

Abstract
TDP-43 is an RNA-binding protein linked to amyotrophic lateral sclerosis (ALS) that is known to regulate the splicing, transport, and storage of specific mRNAs into stress granules. Although TDP-43 has been shown to interact with translation factors, its role in protein synthesis remains unclear, and no in vivo translation targets have been reported to date. Here we provide evidence that TDP-43 associates with futsch mRNA in a complex and regulates its expression at the neuromuscular junction (NMJ) in Drosophila. In the context of TDP-43-induced proteinopathy, there is a significant reduction of futsch mRNA at the NMJ compared with motor neuron cell bodies where we find higher levels of transcript compared with controls. TDP-43 also leads to a significant reduction in Futsch protein expression at the NMJ. Polysome fractionations coupled with quantitative PCR experiments indicate that TDP-43 leads to a futsch mRNA shift from actively translating polysomes to nontranslating ribonuclear protein particles, suggesting that in addition to its effect on localization, TDP-43 also regulates the translation of futsch mRNA. We also show that futsch overexpression is neuroprotective by extending life span, reducing TDP-43 aggregation, and suppressing ALS-like locomotor dysfunction as well as NMJ abnormalities linked to microtubule and synaptic stabilization. Furthermore, the localization of MAP1B, the mammalian homolog of Futsch, is altered in ALS spinal cords in a manner similar to our observations in Drosophila motor neurons. Together, our results suggest a microtubule-dependent mechanism in motor neuron disease caused by TDP-43-dependent alterations in futsch mRNA localization and translation in vivo.
Keywords: microtubule stability, neuromuscular junction, RNA metabolism
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, fatal neurodegenerative disease linked to defects in multiple cellular processes including RNA metabolism (Lagier-Tourenne and Cleveland, 2009; Ling et al., 2013). TDP-43 is an RNA-binding protein that has been shown to harbor disease causative mutations and to associate with pathological aggregates (Neumann et al., 2006; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Janssens and Van Broeckhoven, 2013; Ling et al., 2013). Studies in several animal models indicate that TDP-43 knockdown or overexpression recapitulates several aspects of ALS pathology, including neural degeneration, locomotor dysfunction, and reduced survival (for review, see Yang et al., 2014). Together, these findings suggest that TDP-43 is a central contributor to ALS pathophysiology, and thus understanding both its normal function and its involvement in disease is likely to provide important insights for developing effective therapeutic strategies.
TDP-43 is a member of the heterogeneous nuclear ribonuclear protein (RNP) A/B family and harbors two RNA Recognition Motif (RRM) domains, a Nuclear Localization Signal (NLS), a Nuclear Export Signal (NES), and a prion-like C-terminus domain (Ou et al., 1995; Cushman et al., 2010; King et al., 2012). RNA sequencing and in vitro binding approaches have revealed that TDP-43 binds Uridine Guanine (UG)-rich sequences with high affinity and regulates the expression and splicing of numerous targets, including its own transcript (Buratti and Baralle, 2001; Ayala et al., 2011; Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). TDP-43's long list of predicted targets include cytoskeleton-associated proteins and pre-mRNAs with long introns encoding synaptic proteins (Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). TDP-43 accompanies its targets in RNA granules during microtubule-dependent transport to distal sites, a process that is more robustly affected by ALS-causing mutations compared with wild-type TDP-43 (Alami et al., 2014). These findings suggest that TDP-43's normal roles include regulation of the neuronal and synaptic architecture and function by controlling the expression of specific mRNAs.
Although TDP-43 interacts with several translational regulators, its role in protein synthesis remains poorly understood (Elden et al., 2010; Freibaum et al., 2010; Sephton et al., 2011). Using a combined molecular and genetic approach in a Drosophila model of ALS based on TDP-43 (Estes et al., 2011, 2013), we show that futsch mRNA and protein levels at the neuromuscular junction (NMJ) are significantly reduced. Together with polysome fractionation experiments indicating a shift in futsch mRNA from actively translating polysomes to untranslated RNPs, our data suggest a model whereby in the context of TDP-43 proteinopathy, futsch localization and translation in motor neurons are impaired. futsch bears homology to mammalian MAP1B and has been shown to regulate axonal and dendritic development as well as the organization of microtubules at the synapse (Hummel et al., 2000; Roos et al., 2000). Consistent with our findings in the fly model, MAP1B distribution is altered in ALS spinal cord motor neurons. Furthermore, restoring futsch expression in Drosophila motor neurons is neuroprotective by restoring microtubule stability at the NMJ and reducing the aggregation of TDP-43 in vivo.
Materials and Methods
Drosophila genetics
All Drosophila stocks and crosses were kept on standard yeast/cornmeal/molasses food at 25°C unless otherwise noted. Transgenics expressing human TDP-43 variants with C-terminal tags YFP were generated as described previously (Estes et al., 2011, 2013). D42 Gal4 (Gustafson and Boulianne, 1996) and GMR Gal4 R13 were used to drive expression in motor neurons and retinal neurons, respectively. For controls, w1118 was crossed with the D42 or GMR Gal4 drivers, as appropriate. To manipulate futsch, we obtained the following stocks from Bloomington: P{EP}futschEP1419w1118 and y1v1;P{TRiP.HMS02000}attp40.
Larval turning assays
Assays were performed as described previously (Estes et al., 2011). Briefly, crosses were kept at 22°C, and wandering third instar larvae were placed on a grape juice plate. After a 30 s acclimation period, crawling larvae were gently turned on their backs (ventral side up). They were observed until they turned over (dorsal side up) and made a forward motion. The time it took to complete this task was recorded for ≥30 larvae per genotype. A Student's t test was performed to determine statistical significance.
Life span analysis
All crosses were performed at 22°C. Newly eclosed males were collected and transferred into a new vial every 7 d. Survival plots were generated using the survival and Hmisc packages in R (R Development Core Team, 2013) and Rstudio (R Studio) software. Statistical analysis was done using the log-rank test in R.
Immunohistochemistry and confocal imaging
Larval NMJs were prepared as described previously (Estes et al., 2011, 2013). Briefly, wandering third instar larvae were filleted in HL-3 saline, pinned out on Sylgard dishes, fixed in 3.5% formaldehyde in PBS, pH 7.2, for 30 min, and permeabilized with 0.1% Triton X-100. The blocking agent consisted of 2% BSA and 5% normal goat serum. The following antibodies were used: anti-Futsch at 1:50 (mAb 22C10; DSHB), anti-mouse Alexa Fluor 568 at 1:500 (Invitrogen), and anti-HRP–Cy5 at 1:200 (Invitrogen). Phalloidin-488 (Invitrogen) was used to visualize muscles. Larval muscles 6 and 7, segment A3, were imaged using an LSM 510 confocal microscope (Zeiss). NIH ImageJ was used to quantify Futsch intensity as well as to count bouton numbers and Futsch loops. Measurements were normalized to HRP and muscle areas to account for size variations. “Fuzzy” loops were ascertained based on the lack of well defined Futsch-positive loops compared with controls.
Larval ventral ganglia.
Third instar ventral ganglia were dissected, treated with 1 mg/ml collagenase diluted 1:100 in HL-3 saline with 1.5 mm Ca2+ for 15 min, rinsed with PBS with 0.3% Triton X-100, fixed in 3.5% formaldehyde in PBS, pH 7.2, for 30 min, and permeabilized with 0.3% Triton X-100. The blocking agent contained 2% BSA and 5% normal goat serum. The following antibodies were used: anti-22C10 at 1:50 (DSHB), anti-mouse Alexa Fluor 568 at 1:500 (Invitrogen), goat anti-GFP-FITC at 1:300 (Rockland), and Hoechst 33342 at 1:100 (Invitrogen). Ventral ganglia were imaged with an LSM 510 confocal microscope (Zeiss).
Human tissues.
ALS and control postmortem tissues were obtained from the University of Pittsburgh ALS Tissue Bank, with clinical diagnosis made by board-certified neuropathologists. All human tissues were collected using Institutional Review Board-approved consent documents. Paraffin-embedded tissue sections of lumbar spinal cord and hippocampus from eight ALS (three male/five female) and six non-neurologic disease control (two male/four female) cases were used for this study (see Table 5). The average age of ALS subjects was 58 years old (range, 52–63 years), and the average age of the controls was 55 years old (range, 51–58 years). The average postmortem interval was 4.5 h for ALS subjects and 5.5 h for control subjects. One ALS subject was a familial ALS patient harboring the C9ORF72 repeat expansion [ALS with frontotemporal lobar degeneration (ALS/FTLD)]. Paraffin was removed from all sections by graded ethanol and rehydrated. A hydrogen peroxide/methanol mixture was used to block endogenous peroxidase activity. Antigen retrieval was performed using Target Antigen Retrieval Solution, pH 9.0 (Dako), for 20 min in a steamer. After cooling to room temperature, nonspecific binding sites were blocked using an avidin (Vector Laboratories) and Super Block (Scytek) solution for 1 h. Mouse monoclonal anti-MAP1B (3G5; Abcam) primary antibody was diluted to 1.25 μg/ml in a biotin (Vector Laboratories) and Super Block solution and incubated overnight. After washing, tissue sections were incubated for 1 h in the appropriate biotinylated IgG secondary antibody (1:200; Vector Laboratories) diluted in Super Block. Slides were washed, and immunostaining was visualized using the Vectastain Elite ABC reagent (Vector Laboratories) and Vector NovaRED peroxidase substrate kit (Vector Laboratories). Slides were counterstained with hematoxylin (Sigma-Aldrich). Sections were visualized using a BX40 light microscope (Olympus), and images were acquired using a DS L2 digital camera (Nikon). To determine the percentage of spinal motor neurons containing cytoplasmic MAP1B, we used between two and four nonconsecutive lumbar spinal cord sections from each case (between 10 and 162 motor neurons per case, with ALS cases typically having fewer surviving motor neurons). Motor neurons were identified by morphologic assessment, and only those containing a definite nucleus were included in the analysis. The percentage of motor neurons containing cytoplasmic MAP1B immunostaining was determined independently, by two separate investigators.
Table 5.
Human tissues and quantification of MAP1B in the cytoplasm of spinal cord motor neurons
| Case | Diagnosis | Age (years)/gender | PMT (hours) | Number of MNs counted | Percentage of MNs with MAP1B in cell body |
|---|---|---|---|---|---|
| C1 | Control brain–no pathology | 54/M | 6 | 148 | 0 |
| C2 | Subacute infarcts | 53/F | 4 | 162 | 1.8 |
| C3 | Metastatic carcinoma | 51/F | 5 | 65 | 0 |
| C4 | Metastatic carcinoma | 57/F | 11 | 28 | 0 |
| C5 | Pulmonary fibrosis | 57/M | 2 | 108 | 0 |
| C6 | Control brain–no pathology | 58/F | 5 | 62 | 0 |
| ALS1 | ALS | 62/F | 4 | 32 | 19 |
| ALS2 | ALS | 50/F | 7 | 10 | 30.0 |
| ALS3 | Familial ALS (C9ORF72) | 63/F | 4 | 35 | 0 |
| ALS4 | ALS | 60/F | 4 | 44 | 66 |
| ALS5 | ALS | 59/M | 4 | 121 | 72 |
| ALS6 | ALS | 63/F | 2 | 51 | 59 |
| ALS7 | ALS | 53/M | 8 | 25 | 52 |
| ALS8 | ALS | 55/M | 3 | 37 | 57 |
PMT, Postmortem interval; MN, motor neuron; M, male; F, female. The percentage of spinal cord motor neurons containing MAP1B in the cell body was determined in motor neurons observed in between two and four tissue sections per case. The average percentage of motor neurons containing MAP1B in the cell body was 0.3 ± 0.3% per control case and 44 ± 8.9% per ALS case (±SD).
Cellular fractionations and Western blotting
Cellular fractionations were adapted from a protocol described previously (Estes et al., 2013). Briefly, 25 wandering third instar larvae were homogenized in 250 μl of low-salt (LS) buffer (10 mm Tris, 5 mm EDTA, 10% sucrose, pH 7.5). Homogenates were centrifuged at 2000 × g for 10 s. Fat and cuticle were discarded, and 50 μl of the homogenate was set aside as input. The remaining 200 μl of homogenate was centrifuged at 25,000 × g for 30 min. The supernatant from this step represents the LS fraction. The pellet was further extracted with ionic detergent-containing buffer (10 mm Tris, 5 mm EDTA, 1% Sarkosyl, 10% sucrose, pH 7.5) and centrifuged at 180,000 × g for 20 min. The supernatant from this step represents the Sark fraction. The remaining detergent-insoluble pellet was solubilized in urea-containing buffer (30 mm Tris, 7 m urea, 2 m thiourea, 4% CHAPS, pH 8.5). All buffers contained 2× Complete Protease Inhibitor Cocktail (Roche) supplemented with 0.5 mm phenylmethylsulfonylfluoride (PMSF) to block proteolysis. An equal volume of 2× Laemmli buffer was added to individual fractions, which were resolved on 4–20% SDS-PAGE gradient precast gels (Bio-Rad) and transferred to a PVDF membrane (Millipore) for Western blotting. Protein concentrations were determined using Qubit (Invitrogen), and 20 μg of protein was loaded. The following primary antibody was used: rabbit anti-GFP at 1:6000 (Invitrogen). The secondary antibody was goat anti-rabbit-conjugated HRP at 1:1000 (Thermo Fisher Scientific). Proteins were detected using SuperSignal West Femto Substrate (Thermo Fisher Scientific) and quantified using NIH ImageJ software. TDP-43 levels in individual fractions were normalized to input. The Fisher's exact test was used to determine statistical significance.
Acetylated tubulin.
Wandering third instar larvae were homogenized in 2× Laemmli buffer, and Western blot procedures were used as described above. The following primary antibodies were used: mouse anti-acetylated α-tubulin (6-11B-1) at 1:1000 (Santa Cruz Biotechnology) and mouse anti-tubulin β clone KMX-1 at 1:1000 (Millipore). Secondary antibodies, detection, and quantification methods were as described above.
Futsch.
Ventral ganglia or body-wall muscles were dissected using standard procedures and homogenized in 2× Laemmli buffer. Samples were resolved on 4–20% SDS-PAGE gradient gels (Bio-Rad) and transferred overnight on nitrocellulose membrane (Bio-Rad). The following primary antibodies were used: 1:1000 mouse anti-22C10 (DSHB) and 1:10,000 rabbit anti-β-Actin (Cell Signaling). Secondary antibodies, detection, and quantification methods were as described above.
RNA immunoprecipitations and RT-PCR experiments
Drosophila immunoprecipitations.
Heads from GMR>TDPWT or GMR>TDPG298S flies were collected, frozen, ground into fine powder, and resuspended in 3 ml of lysis buffer [50 mm HEPES, pH 7.4, 0.5% Triton X-100, 150 mm NaCl, 30 mm EDTA, 7× Protease Inhibitor Cocktail (Roche), 40 U/μl Rnasin Plus (Promega), 0.2 mm PMSF]. After homogenization, the cell lysate was cleared of debris by centrifugation at 7000 rpm for 10 min at 4°C. Ten micrograms of rabbit α-GFP polyclonal antibody (Invitrogen) were added to lysate and incubated for 2 h at 4°C. Next, Dynabeads Protein A was added and incubated for 2 h at 4°C. Ten micrograms of rabbit IgG (Roche) were used as a control. The Dynabeads Protein A immune complexes were washed three times for 5 min each in wash buffer (50 mm HEPES, pH 7.4, 0.5% Triton X-100, 250 mm NaCl, 30 mm EDTA, 40 U/μl Rnasin Plus). The immune complexes were collected, resuspended in 1× Laemmli buffer (0.0625 m Tris HCl, 2% SDS, 10% glycerol, 0.002% bromophenol blue), and analyzed by immunoblotting using standard procedures. Protein concentrations were calculated using Qubit, and equal amounts of protein (20 μg) were loaded. Chicken α-GFP at 1:10,000 (Abcam) was used to probe GFP TDP-43.
RT-PCR.
After RNA immunoprecipitation, RNA was isolated from total lysate, IgG control immunoprecipitation (IP), or immunoprecipitated TDP-43 complex using an RNeasy kit (Qiagen) with on-column DNase treatment. One microgram of RNA was used to perform first-strand cDNA synthesis with a Superscript III cDNA synthesis kit (Invitrogen). RT-PCRs were performed using GoTaq Flexi DNA Polymerase (Promega). Equal amounts of cDNA (100 ng) were used for each RT-PCR. The following primers were used: GAPDH, forward CCGCAGTGCTTGTTTTGCT and reverse TATGGCCGAACCCCAGTTG; Futsch 6/7 exon–exon junction, forward TAACATGCTTGTTGACGGCG and reverse CCCTTGCGGTCAGGTACATT.
Polysome fractionations
One hundred fifty milligrams of adult frozen flies were ground and homogenized in 1 ml of buffer containing 25 mm Tris-HCl, pH 7.5, 250 mm NaCl, 250 mm NH4Cl, 50 mm MgCl2, 250 mm sucrose, 1 mg/ml cycloheximide, 100 U/ml Rnasin, 1% Triton, 0.5% β-mercaptoethanol). Polysomes were separated by centrifugation on 15–50% sucrose gradients at 188,777 rcf and at 4°C for 2 h, 35 min; and fractionated using a flow-through spectrophotometer (Brandel) with a 254 nm detector. RNA was precipitated from fractions using Trizol (Invitrogen). Protein was precipitated using trichloroacetic acid and acetone, after which the pellet was dissolved in 100 μl of 0.5 M Tris, pH.11, and 3% SDS. Ten micrograms of protein were loaded in each lane, subjected to SDS-PAGE and Western blotting as described for cellular fractionation experiments. S6 ribosomal protein was detected using mAb #2317 (Cell Signaling) at 1:500. Secondary antibodies, detection, and quantification methods were as described above.
qRT-PCR
Total RNA was prepared from wandering third instar larvae, body-wall muscles, or ventral ganglia using the RNeasy kit (Qiagen) with on-column DNase treatment. First-strand cDNA synthesis was performed with a Superscript III cDNA synthesis kit (Invitrogen). Quantitative PCRs (qPCRs) were conducted with SYBR Select Master Mix (Applied Biosystems) and an ABI 7300 Real Time PCR System (Applied Biosystems). These experiments were performed with three different primer sets spanning different exon–exon junctions, and similar results were obtained (see Table 3). Primers used are listed above as well as the following additional primers for whole larvae: 5/6 exon–exon junction, forward AGAGCACGACGTACAAAGGG and reverse ATGGGCGCGAGGTAATCAAT; 8/9 exon–exon junction, forward GCACCACTCCCAAGAAGGAA and reverse GGTGACCTCCAACTCCTTGTC. Samples were prepared and run in triplicate. Fold differences were calculated by standard ΔΔCT methods (Pfaffl, 2001).
Table 3.
futsch mRNA levels quantified with different primer sets
| D42>w1118 | D42>TDPWT | D42>TDPG298S | |
|---|---|---|---|
| Futsch 5/6 | 1.0 | 1.02 | 1.12 |
| Futsch 6/7 | 1.0 | 0.97 | 1.02 |
| Futsch 8/9 | 1.0 | 1.03 | 0.92 |
futsch mRNA levels in whole larvae expressing TDP-43 using three different primer sets spanning different exon–exon junctions along the futsch transcript.
qPCR for polysome fractions.
After polysome fractionation and RNA precipitation with Trizol (Invitrogen), first-strand cDNA synthesis was performed with a Superscript III cDNA synthesis kit (Invitrogen), and qPCR was performed as described above with GAPDH and Futsch 6/7 primers. Fractions were normalized to their respective inputs.
qPCR for RNA immunoprecipitations.
After RNA immunoprecipitations (RIPs), RNA isolation and cDNA synthesis were performed as above. qPCR was performed as above with Futsch 6/7 primers and the following TDP-43 primers: forward ACAACCGAACAGGACCTG and reverse GGCTCATCTTGGCTTTGC. TDP-43 was used as the reference mRNA for calculating fold change as GAPDH is not present in the TDP-43 complex and TDP-43 has previously been shown to bind its own mRNA (Ayala et al., 2011).
Statistical analyses
The Student's t test was used for statistical analysis of larval turning experiments, neuromuscular junction analyses including protein levels, bouton and satellite bouton counts, fuzzy Futsch loops quantifications, and acetylated tubulin Western blot analyses. The Fisher's exact test was used for cellular fractionation experiments as we compared cellular fractions across genotypes. The log-rank test was used for survival analyses.
Results
Futsch mRNA associates with TDP-43 in a complex in vivo
Recently, there has been a concerted effort by several laboratories to identify RNA targets of TDP-43 using various molecular and computational approaches (Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). The challenge however, remains to identify which of those potential targets act as effectors of toxicity in vivo. Given that the synaptic morphological defects we have previously reported in a Drosophila model of ALS based on TDP-43 resemble aspects of those caused by loss of function for futsch/MAP1B (Roos et al., 2000; Estes et al., 2013), a putative RNA target that harbors TDP-43-binding sites (Polymenidou et al., 2011), we set out to determine whether futsch mRNA is an in vivo target of TDP-43. We hypothesized that TDP-43 may sequester its mRNA targets into RNA protein complexes (RNPs) and hinder their normal cellular activities, thus leading to observed ALS-like phenotypes. To test this hypothesis, we performed RIPs using antibodies against GFP to pull down TDP-43 YFP variants (wild type and G298S) expressed in Drosophila neurons using the GMR Gal driver. An irrelevant IgG antibody was used as a negative control (Fig. 1A). Next, we performed RT-PCR and found that futsch mRNA was present in the TDP-43 complex compared with the total lysate (Fig. 1B) and was not detected in the IgG control RIP (Fig. 1B). In addition, specificity was confirmed by the presence of GAPDH mRNA in input lysate but not TDP-43 or IgG RIP. Subsequent qRT-PCR experiments confirmed that futsch mRNA was enriched in the TDP-43 complex compared with total lysate (Fig. 1C), which is consistent with previous reports that MAP1B, the mammalian homolog of futsch, also associates with TDP-43 in RNA protein complexes in mammalian cells and brains (Sephton et al., 2011). These results indicate that futsch mRNA associates with both wild-type and disease-variant TDP-43 RNP complexes and suggest that futsch may be an RNA target of TDP-43 in vivo.
Figure 1.

Futsch mRNA associates with TDP-43 in a complex. A, RIP of TDP-43 variants in Drosophila heads. Genotypes and antibodies used for RIP are indicated at the top. The antibody used for Western blot analysis is indicated on the right. B, RT-PCR of immunoprecipitated TDP-43 complexes. Genotypes and antibodies used for IP as indicated in A. mRNA is indicated on the right. C, qPCR of futsch mRNA in immunoprecipitated TDP-43 complexes.
TDP-43 alters futsch mRNA localization and inhibits Futsch expression post-transcriptionally
To determine whether TDP-43 regulates the expression of futsch mRNA in vivo, we first expressed TDP-43 in motor neurons using the motor neuron-specific D42 Gal4 driver (Gustafson and Boulianne, 1996). We then used confocal microscopy to examine Futsch protein levels in the cell bodies of motor neurons in the ventral ganglion and found a marked increase compared with controls (compare Fig. 2, C,D, A,B). In contrast, there was a significant reduction in Futsch protein levels at the NMJ (51% decrease for TDP-43WT, Pvalue = 5.48E-11, Fig. 2G,H,L; 43% decrease for TDP-43G298S, Pvalue = 2.17E-08, data not shown) compared with controls (Fig. 2E,F,L). Consistent with our imaging results, Western blots showed a significant increase in Futsch protein in ventral ganglia (63% for TDP-43WT, Pvalue = 0.01; 61% for TDP-43G298S, Pvalue = 0.01; Fig. 2M,O; Table 1) and a significant reduction at the NMJ (47% decrease for TDP-43WT, Pvalue = 0.001; 44% decrease for TDP-43G298S, Pvalue = 0.001; Fig. 2N,P; Table 1) compared with controls. Notably, we found an increase in Futsch protein at the NMJ compared with ventral ganglia in w1118 controls (Fig. 2M,N; Table 1). These data indicate TDP-43-dependent alterations in the localization of Futsch, with more protein in motor neuron cell bodies and less at the NMJ, suggesting a possible defect in mRNA transport and/or translation. To test this, we quantified futsch mRNA by qPCR and found an increase in futsch transcript levels (3.99-fold for TDP-43WT and 3.15-fold for TDP-43G298S; Fig. 2J; Table 2) in the ventral ganglion (motor neuron cell bodies), whereas at the NMJ there was a significant decrease (40% decrease for TDP-43WT and 34% decrease for TDP-43G298S; Fig. 2K; Table 2) compared with controls. Notably, the reduction in Futsch protein levels quantified by Western blot (or confocal microscopy) is more pronounced than the decrease in futsch mRNA levels as determined by qPCR at the NMJ (compare Fig. 2, J,K, O,P; Tables 1, 2). These data suggest that TDP-43 alters futsch mRNA localization and also inhibits its expression post-transcriptionally. Indeed, futsch mRNA quantification by qPCR from whole larvae showed that neither TDP-43WT nor TDP-43G298S have an effect on steady-state transcript levels compared with controls (Fig. 2I; Table 3). Although at this time we cannot eliminate the possibility that TDP-43 controls the stability of futsch mRNA and/or protein degradation, together our data indicate that TDP-43 alters the localization of futsch mRNA and protein and suggest a role in mRNA transport and translation at the NMJ.
Figure 2.

TDP-43 inhibits Futsch expression post-transcriptionally, and futsch mRNA localization and translation are inhibited in the context of TDP-43. A–D, Larval ventral ganglia of D42>w1118 controls (A, B) and larvae expressing TDP-43WT (C, D) in motor neurons immunostained for Futsch and Hoescht. E–H, Larval neuromuscular junctions of D42>w1118 controls (E, F) and larvae expressing TDP-43WT (G, H) in motor neurons immunostained for Futsch and the neuronal membrane marker HRP. I–K, qPCR of futsch mRNA in the context of TDP-43WT and TDP-43G298S overexpression versus controls in whole larvae (I), ventral ganglia (VG; J), or neuromuscular junctions (K). L, Quantification of Futsch protein intensity at the neuromuscular junction normalized to HRP and muscle areas. All images were acquired using identical parameters. M, N, Western blots for Futsch levels in VGs (M) and NMJs (N) of TDP-43-expressing larvae. Genotypes are indicated at the bottom. Actin was used as a loading control. O, P, Quantification of Futsch protein levels from Western blots represented as a ratio to D42>w1118 controls. Q, TDP-43WT and TDP-43G298S associate with RNPs and actively translating polysomes. R–T, qPCR of futsch mRNA in input (R), RNPs (S), and polysomes (T) in the context of TDP-43WT and TDP-43G298S compared with controls. **p < 0.05; ***p < 0.001, Student's t test. Scale bars: A, 20 μm; E, 55 μm.
Table 1.
TDP-43 expression alters Futsch protein localization
| D42>w1118 (absolute value; ratio to w1118 VG) | D42>TDPWT (absolute value; ratio to w1118) | D42>TDPG298S (absolute value; ratio to w1118) | |
|---|---|---|---|
| VG | 1.20 ± 0.07; 1.0 | 1.96 ± 0.15; 1.63 (p = 0.01) | 1.94 ± 0.16; 1.61 (p = 0.01) |
| NMJ | 1.72 ± 0.09; 1.4 | 0.92 ± 0.04; 0.53 (p = 0.001) | 0.96 ± 0.06; 0.56 (p = 0.001) |
Futsch protein levels normalized to actin at the ventral ganglia (VG) and NMJ. Genotypes are as indicated. Mean ± SEM and p values are as shown. See also Fig. 8. Western blots were performed as described in Materials and Methods.
Table 2.
TDP-43 expression alters futsch mRNA localization
| D42>w1118 | D42>TDPWT (ratio to w1118) | D42>TDPG298S (ratio to w1118) | |
|---|---|---|---|
| VG | 1.0 | 3.99 | 3.15 |
| NMJ | 1.4 | 0.60 | 0.66 |
futsch mRNA levels at the ventral ganglia (VG) and NMJ in TDP-43-expressing larvae are represented as fold change compared with w1118 controls. w1118 futsch mRNA levels at the NMJ are represented as fold change compared with w1118 VG.
Futsch mRNA translation is inhibited in the context of TDP-43 proteinopathy in motor neurons
To further probe TDP-43's role in translation, we performed polysome fractionation experiments from whole adult flies expressing TDP-43 in motor neurons (D42>TDP-43). These experiments show that TDPWT and TDPG298S associate with both untranslated fractions (RNPs and ribosomal subunits) and actively translating polyribosomes (Fig. 2Q; Table 4). Next, we performed qRT-PCR to quantify the levels of futsch transcript in input compared with RNPs and polysome fractions. As previously seen in larvae, we found that TDP-43WT and TDP-43G298S have no effect on the input levels of futsch mRNA in adult flies (D42>TDPWT and D42>TDPG298S) compared with controls (D42>w1118; Fig. 2R). In contrast, we found a 4.7- and 2.9-fold increase in futsch mRNA levels in the RNP fraction of TDP-43WT- and TDP-43G298S-expressing flies, respectively, compared with w1118 controls (Fig. 2S) and a corresponding decrease in futsch mRNA in the polysome fraction of TDP-43-expressing flies compared with controls (2.9-fold/66% decrease for both TDP-43WT and TDP-43G298S compared with w1118 controls; Fig. 2T). These data indicate that futsch mRNA undergoes a shift from the actively translated to the untranslated fractions in a TDP-43-dependent manner and demonstrate that futsch mRNA is translationally repressed by TDP-43 in motor neurons.
Table 4.
Quantification of TDP-43 distribution in polysome fractions
| TDP-43WT (absolute value; ratio to input) | TDP-43G298S (absolute value; ratio to input) | |
|---|---|---|
| Input | 1.04 ± 0.02 | 1.15 ± 0.12 |
| RNP | 1.05 ± 0.02; 1.01 ± 0.001 | 1.14 ± 0.12; 0.99 ± 0.003 |
| 40S/60S | 0.97 ± 0.06; 0.93 ± 0.04 | 1.05 ± 0.05; 0.93 ± 0.07 |
| 80S (monosome) | 0.83 ± 0.05; 0.80 ± 0.03 | 0.88 ± 0.03; 0.78 ± 0.05 |
| 2 ribosomes | 0.90 ± 0.04; 0.87 ± 0.02 | 0.85 ± 0.01; 0.75 ± 0.07 |
| 3 ribosomes | 0.85 ± 0.04; 0.82 ± 0.03 | 0.85 ± 0.03; 0.76 ± 0.08 |
| 4 ribosomes | 0.84 ± 0.05; 0.80 ± 0.03 | 0.84 ± 0.08; 0.74 ± 0.04 |
| 5 ribosomes | 0.85 ± 0.03; 0.82 ± 0.02 | 0.83 ± 0.08; 0.73 ± 0.04 |
| 6 ribosomes | 0.84 ± 0.01; 0.81 ± 0.01 | 0.82 ± 0.08; 0.72 ± 0.07 |
Quantification of TDP-43 protein distribution throughout the polysome gradient relative to S6 ribosomal protein distribution.
Futsch is neuroprotective in the context of TDP-43 overexpression
Given the known role of futsch in the development and maintenance of synaptic contacts at the NMJ, its post-transcriptional regulation by TDP-43, and the anatomical and functional neuromuscular defects caused by TDP-43 in our Drosophila model of ALS (Hummel et al., 2000; Roos et al., 2000; Estes et al., 2013), we hypothesized that restoring futsch expression in the context of TDP-43 may be neuroprotective. To test this possibility, we measured the effects of futsch dosage on survival and larval turning phenotypes caused by TDP-43 in motor neurons (Fig. 3). Although futsch knockdown by RNAi in the context of TDP-43 had no statistically significant effect on larval turning (Fig. 3A), there was 100% pupal lethality, indicating that viability was affected (Figs. 3B,C). A possible explanation for this differential effect on locomotor function versus life span is that even though the efficiency of the RNAi knockdown is high (92% reduction in protein expression locally at the NMJ; data not shown), locomotor defects elicited by TDP-43 overexpression are already so severe that a further reduction in futsch levels has no additional impact. In contrast, overexpression of futsch (futsch OE, 26% over endogenous levels by Western; data not shown) suppressed TDP-43 toxicity, as indicated by a significant decrease in larval turning time (Fig. 3A), and increased life span (25 d for TDPWT futsch OE compared with 22 d for TDPWT, Pvalue = 0.0009, Fig. 3B; 24 d for TDPG298S futsch OE compared with 18 d for TDPG298S, Pvalue = 0.001, Fig. 3C). futsch knockdown by RNAi alone resulted in a small but significant life span reduction (35 d for futschRNAI vs 40 d for w1118 controls; Pvalue = 0.001), whereas futsch OE did not have a significant effect (38 d; Pvalue = 0.35) compared with w1118 controls. These findings demonstrate that futsch is neuroprotective and can mitigate TDP-43-dependent phenotypes including locomotor dysfunction and life span reduction.
Figure 3.

Futsch is neuroprotective in the context of TDP-43 overexpression. A, D42 Gal4 expression of TDP-43 WT or G298S results in impaired larval turning behavior, which is not significantly altered by futsch RNAi but is rescued by futsch OE. B, C, D42 Gal4 expression of TDP-43 variants, WT (B) and G298S (C), results in reduced adult survival time, which is rescued by futsch OE. FutschRNAi shifts the lethality caused by TDP-43 to pupal stage (see Results). The Student's t test was used to calculate larval turning time p values. The log-rank test was used to calculate survival p values. *p < 0.05; **p < 0.01; ***p < 0.001.
Futsch mitigates architectural defects at the neuromuscular junction by increasing microtubule stability
Given its known structural role at the neuromuscular synapse (Hummel et al., 2000; Roos et al., 2000), we hypothesized that futsch may also alleviate morphological abnormalities caused by TDP-43 at the larval NMJ (Estes et al., 2013). As reported previously, we found that overexpression of TDP-43WT or TDP-43G298S in motor neurons results in a significant reduction in synaptic bouton numbers/muscle area compared with w1118 controls (Pvalue = 0.004 for TDP-43WT and Pvalue = 0.003 for TDP-43G298S; Fig. 4G; Estes et al., 2013). After futsch overexpression, we found that bouton numbers were restored to control levels (Pvalue = 8.7E-05 for TDP-43WT and Pvalue = 0.0004 for TDP-43G298S; Fig. 4G). Although futsch overexpression alone did not have a significant effect on the number of synaptic boutons compared with w1118 controls (Fig. 4G), it did result in longer presynaptic terminals, consistent with Futsch's role in microtubule stability.
Figure 4.
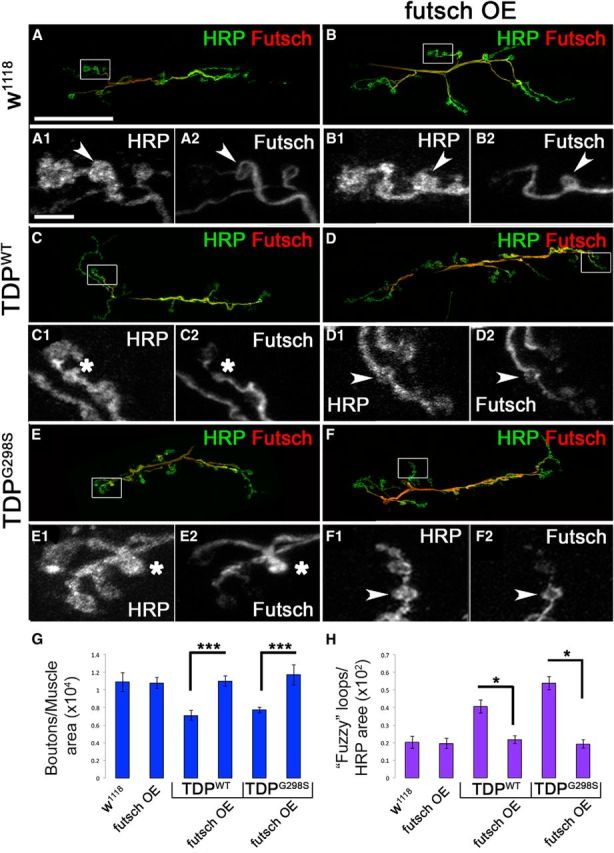
Futsch is neuroprotective at the Drosophila neuromuscular junction. A, C, E, Larval neuromuscular junctions of D42>w1118 controls (A) and larvae expressing TDP-43WT (C) or TDP-43G298S (E) in motor neurons immunostained for Futsch and the neuronal membrane marker HRP. B, D, F, Larval neuromuscular junctions of Futsch OE in a TDP-43WT (D) and TDP-43G298S (F) background compared with Futsch OE controls (B) immunostained for Futsch and the neuronal membrane marker HRP. All images were acquired using identical parameters. A1–F1, High-magnification views of HRP immunostaining shown in A–F. The white arrowhead or asterisk indicates bouton of interest. A2–F2, High-magnification views of Futsch immunostaining shown in A–F. White arrowheads indicate fully formed Futsch loops in the bouton of interest, and white asterisks indicate fuzzy Futsch staining in the bouton of interest. G, Quantification of bouton number normalized to muscle areas. H, Quantification of fuzzy Futsch loops normalized to HRP areas. ***p < 0.001, *p < 0.05, Student's t test. Scale bars: A, 35 μm; A1, 5 μm.
While performing these studies, we noticed marked differences in the distribution of Futsch protein at the NMJ synapse in the context of TDP-43. It has been previously reported that when synaptic boutons stabilize, they harbor stable microtubules shaped as clearly defined, Futsch positive loops (Ruiz-Canada et al., 2004; Fig. 4A,B,D,F, insets A2,B2,D2,F2). In contrast, TDP-43 overexpression in motor neurons led to the appearance of fuzzy Futsch loops that were either misshapen or lacked a clearly defined loop structure (compare Fig. 4, A,B, C,E). Quantification of Futsch-positive loops (Fig. 4C,E, insets C2,E2) showed a significant increase in fuzzy loops in TDP-43WT- and TDP-43G298S-expressing larvae compared with controls (Pvalue = 0.0006 for TDP-43WT and Pvalue = 1.26E-06 for TDP-43G298S; Fig. 4H). After co-overexpression of futsch and TDP-43 (Fig. 4D,F), the number of fuzzy Futsch loops decreased significantly to approximately control numbers (Pvalue = 0.0003 for TDP-43WT and Pvalue = 6.59E-07 for TDPG298S; Fig. 4H). In addition, co-overexpression of futsch and TDP-43 led to a restoration of Futsch protein levels at the NMJ to that of controls (data not shown). These results suggest that in the context of TDP-43 toxicity, microtubule disorganization may occur within synaptic boutons, leading to the observed morphological and functional defects (Estes et al., 2011, 2013). Indeed, we found a significant increase in the number of satellite boutons (which are associated with defects in synaptic growth as a result of microtubule instability at the NMJ) in both TDP-43WT- and TDP-43G298S-expressing larvae compared with controls (Fig. 5A–C,H). After overexpression of futsch in a TDP-43 background, the number of satellite boutons was restored to control levels (Fig. 5D–F,H). To further investigate whether the reduction of satellite boutons after Futsch OE was indeed caused by increased microtubule stability, we quantified the levels of acetylated tubulin, a marker of stable microtubules by Western blot analysis. Stable microtubules contain more acetylated α subunits without a change in total number of α and β subunits. We found that acetylated tubulin levels were reduced in larvae expressing TDP-43WT or TDP-43G298S, when normalized to β-tubulin and compared with controls (Fig. 5G,I), which is consistent with unstable microtubules. Conversely, futsch OE in the context of TDP-43WT and TDP-43G298S restored acetylated tubulin levels back to those of controls (Fig. 5G,I). As expected, Futsch OE alone had increased levels of acetylated tubulin compared with controls (Fig. 5G,I) and is consistent with its known role in promoting microtubule stability. Together, these data indicate that Futsch overexpression in the context of TDP-43 rescues synaptic growth abnormalities by increasing microtubule stability. Given our findings of translational dysregulation (Fig. 2) and the suggested role of TDP-43 in local translation at synapses (Wang et al., 2008; Fallini et al., 2012; Alami et al., 2014), an attractive scenario is that the reduction of well defined Futsch-positive loops and microtubule stability within synaptic boutons of TDP-43-expressing larvae could be the result of defects in localized futsch mRNA protein synthesis.
Figure 5.

Futsch overexpression increases microtubule stability at the NMJ. A–C, Larval neuromuscular junctions of D42>w1118 controls (A) and larvae expressing TDP-43WT (B) or TDP-43G298S (C) in motor neurons immunostained for the neuronal membrane marker HRP. Arrowheads indicate satellite boutons. D–F, Larval neuromuscular junctions of Futsch OE in a TDP-43WT (E) and TDP-43G298S (F) background compared with Futsch OE controls (D) immunostained for the neuronal membrane marker HRP. Arrowheads indicate satellite boutons. G, Western blot analysis showing acetylated tubulin levels. Genotypes are indicated at the top. Antibodies are indicated on the left. Tubulin was used as a loading control. H, Quantification of satellite bouton number normalized to total boutons. I, Quantification of relative protein levels from Western blot analysis. *p < 0.05; **p < 0.01; ***p < 0.001, Student's t test. Scale bar, 5 μm.
TDP-43 aggregates are significantly decreased by futsch overexpression
One of the suggested mechanisms for the pathophysiology of ALS is the formation and persistence of cellular, proteinaceous aggregates. Indeed, we have previously reported that a small fraction of TDP-43 associates with aggregates defined as urea-insoluble complexes (Estes et al., 2013); thus, we hypothesized that at the molecular level, futsch overexpression may be neuroprotective by decreasing the amount of insoluble TDP-43. To test this, we performed cellular fractionations from larvae overexpressing futsch or TDP-43, individually or together in motor neurons, and quantified the amount of TDP-43 present in low-salt-, Sarkosyl-, and urea-insoluble fractions. These experiments showed that overexpression of futsch significantly reduced the amount of TDP-43 (both TDP-43WT and TDP-43G298S) in the urea-insoluble fraction (Fig. 6). Additionally, futsch overexpression significantly increased the amount of TDP-43G298S present in the soluble (LS) fraction (Fig. 6B,D). These findings are consistent with the notion that futsch overexpression is neuroprotective by reducing the ability of TDP-43 to form insoluble aggregates (see Fig. 8 for model).
Figure 6.

TDP-43 aggregates are significantly decreased by Futsch OE. A, B, Solubility studies using third instar larvae show the distribution of TDP-43 variants WT (A) and G298S (B) in LS, Sarkosyl (Sark), and urea fractions alone in the context of Futsch overexpression (Futsch OE). C, D, Quantification of WT (C) or G298S (D) TDP-43 levels in LS, Sark, and urea fractions normalized to input. *p < 0.05; ***p < 0.001, Fisher's exact test.
Figure 8.

Model of futsch mRNA regulation by TDP-43. A, In controls, TDP-43 transports futsch mRNA to synapses where translation occurs, resulting in stabilized synaptic connections. B, In the ALS model, TDP-43 alters the localization of futsch mRNA, increasing its concentration in the cell body and reducing its concentration at the NMJ. Additionally, TDP-43 negatively regulates Futsch expression post-transcriptionally at the level of translation, resulting in decreased Futsch protein levels in both the cell body and at the NMJ relative to mRNA concentrations. Subsequently, this leads to decreased microtubule stability, increased aggregation of TDP-43, and reduced NMJ size, locomotor function, and life span. C, After Futsch overexpression in the context of TDP-43, futsch mRNA localization and Futsch protein levels are restored subsequently, resulting in a rescue of microtubule stability, TDP-43 solubility, as well as NMJ size, locomotor function, and life span.
Futsch/MAP1B localization is altered in ALS spinal cords
Our findings in the fly predict that the expression or subcellular distribution of MAP1B is altered in motor neurons of ALS patients. To test this, we used immunohistochemistry to examine the subcellular localization of MAP1B in the lumbar spinal cord and hippocampus from six non-neurologic disease controls and eight ALS subjects with TDP-43 pathology. Consistent with our findings in the fly, these data indicate that MAP1B accumulates in spinal cord motor neuron cell bodies in seven of eight ALS subjects (compare Fig. 7, B, A) compared with controls. We determined the percentage of spinal cord motor neurons containing MAP1B in the cell body for each subject, with MAP1B in the cell body of 0.3 ± 0.3% motor neurons in non-neurologic controls and 44.3 ± 8.9% in ALS cases (Table 5). MAP1B was not observed in the cell body of spinal motor neurons in the familial ALS patient that harbored the C9ORF72 repeat expansion. We did not detect an obvious alteration in MAP1B immunoreactivity in the spinal cord white matter (data not shown). We also examined the subcellular distribution of MAP1B in the hippocampus. MAP1B was detected in the cell body and neurites of neurons within the hippocampus of all control subjects (Fig. 7C,E,G,I). A similar pattern of MAP1B immunostaining was observed in ALS hippocampal tissues (Fig. 7D,F,H,J), albeit increased staining in some dystrophic neurites throughout the hippocampus in ALS patients and more intense cell body and neurite immunoreactivity within dentate granule cells of the ALS/FTLD patient with the C9ORF72 repeat expansion (Fig. 7F,J).
Figure 7.
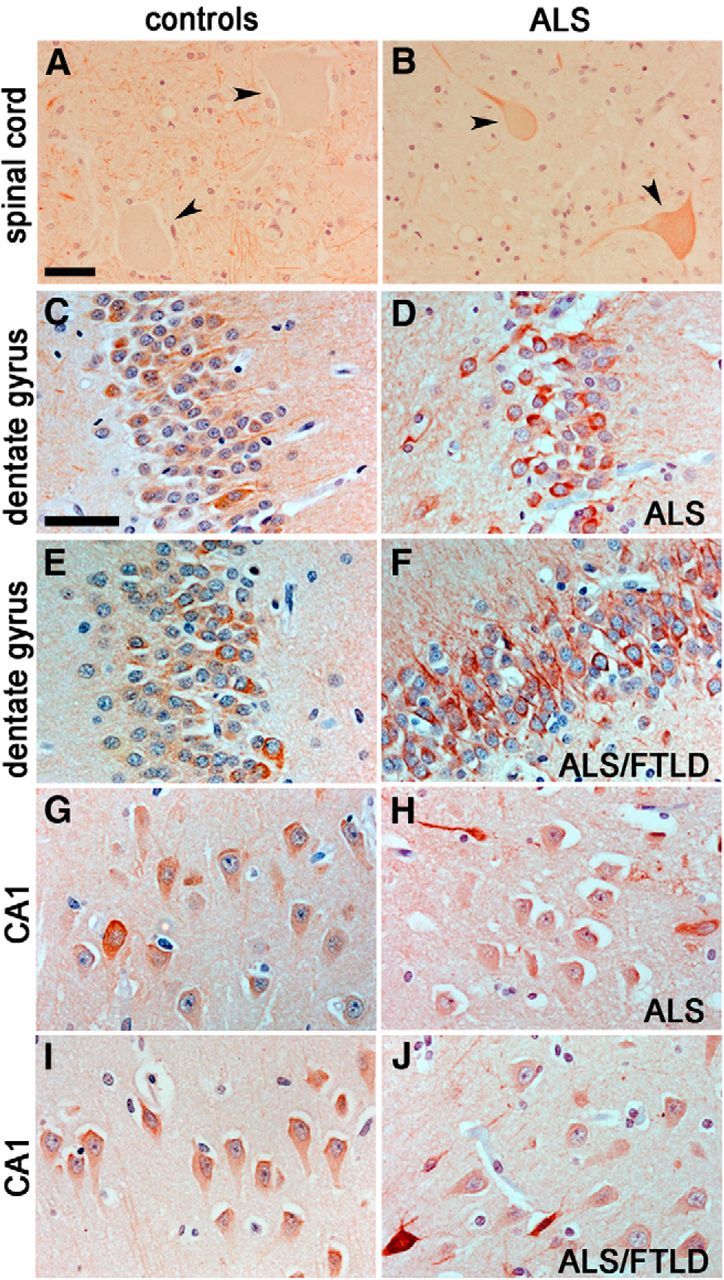
MAP1B localization in ALS patient samples. A, B, MAP1B immunostaining in control (A) and ALS (B) lumbar spinal cords. Note the cell body accumulation of MAP1B in ALS tissues (compare arrowheads). C–J, MAP1B immunostainings in control (C, E, G, I) and ALS (D, F, H, J) dentate gyrus (DG) and CA1 regions of the hippocampus. Note the additional cell body and proximal neurite accumulation of MAP1B in the DG of the ALS/FTLD subject (F). Scale bars: A, 40 μm; C, 30 μm.
Discussion
Here we show that TDP-43, an RNA-binding protein linked to a significant fraction of ALS cases, associates with futsch mRNA in a complex in vivo and regulates its localization and translation in Drosophila motor neurons. Using polysome fractionations, we show that wild-type and disease-associated mutant TDP-43 cofractionate with both the untranslated fractions, namely RNPs and ribosomal subunits, and actively translating polyribosomes. Our results add translation regulation to TDP-43's plethora of known roles in RNA processing, such as transcription, splicing, and mRNA transport (Polymenidou et al., 2011; Alami et al., 2014), and suggest that TDP-43 contributes to the pathophysiology of ALS via multiple RNA-based mechanisms.
To our knowledge, these data provide the first in vivo demonstration that TDP-43 associates with polysomes and regulates the translation of futsch mRNA. Our findings are consistent with previous reports that futsch mRNA coimmunoprecipitates with Drosophila TBPH, and the mammalian homolog, MAP1B, is a candidate target of TDP-43 in mouse brains (Godena et al., 2011; Sephton et al., 2011). Our results showing a decrease in Futsch levels at the NMJ and an increase in Futsch levels in motor neuron cell bodies suggest a model whereby futsch/MAP1B mRNA may not be properly transported into axons (see Fig. 8 for model). This is substantiated by qPCR from ventral ganglia where futsch mRNA is found at higher levels than at the NMJ compared with controls. Although we cannot exclude the possibility that TDP-43 regulates futsch mRNA stability, given the more pronounced reduction in protein versus transcript levels at the NMJ compared with cell bodies and the shift to untranslated fractions in polysomes, these data suggest TDP-43-dependent defects in futsch/MAP1B mRNA transport and protein expression at the NMJ. Although the underlying cause remains unknown, Futsch protein levels are also reduced at the NMJ in the absence of TBPH (the Drosophila homolog of TDP-43; Godena et al., 2011), suggesting a loss-of-function mechanism for the disease. Interestingly, there is a marked increased in futsch mRNA in motor neuron cell bodies in ventral ganglia relative to Futsch protein levels. This suggests that TDP-43 also regulates the expression of Futsch in motor neuron cell bodies possibly through sequestration into RNP complexes. Although we could not detect cytoplasmic TDP-43 puncta in vivo, a plausible scenario is that TDP-43-containing complexes are below the limit of resolution of the confocal microscope, as suggested by cellular fractionation experiments indicating the presence of cytoplasmic TDP-43 (data not shown and Estes et al., 2013). Since futsch is the Drosophila homolog of MAP1B and MAP1B mRNA has been identified in TDP-43-containing RNP complexes in mouse models (Sephton et al., 2011), these results predict that MAP1B and microtubule-based processes may also be affected in ALS patient tissues. Indeed, similar to our results in the fly, immunohistochemistry experiments reveal a significant accumulation of MAP1B in motor neuron cell bodies in ALS spinal cords compared with controls but not in the hippocampus. Although these alterations may be the result of ongoing neurodegeneration, the remarkable similarities with the fly model suggest that comparable defects in transport and translation processes may occur in the human disease. It is interesting to note that spinal cord motor neurons in the patient with the C9ORF72 repeat expansion failed to contain MAP1B immunostaining in the cell body, suggesting altered pathogenic mechanisms from sporadic ALS patients, though further studies with an increased number of patients within this repeat expansion are required to confirm this finding.
Interestingly, Futsch protein expression is similarly inhibited by wild-type or mutant TDP-43, supporting a scenario in which MAP1B dysregulation may be a shared feature of ALS cases with TDP-43-positive pathology, regardless of etiology. It is possible that other targets, which remain to be identified, are regulated in a variant-dependent manner. Indeed, RNA sequencing experiments from wild-type and mutant polysome fractions indicate several distinct, up- and down-translated mRNAs (S. G. Daniel and D. C. Zarnescu, unpublished observations). Perhaps TDP-43's role in translation is not surprising as it has previously been shown to associate with stress granules (SGs), which are known to sequester mRNAs and inhibit their translation during environmental stress (Colombrita et al., 2009; Liu-Yesucevitz et al., 2010; Dewey et al., 2011; McDonald et al., 2011). Although TDP-43 does not seem to be required for SG formation, ALS-linked mutations in TDP-43 were shown to alter the dynamics of SG assembly and disassembly (Dewey et al., 2011; McDonald et al., 2011). This is consistent with our previous findings that the molecular mobility of wild-type TDP-43 differs from that of the mutant variants in primary motor neurons (Estes et al., 2013). Furthermore, TDP-43-containing cytoplasmic aggregates can “evolve” from paraquat-induced SG (Parker et al., 2012). Together, these findings suggest a scenario whereby, in response to stress, possibly caused by aging or environmental factors, TDP-43 localizes to cytoplasmic SGs that in turn lead to altered ribostasis (Ramaswami et al., 2013), including abnormal futsch mRNA expression as demonstrated by our experiments.
Using genetic interaction approaches, we show that futsch is a physiologically significant RNA target of TDP-43 and can alleviate locomotor dysfunction and increase life span. At the structural level, Futsch protein shares homology with mammalian MAP1B as well as neurofilaments, which do not exist per se in Drosophila (Hummel et al., 2000). Interestingly, TDP-43 was shown to bind, transport, and sequester neurofilament light (NEFL) mRNA into SG, which contributes to ALS-like phenotypes in motor neurons (Strong et al., 2007; Volkening et al., 2009; Alami et al., 2014). Given Futsch's known requirement in axonal and dendritic development and the organization of microtubules at the synapse (Hummel et al., 2000; Roos et al., 2000), our findings suggest that these processes may be involved in the pathophysiology of ALS. Indeed, recent studies in SOD1- and TDP-43-based models of ALS demonstrate an impairment in microtubule-based axonal transport (Magrane et al., 2013; Robberecht and Philips, 2013).
Consistent with previous studies in which tubulin acetylation has been shown to rescue transport defects in neurodegeneration (Dompierre et al., 2007), our data show that TDP-43 leads to reduced levels of acetylated tubulin, and this is rescued by futsch overexpression. Other TDP-43 targets such as HDAC6, which is regulated by TDP-43 at the level of transcription, are also linked to microtubule stability (Fiesel et al., 2011), providing additional support to the notion that microtubule stability is an important factor mediating TDP-43 toxicity. It is possible that microtubule stability is regulated locally by an interplay between Futsch and HDAC6 at the NMJ. Additionally, microtubule integrity and stress granules have been intricately linked. It has been demonstrated that microtubule integrity is important for the transportation and disassembly of stress granules (Nadezhdina et al., 2010). Also, SGs can be cleared by autophagy (Buchan et al., 2013), and microtubules are used to transport autophagosomes to the microtubule organizing center where they fuse with lysosomes (Mackeh et al., 2013; Nixon, 2013). Notably, microtubule stability can promote transport by association of motors that preferentially bind acetylated microtubules (Conde and Caceres, 2009). Futsch is often associated with the stabilization of microtubules at the synapse through the formation of loop structures (Roos et al., 2000) as we have also seen in the context of TDP-43. Therefore, restoration of Futsch levels and microtubule stability may mitigate TDP-43 toxicity through multiple mechanisms, including restoring transport of target mRNAs essential for synaptic stability and transport, disassembly, and/or clearance of stress granules, which in turn may interfere with aggregate formation as suggested by the observed decrease in insoluble TDP-43. In conclusion, we identify futsch as a disease-relevant and functionally significant post-transcriptional target of TDP-43. Given the role of futsch/MAP1B in microtubule and synaptic stabilization, our findings point to microtubule-based processes as targets for the development of therapeutic strategies for TDP-43 proteinopathies.
Footnotes
This work was supported by NIH Grants NS078429 (D.C.Z.) and NS061867 (R.B.) and MDA Grant 255293 (D.C.Z.). Additional funds were provided by the Himelic Family Foundation, the Neuroscience Graduate Interdisciplinary Program at the University of Arizona (A.N.C.), and the Beckman Foundation (A.P.). The 22C10 monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by the Department of Biology, The University of Iowa (Iowa City, IA). We thank members of the Zarnescu laboratory and Ross Buchan for helpful suggestions on this manuscript.
The authors declare no competing financial interests.
References
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendaño-Vázquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P, 3rd, Good SK, Johnson BA, Herz J, Yu G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 2011;31:1098–1108. doi: 10.1128/MCB.01279-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes PS, Boehringer A, Zwick R, Tang JE, Grigsby B, Zarnescu DC. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum Mol Genet. 2011;20:2308–2321. doi: 10.1093/hmg/ddr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes PS, Daniel SG, McCallum AP, Boehringer AV, Sukhina AS, Zwick RA, Zarnescu DC. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis Model Mech. 2013;6:721–733. doi: 10.1242/dmm.010710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet. 2012;21:3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiesel FC, Schurr C, Weber SS, Kahle PJ. TDP-43 knockdown impairs neurite outgrowth dependent on its target histone deacetylase 6. Mol Neurodegener. 2011;6:64. doi: 10.1186/1750-1326-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9:1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godena VK, Romano G, Romano M, Appocher C, Klima R, Buratti E, Baralle FE, Feiguin F. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS One. 2011;6:e17808. doi: 10.1371/journal.pone.0017808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson K, Boulianne GL. Distinct expression patterns detected within individual tissues by the GAL4 enhancer trap technique. Genome. 1996;39:174–182. doi: 10.1139/g96-023. [DOI] [PubMed] [Google Scholar]
- Hummel T, Krukkert K, Roos J, Davis G, Klämbt C. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 2000;26:357–370. doi: 10.1016/S0896-6273(00)81169-1. [DOI] [PubMed] [Google Scholar]
- Janssens J, Van Broeckhoven C. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum Mol Genet. 2013;22:R77–R87. doi: 10.1093/hmg/ddt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5:e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackeh R, Perdiz D, Lorin S, Codogno P, Poüs C. Autophagy and microtubules–new story, old players. J Cell Sci. 2013;126:1071–1080. doi: 10.1242/jcs.115626. [DOI] [PubMed] [Google Scholar]
- Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2013;23:1413–1424. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. 2011;20:1400–1410. doi: 10.1093/hmg/ddr021. [DOI] [PubMed] [Google Scholar]
- Nadezhdina ES, Lomakin AJ, Shpilman AA, Chudinova EM, Ivanov PA. Microtubules govern stress granule mobility and dynamics. Biochim Biophys Acta. 2010;1803:361–371. doi: 10.1016/j.bbamcr.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, Kanninen KM, White AR. Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem Int. 2012;60:415–424. doi: 10.1016/j.neuint.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klämbt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/S0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Ruiz-Canada C, Ashley J, Moeckel-Cole S, Drier E, Yin J, Budnik V. New synaptic bouton formation is disrupted by misregulation of microtubule stability in aPKC mutants. Neuron. 2004;42:567–580. doi: 10.1016/S0896-6273(04)00255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, Moore MJ, Yu G. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286:1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–327. doi: 10.1016/j.mcn.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Zupunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS) Brain Res. 2009;1305:168–182. doi: 10.1016/j.brainres.2009.09.105. [DOI] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, Tan W, Salameh J, McKenna-Yasek DM, Smith T, Peng L, Moore MJ, Brown RH, Jr, Cai H, Xu Z. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2014;111:E1121–E1129. doi: 10.1073/pnas.1322641111. [DOI] [PMC free article] [PubMed] [Google Scholar]