2 protein, because of increased availability of other subunits, such as nicastrin, Aph-1, and Pen-2 in the absence of PS1. Similarly, more PS2 was detected in cortical lysates of PS1 cKO;PS2+/− mice (Fig. 6), suggesting that neurodegeneration detected in these mice may be otherwise more severe (Fig. 1). Furthermore, a recent report indicated that PS2 containing γ-secretase in microglia plays a role in protection from neuronal cell death by suppressing neuroinflammatory responses (Jayadev et al., 2010). Although this may explain the severe neurodegeneration and gliosis observed in PS cDKO mice, in which PS2 is eliminated, it cannot explain similarly severe neurodegeneration and gliosis observed in Nct cKO mice, in which PS2 expression is intact in microglia (Tabuchi et al., 2009). Although one copy of PS2 is insufficient to prevent age-dependent neurodegeneration, one copy of PS1 in germ-line PS1+/−;PS2−/− mice (Tournoy et al., 2004) or conditional PS1floxed/+;PS2−/−;Cre mice (D. Xia and J.S., unpublished data) is sufficient to prevent neurodegeneration in the brain, although defects in the peripheral systems were reported, including benign skin hyperplasia, splenomegaly, and leukocytosis (Qyang et al., 2004; Tournoy et al., 2004). These results are consistent with more prominent role of PS1 in maintaining γ-secretase activity and brain functions.
In summary, we demonstrate that, during aging, survival of excitatory neurons in the cerebral cortex is dependent on the dosage of the presenilin genes. These excitatory neurons are particularly vulnerable in AD and normally express high levels of the PS genes. Partial to complete loss of presenilins results in progressively more severe neuronal degeneration and earlier onset of increased apopototic cell death. The milder neurodegeneration and the later onset of apoptosis in PS1 cKO;PS2+/− mice are more reminiscent of the progressive neurodegeneration in AD, in contrast to the striking and earlier onset of neurodegeneration in PS cDKO mice. This is the first experimental evidence showing that partial loss of presenilins is sufficient to cause neuronal cell death in the mouse brain during aging. Considering the fact that FAD mutations in the PSEN genes are loss-of-function mutations (Levitan et al., 1996; Song et al., 1999; Moehlmann et al., 2002; Seidner et al., 2006; Shen and Kelleher, 2007; Heilig et al., 2010) and that expression and activity of presenilins/γ-secretase is attenuated in aged mouse brains (Placanica et al., 2009), the current study provides additional experimental support for the loss of PS function underlying neurodegeneration in FAD and AD.
Footnotes
This work was supported by National Institutes of Health Grants R01NS041783 and R01NS042818 (J.S.). We thank H. Zhao for breeding and genotyping the mice and other laboratory members for helpful discussions.
The authors declare no competing financial interests.
References
- Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem. 2004;279:46907–46914. doi: 10.1074/jbc.M409544200. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Reversé D, Caluwaerts N, Ris L, Kuipéri C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002;22:3445–3453. doi: 10.1523/JNEUROSCI.22-09-03445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13:2801–2810. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of Alzheimer's presenilin-1 and presenilin-2. Proc Natl Acad Sci U S A. 2004;101:8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127:2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- Heilig EA, Xia W, Shen J, Kelleher RJ., 3rd A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J Biol Chem. 2010;285:22350–22359. doi: 10.1074/jbc.M110.116962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Rodríguez JJ, Verkhratsky A. Neuroglia in neurodegeneration. Brain Res Rev. 2010;63:189–211. doi: 10.1016/j.brainresrev.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Ho A, Shen J. Presenilins in synaptic function and disease. Trends Mol Med. 2011;17:617–624. doi: 10.1016/j.molmed.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayadev S, Case A, Eastman AJ, Nguyen H, Pollak J, Wiley JC, Moller T, Morrison RS, Garden GA. Presenilin 2 is the predominant gamma-secretase in microglia and modulates cytokine release. PLoS One. 2010;5:e15743. doi: 10.1371/journal.pone.0015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Shen J. Presenilins are required for maintenance of neural stem cells in the developing brain. Mol Neurodegener. 2008;3:2. doi: 10.1186/1750-1326-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MT, Chen E, Crouthamel MC, DiMuzio-Mower J, Xu M, Huang Q, Price E, Register RB, Shi XP, Donoviel DB, Bernstein A, Hazuda D, Gardell SJ, Li YM. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J Biol Chem. 2003;278:22475–22481. doi: 10.1074/jbc.M300974200. [DOI] [PubMed] [Google Scholar]
- Lee SH, Sharma M, Südhof TC, Shen J. Synaptic function of nicastrin in hippocampal neurons. Proc Natl Acad Sci U S A. 2014;111:8973–8978. doi: 10.1073/pnas.1408554111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu YH, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci U S A. 2002;99:8025–8030. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/S0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Placanica L, Zhu L, Li YM. Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One. 2009;4:e5088. doi: 10.1371/journal.pone.0005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qyang Y, Chambers SM, Wang P, Xia X, Chen X, Goodell MA, Zheng H. Myeloproliferative disease in mice with reduced presenilin gene dosage: effect of gamma-secretase blockage. Biochemistry. 2004;43:5352–5359. doi: 10.1021/bi049826u. [DOI] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacmias B, Piacentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, St George-Hyslop PH. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42:23–36. doi: 10.1016/S0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidner GA, Ye Y, Faraday MM, Alvord WG, Fortini ME. Modeling clinically heterogeneous presenilin mutations with transgenic Drosophila. Curr Biol. 2006;16:1026–1033. doi: 10.1016/j.cub.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/S0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci U S A. 1999;96:6959–6963. doi: 10.1073/pnas.96.12.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C. A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J Biol Chem. 1999;274:28669–28673. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Chen G, Südhof TC, Shen J. Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J Neurosci. 2009;29:7290–7301. doi: 10.1523/JNEUROSCI.1320-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournoy J, Bossuyt X, Snellinx A, Regent M, Garmyn M, Serneels L, Saftig P, Craessaerts K, De Strooper B, Hartmann D. Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Hum Mol Genet. 2004;13:1321–1331. doi: 10.1093/hmg/ddh151. [DOI] [PubMed] [Google Scholar]
- Wines-Samuelson M, Shen J. Presenilins in the developing, adult, and aging cerebral cortex. Neuroscientist. 2005;11:441–451. doi: 10.1177/1073858405278922. [DOI] [PubMed] [Google Scholar]
- Wines-Samuelson M, Handler M, Shen J. Role of presenilin-1 in cortical lamination and survival of Cajal-Retzius neurons. Dev Biol. 2005;277:332–346. doi: 10.1016/j.ydbio.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Wines-Samuelson M, Schulte EC, Smith MJ, Aoki C, Liu X, Kelleher RJ, 3rd, Shen J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS One. 2010;5:e10195. doi: 10.1371/journal.pone.0010195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Yamaguchi H, Lai FA, Shen J. Presenilins regulate calcium homeostasis and presynaptic function via ryanodine receptors in hippocampal neurons. Proc Natl Acad Sci U S A. 2013;110:15091–15096. doi: 10.1073/pnas.1304171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Shen J. Histological analysis of neurodegeneration in the mouse brain. Methods Mol Biol. 2013;1004:91–113. doi: 10.1007/978-1-62703-383-1_8. [DOI] [PubMed] [Google Scholar]
- Yonemura Y, Futai E, Yagishita S, Suo S, Tomita T, Iwatsubo T, Ishiura S. Comparison of presenilin 1 and presenilin 2 gamma-secretase activities using a yeast reconstitution system. J Biol Chem. 2011;286:44569–44575. doi: 10.1074/jbc.M111.270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/S0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Zhang C, Ho A, Kirkwood A, Südhof TC, Shen J. Inactivation of presenilins causes pre-synaptic impairment prior to post-synaptic dysfunction. J Neurochem. 2010;115:1215–1221. doi: 10.1111/j.1471-4159.2010.07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]