

Abstract
Parkinson's disease (PD), the second most prevalent neurodegenerative disorder, is characterized by the degeneration of dopamine (DA) neurons and age-dependent formation of protein inclusions that contain the α-synuclein (α-syn) protein. RNA interference (RNAi) screening using Caenorhabditis elegans identified RTCB-1, an uncharacterized gene product, as one of several significant modifiers of α-syn protein misfolding. RTCB-1 is the worm ortholog of the human HSPC117 protein, a component of RNA trafficking granules in mammalian neurons. Here we show that RTCB-1 protects C. elegans DA neurons from age-dependent degeneration induced by human α-syn. Moreover, neuronal-specific RNAi depletion of rtcb-1 enhanced α-syn-induced degeneration. Similar results were obtained when worms were exposed to the DA neurotoxin 6-hydroxydopamine. HSPC117 has been characterized recently as an essential subunit of the human tRNA splicing ligase complex. tRNA ligases have alternative functions in RNA repair and nonconventional mRNA splicing events. For example, in yeast, unconventional splicing of HAC1, a transcription factor that controls the unfolded protein response (UPR), is mediated by a tRNA ligase. In C. elegans, we demonstrate that RTCB-1 is necessary for xbp-1 (worm homolog of HAC1) mRNA splicing. Moreover, using a RNA ligase-dead mutant, we determine that the ligase activity of worm RTCB-1 is required for its neuroprotective role, which, in turn, is mediated through XBP-1 in the UPR pathway. Collectively, these studies highlight the mechanistic intersection of RNA processing and proteostasis in mediating neuroprotection.
Keywords: alpha-synuclein, C. elegans, dopamine, neuroprotection, Parkinson's, RNA
Introduction
The pathogenesis of Parkinson's disease (PD) involves the loss of dopamine (DA) neurons in the midbrain and manifests through mechanisms such as mitochondrial defects and protein misfolding, leading to endoplasmic reticulum (ER) stress and proteasome dysfunction (Yacoubian and Standaert, 2009). Another characteristic of PD is the accumulation of α-synuclein (α-syn) into inclusions termed Lewy bodies (Dickson et al., 2009). Mutation or multiplication of the wild-type (WT) α-syn locus causes misfolding of α-syn protein, leading to aggregation (Uversky, 2007). Discerning molecular mechanisms underlying neurodegeneration is a prerequisite for identifying functional modifiers with translational potential for PD.
We have established previously Caenorhabditis elegans as an animal model to investigate PD, wherein overexpression of WT human α-syn driven under a DA neuron-specific promoter causes age- and dose-dependent neurodegeneration (Cao et al., 2005; Hamamichi et al., 2008). This correlates with human genetics, in which multiplication of the WT α-syn locus leads to familial PD (Singleton et al., 2003). Hamamichi et al. (2008) conducted a large-scale RNA interference (RNAi) screen in C. elegans that identified several genetic modifiers of α-syn toxicity, including one uncharacterized open-reading frame, F16A11.2, now annotated as rtcb-1.
RTCB-1, encoding a 505 aa protein, contains a highly conserved RtcB domain, common to the RtcB family of proteins present in eukaryotes, bacteria, and archaebacteria. RtcB proteins are atypical RNA ligases with broad substrate specificity implicated in tRNA splicing and repair (Englert et al., 2011; Tanaka et al., 2011; Chakravarty et al., 2012). Human HSPC117 can bind to AU-rich elements within mRNA (Rousseau et al., 2002) and was characterized recently as an essential subunit of the tRNA splicing ligase complex (Popow et al., 2011). Although HSPC117 is ubiquitously expressed in human tissues, a functional role in neurons was suggested by its presence in RNA transport granules in mouse brain extracts (Kanai et al., 2004).
Besides tRNA splicing, RNA ligases have been demonstrated to function in RNA repair and nonconventional mRNA splicing events. One such event is the unconventional splicing of HAC1 mRNA, encoding a yeast transcription factor that controls the unfolded protein response (UPR). Interestingly, the bacterial homolog of RTCB-1, RtcB, can functionally complement yeast tRNA ligase splicing activity (Sidrauski et al., 1996; Tanaka et al., 2011). The UPR pathway is conserved from yeast to mammals (Iwawaki et al., 2001), in which XBP1, a homolog of HAC1, is cleaved by IRE1 endonuclease (Shen et al., 2001). During ER stress, the unspliced XBP1 transcript is alternatively spliced to produce an active version of XBP1 mRNA (Calfon et al., 2002). XBP1 mediates transcription of chaperone proteins such as BiP/Grp78 and facilitates the exit and degradation of misfolded proteins via ER-associated protein degradation (ERAD). In C. elegans, XBP-1 activates UPR genes involved in folding, secretion, ER biogenesis, and autophagy. There are no homologs of yeast tRNA ligase in either mammals or C. elegans, thus the factor responsible for splicing of XBP1 in metazoan systems has remained unknown (Wang and Shuman, 2005). Here we report RTCB-1 as a functional mediator of xbp-1 splicing and describe its role in neuroprotection.
Materials and Methods
Plasmid construction.
rtcb-1 and xbp-1 were amplified from genomic DNA isolated from N2 Bristol nematodes using Phusion high-fidelity polymerase. An N-terminal FLAG tag sequence was added to rtcb-1/xbp-1 cDNA during the PCR amplification process. Using Gateway Technology (Life Technologies), PCR-amplified constructs were cloned into plasmid entry vector pDONR221 by BP reaction, and the constructs were further cloned into Gateway expression vector pDEST–DAT-1 (Cao et al., 2005). To generate the point mutant RTCB-1 C122A, TagMaster site-directed mutagenesis kit (GM Biosciences) was used. The constructs were verified by DNA sequencing.
C. elegans strains.
Nematodes were maintained using standard procedures (Brenner, 1974). We obtained the following strains from the Caenorhabditis Genetics Center: rtcb-1(gk451) and xbp-1(zc12). Strain BY250 [vtIs7 (Pdat-1::GFP)] was a generous gift from Randy Blakely (Vanderbilt University, Nashville, TN). The isogenic strain UA44 [baIn11 (Pdat-1::α-syn, Pdat-1::GFP)] expresses α-syn and GFP in the DA neurons. UA196 [sid-1(pk3321); baIn33 (Pdat-1::sid-1, Pmyo-2::mCherry); baIn11], UA197 [sid-1(pk3321); uIS69 [Punc-119::sid-1, Pmyo-2::mCherry); baIn11], and UA202 [sid-1(pk3321); baIn36 (Pdat-1::sid-1, Pmyo-2::mCherry); vtIs7(Pdat-1::GFP)] were generated as described previously (Harrington et al., 2012). UA196 expresses α-syn, GFP, and SID-1 in the DA neurons. UA202 expresses only GFP and SID-1 in the DA neurons. Both are sensitive to RNAi specifically in the DA neurons. UA197 is a pan-neuronal specific RNAi strain expressing α-syn and GFP in DA neurons and SID-1 in all neurons. Three independent stable transgenic lines were generated by injecting Pdat-1:: rtcb-1, along with a phenotypic marker (Punc-54::mCherry), into UA44 hermaphrodites. These lines express RTCB-1 in the DA neurons and mCherry in the body wall muscle cells, as a phenotypic marker. One of the stable lines, with the most significant level of neuroprotection against α-syn-induced degeneration (data not shown), was further integrated into the C. elegans genome by UV irradiation (Inoue and Thomas, 2000) to create strain UA266 [baIn48 (Pdat-1:: rtcb-1; Punc-54::mCherry); baIn11]. The isolated homozygous integrated line was used for neuroprotection analysis in Figure 1A. UA266 was further crossed into strain UA197 to generate UA267 [baIn48; baIn11; sid-1(pk3321); uIS69], which was used for neuroprotection analysis in Figure 4A. sid-1(pk3321) homozygosity was maintained as described previously (Harrington et al., 2012). This strain expresses α-syn, GFP, and RTCB-1 in the dopaminergic neurons and is highly selective for RNAi in the neurons. To create UA270 {baEx159 [Pdat-1::xbp-1u; rol-6(su1006)]; sid-1(pk3321); uIS69; baIn11}, analyzed in Figure 4B, Pdat-1::xbp-1u and pRF4 rol-6(su1006) were injected into UA197. Strain UA269, analyzed in Figure 5, consists of {baEx158[Pdat-1::rtcb-1(C122A), rol-6(su1006)]; baIn11}.
Figure 1.

RTCB-1 is neuroprotective against α-syn or 6-OHDA-induced toxicity. A, RTCB-1 overexpression protects C. elegans DA neurons from α-syn-induced neurodegeneration at 7- and 10-d worm stages compared with the α-syn control. B, C, Representative images of the neuroprotection assay described in A. B, Worm expressing GFP and α-syn specifically in the six anterior DA neurons with four neurons intact (arrowheads) and two neurons degenerated (arrows). C, Worm coexpressing RTCB-1 shows neuroprotection with all six neurons protected (arrowheads). D, RNAi depletion of rtcb-1 enhances DA neurodegeneration caused by α-syn at 4 and 6 d worm stages when compared with EV α-syn control. E, F, Representative worm images of the RNAi experiment described in D. E, Representative RNAi-treated (EV) worm expressing α-syn missing two neurons (arrows). F, RNAi knockdown of rtcb-1 shows increased degeneration with loss of all five of six anterior DA neurons (arrows). G, RNAi depletion of rtcb-1 accelerates DA neurodegeneration caused by 6-OHDA at 24, 48, and 72 h after 1 h 6-OHDA treatment when compared with 6-OHDA-treated EV control. H, Exemplar worm exposed to 6-OHDA, in this case missing three DA neurons (arrows). I, A worm treated with rtcb-1 RNAi and 6-OHDA is missing all six neurons, indicative of enhanced degeneration (arrows). Data are reported as the mean ± SD, n = 30 worms per trial for a total of three replicates. *p < 0.05, Student's t test. Scale bar, 20 μm.
Figure 4.
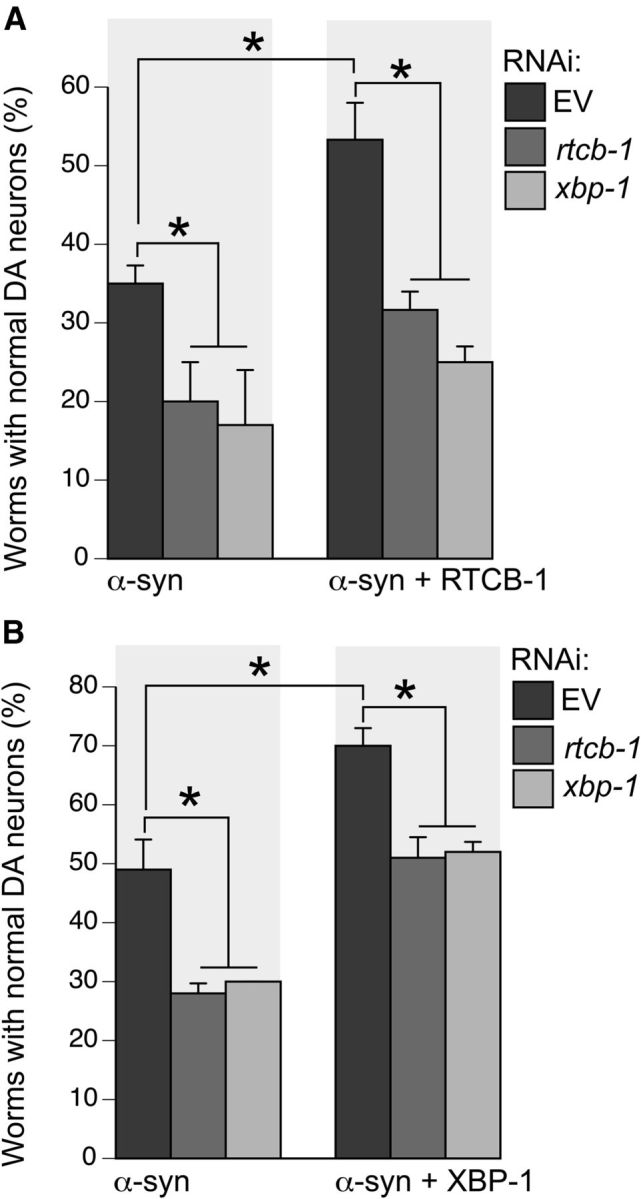
RTCB-1-induced neuroprotection against α-syn is mediated by XBP-1 signaling. A, Pan-neuronal-specific knockdown of rtcb-1 or xbp-1 enhances α-syn-induced neurodegeneration when compared with the EV control. These worms expressing α-syn and GFP were then crossed with worms expressing RTCB-1. Overexpression of RTCB-1 protects DA neurons from α-syn-induced toxicity in worms treated with RNAi EV. RNAi knockdown of rtcb-1 or xbp-1 failed to protect neurons from α-syn-induced degeneration in an RTCB-1 overexpression background. B, Overexpression of XBP-1 protects DA neurons from α-syn-induced toxicity in worms treated with RNAi EV when compared with α-syn control. Pan-neuronal-specific knockdown of rtcb-1 blocks the neuroprotective phenotype of XBP-1. DA neuron analyses were performed at day 6 after hatching. Data are reported as the mean ± SD, n = 30 worms per trial for a total of three replicates. *p < 0.05, one-way ANOVA.
Figure 5.

Overexpression of rtcb-1 (C122A) in DA neurons is not protective from α-syn-induced neurodegeneration. Analysis of worm RTCB-1 mutant at days 7 and 10 failed to show any significant protection of DA neurons when compared with α-syn control, yet overexpression of WT RTCB-1 was protective.
RNAi treatments.
Bacterial RNAi feeding constructs of rtcb-1 and xbp-1 were obtained from the Ahringer C. elegans library (Kamath et al., 2003); they were isolated and grown overnight in Luria broth media containing 100 μg/ml ampicillin. Nematode growth medium (NGM) plates containing 0.25% β-d-lactose were seeded with 250 μl of RNAi culture and allowed to dry overnight. Ten dauer or L1-stage worms [N2, or neuron-specific RNAi worm strains (UA196, UA197, UA266 or UA267, UA270] were transferred to the plates and grown at 20°C until adulthood. Adult worms were then transferred to corresponding freshly made RNAi plates and allowed to lay eggs for 6 h to synchronize. For neurodegeneration assays, the DA neurons in the F1 progeny of the RNAi-treated worms were scored at days 4, 6, 7 or day 10 after hatching, as described below. For quantitative PCR (qPCR) studies, RNAi-treated late L4 (larval stage) N2 and/or UA202 worms were treated with 10 μg/ml tunicamycin for 6 h and then collected for RNA isolation, as described below.
C. elegans neurodegeneration assay.
Worms were analyzed for DA neurodegeneration as described previously (Cao et al., 2005). Briefly, worms (strains UA196, UA197, UA266, or UA267) were synchronized, grown at 20°C, and scored for α-syn-induced DA neurodegeneration at days 4, 6, 7 or day 10 after hatching. Worms were considered normal when all six anterior DA neurons [four CEP (cephalic) and two ADE (anterior deirid)] were present without any visible signs of degeneration. If a worm displayed degeneration in at least one of the six neurons, it was scored as exhibiting degeneration. In total, at least 90 adult worms were analyzed for each independent transgenic line or RNAi treatment. 6-Hydroxydopamine (6-OHDA) assay was performed as described previously (Nass et al., 2002). Briefly, L4 worms (UA202) were washed with ddH2O three times and treated with 30 mm 6-OHDA (Tocris Bioscience) containing 1 mm ascorbic acid, followed by gentle agitation for 1 h. Subsequently, the worms were again washed and put onto freshly made RNAi plates until analysis.
RNA isolation from C. elegans.
For each independent sample, total RNA was isolated from 50 young adult hermaphrodite worms as described previously (Hamamichi et al., 2008). Briefly, the worms were washed three times with M9 buffer (3 g of KH2PO4, 12.8 g of Na2HPO4, 5 g of NaCl, 1 ml of 1 m MgSO4, and H2O to 1 L) and then frozen at −80°C until use. After thawing, 500 μl of TRI Reagent (Molecular Research Center) was added to the samples, followed by brief vortex and incubated at room temperature (RT) for 10 min. The samples were then subjected to repeated freeze–thaw cycling for four times in liquid N2. The phases were separated by adding 50 μl of 1-bromo-3-chloropropane (Acros Organics) with 15 s of vortexing. The samples were then incubated at RT for 15 min and centrifuged at 13,000 × g at 4°C for 15 min. The supernatant was then transferred to an RNase-free microcentrifuge tube, mixed with 1.5 μl of glycoblue (Ambion) and 250 μl of −20°C chilled isopropanol, and then stored overnight at −20°C. After incubation, the sample was centrifuged at 13,000 × g at 4°C for 15 min, and the supernatant was discarded. The pellet was washed with 500 μl of RNase-free ethanol (75%), air dried for 5 min, and resuspended in 10 μl of nuclease-free water. Samples were then treated with 1 μl of DNase I and 1 μl of DNase I buffer (Promega) at 37°C for 15 min and then for 10 min with 1 μl of DNase stop at 65°C. Total RNA was quantitated using a Nanodrop and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad) following the protocols of the manufacturer.
Real-time qPCR.
qPCR reactions were performed using IQ SYBR Green Supermix (Bio-Rad) with the CFX96 Real-Time System (Bio-Rad) as described previously (Thompson et al., 2014). The following previously described primer sequences were used: spliced xbp-1, total xbp-1, and hsp-4 (Richardson et al., 2010); and tba-1, ama-1, and gpd-2 (Hoogewijs et al., 2008). For the remaining primers, full-length gene sequences were obtained from WormBase, and primers were designed by the Primer3 software and evaluated for potential secondary structures of the amplicon by MFOLD software. MFOLD analysis was performed by adjusting the values to 50 mm Na+, 3 mm Mg2+, and 60°C annealing temperature.
The following C. elegans primers were used: spliced xbp-1 forward, TGCCTTTGAATCAGCAGTGG; spliced xbp-1 reverse, ACCGTCTGCTCCTTCCTCAATG; unspliced xbp-1 forward, AGAAGTCGTCGGTGAGGTTG; unspliced xbp-1 reverse, CCTGTTCCCACTGCTGAG; total xbp-1 forward, CCGATCCACCTCCATCAAC; total xbp-1 reverse, ACCGTCTGCTCCTTCCTCAATG; hsp-4 forward, AGTTGAAATCATCGCCAACG; hsp-4 reverse, GCCCAATCAGACGCTTGG; rtcb-1 forward, GGAAGTCGAGGACTTGGACA; rtcb-1 reverse, GTTAACCCAGGCGAAGTTTG; tba-1 forward, GTACACTCCACTGATCTCTGCTGACAAG; tba-1 reverse, CTCTGTACAAGAGGCAAACAGCCATG; ama-1 forward, CCTACGATGTATCGAGGCAAA; ama-1 reverse, CCTCCCTCCGGTGTAATAATG; gpd-2 forward, CTCCATCGACTACATGGTCTACTTG; gpd-2 reverse, AGCTGGGTCTCTTGAGTTGTAGAC.
PCR efficiency was calculated from standard curves that were generated using serial dilutions of cDNA of all samples. All targeted genes were measured in triplicate, and three independent biological replicates were tested for each sample. No amplification was detected in no template and no reverse transcriptase controls. The Cq quantification cycle values recorded by CFX Manager Software version 3.0 (Bio-Rad) were exported into qBasePLUS version 2.6 (Biogazelle) for determining reference target stability (GeNorm M <0.5, CV <0.2). ama-1, tba-1, and gpd-2 were used as internal controls. Relative mRNA expression levels were normalized using these reference control genes.
Tunicamycin treatment of hsp-4::GFP worms.
RNAi-treated [empty vector (EV), rtcb-1 or xbp-1] late L4-stage animals were transferred to NGM plates spread with gradient concentrations of tunicamycin: 0, 2, 4, 8, 12, and 16 μg/ml for an exposure time of 6 h, followed by hsp-4::GFP analysis, as described in the next section. Tunicamycin (Calbiochem) was made at a stock concentration of 1 mg/ml in DMSO.
Imaging and statistics.
Fluorescent microscopy was performed using a Nikon Eclipse E800 epifluorescence microscope equipped with an Endow GFP or Texas Red HYQ filter cube (Chroma Technology). A Cool Snap CCD camera (Photometrics) driven by MetaMorph software (Molecular Devices) was used to acquire images. Using established methods for hsp-4::GFP transcriptional fusion reporter analysis (Chen et al., 2010), each animal was imaged in a consistent anatomical region (at the intestinal region directly behind the pharynx) at the same magnification and exposure intensity. The pixel intensity was quantified within a 100 × 100 μm box drawn in the same region and compiled across three replicates. For statistical analyses of all the datasets, either a Student's t test or one-way ANOVA followed by a Tukey's post hoc was used.
Results
RTCB-1 rescues age-dependent DA neuron loss induced by α-syn overexpression in C. elegans
Previously, a large RNAi screen revealed that depletion of rtcb-1 caused enhanced α-syn misfolding in C. elegans body wall muscle cells (Hamamichi et al., 2008). Alignment of amino acid sequences between metazoan RtcB orthologs reveals the strongly conserved nature of these proteins (Popow et al., 2011). A sequence alignment of human HSPC117 with C. elegans RTCB-1 shows 73% amino acid sequence identity.
We wanted to determine whether RTCB-1 would protect DA neurons from age-dependent decline from α-syn-induced neurodegeneration. Transgenic animals expressing C. elegans rtcb-1 cDNA under the control of DA neuron-specific promoter (Pdat-1::rtcb-1) were injected into worms expressing both α-syn and GFP in DA neurons. Overexpression of α-syn alone resulted in significant degeneration of DA neurons whereby only 30 and 14% of the population displayed normal neurons at day 7 and day 10 of the worm lifespan, respectively, indicating an age-dependent degeneration, as reported previously (Hamamichi et al., 2008). Animals coexpressing RTCB-1 and α-syn exhibited significant rescue of DA neurodegeneration at both time points (Fig. 1A). Representative images depict the neuroprotective effect conferred by RTCB-1 in vivo (Fig. 1B vs C).
DA neuronal-specific depletion of rtcb-1 enhanced α-syn- and 6-OHDA-induced degeneration
As might be expected for a ubiquitous gene product associated with critical cellular processes, a putative C. elegans rtcb-1 homozygous null mutant causes sterility. Therefore, to further investigate the neuroprotective properties of RTCB-1, we used a method of neuron-selective RNAi knockdown specifically in DA neurons of C. elegans to assay for neurodegeneration using a DA neuronal-specific RNAi strain expressing α-syn and GFP in the DA neurons (Harrington et al., 2012). Loss of RTCB-1 further enhanced DA degeneration caused by α-syn at day 4 and day 6 post hatching, when compared to RNAi EV control (Fig. 1D). Figure 1, E and F, shows representative images of an RNAi-treated worm expressing α-syn and GFP in the DA neurons (EV control) and an rtcb-1 RNAi-treated worm expressing α-syn and GFP, exhibiting enhanced DA degeneration.
To determine whether the neuroprotective effect of RTCB-1 was specific to α-syn, we examined vulnerability to a neurotoxin, 6-OHDA, in worms. Selective degeneration of DA neurons occurs in C. elegans after exposure to 6-OHDA (Nass et al., 2002). Using another DA neuronal-specific RNAi-sensitive strain, expressing only GFP in DA neurons, worms were knocked down with dsRNA targeting rtcb-1 or EV and subjected to 6-OHDA treatment. Animals treated with 6-OHDA and EV RNAi exhibited degeneration of DA neurons at 24, 48, and 72 h after exposure (Fig. 1G). Cotreatment with rtcb-1 RNAi caused additional enhancement in DA degeneration at all time points evaluated (Fig. 1G). Representative images of C. elegans DA neurons as a result of 6-OHDA exposure with EV or rtcb-1 RNAi treatment are shown in Figure 1, H and I.
RTCB-1 is required for xbp-1 mRNA splicing
According to a recent study in Saccharomyces cerevisiae, the bacterial homolog RtcB acts as an RNA repair enzyme and has the capacity to replace the function of Trl1 (yeast RNA ligase) as a catalyst of unconventional HAC1 mRNA splicing during the UPR (Tanaka et al., 2011). Similar unconventional splicing has also been described for the gene XBP1 in humans, mice, C. elegans, and Drosophila melanogaster (Hooks and Griffiths-Jones et al., 2011). The unconventional splicing of XBP1 is catalyzed by an endonuclease, IRE1, and an undescribed RNA ligase. Based on these studies, we hypothesized that RTCB-1 could represent the RNA ligase required for xbp-1 splicing in C. elegans. Spliced xbp-1 level increases in response to tunicamycin in WT (N2) worms, and the level of xbp-1 is reduced in xbp-1 RNAi worms under both basal and tunicamycin-treated conditions (Chen et al., 2010). To detect a role for RTCB-1 in xbp-1 splicing, we performed RNAi knockdown of rtcb-1 in N2 worms and measured spliced (xbp-1s), unspliced (xbp-1u), and total (xbp-1t; i.e., xbp-1s + xbp-1u) levels of xbp-1 mRNA, using real-time qPCR. Under tunicamycin-treated conditions, knockdown of rtcb-1 significantly lowered xbp-1s and xbp-1t mRNA expression compared with control (Fig. 2A). Notably, xbp-1u levels were unaffected in RNAi versus control samples. rtcb-1 primers were used to ensure its knockdown in these treated worms (Fig. 2A). Under nonstressed (no tunicamycin) conditions, knockdown of rtcb-1 also significantly lowers the basal level of xbp-1s compared with EV control (Fig. 2B).
Figure 2.

RTCB-1 regulates xbp-1 splicing in C. elegans. qPCR-based detection of xbp-1s, xbp-1u, xbp-1t, or rtcb-1 mRNA expression in RNAi-treated and mutant worm strains. A, Tunicamycin (10 μg/ml)-treated N2 worms depleted for rtcb-1 RNAi display suppressed spliced and total levels of xbp-1 when compared with EV control. The unspliced levels of xbp-1 are similar in both RNAi treatments. Primers specific to rtcb-1 were used to confirm knockdown. B, A comparison of spliced xbp-1 mRNA levels in RNAi-treated N2 worms in the absence and presence of tunicamycin exposure. C, Likewise, heterozygous rtcb-1 mutant worms (strain VC1094) treated with tunicamycin showed a significant decrease in xbp-1s and xbp-1t when compared with N2 WT worms. D, DA neuron-specific RNAi knockdown of rtcb-1 also significantly decreased xbp-1s levels in worms (strain UA202) treated with tunicamycin. We confirmed specific knockdown using primers specific to rtcb-1. Relative mRNA expression levels were normalized to the control. Error bars represent ± SD from three different experiments. *p < 0.05, Student's t test.
We further confirmed these results using an rtcb-1 mutant strain, which as a homozygote is sterile and has limited viability; thus, we maintained and analyzed it as a heterozygote. The rtcb-1 mutant gk451 consists of a 370 bp deletion spanning the promoter and the first exon, most likely resulting in a nonfunctional protein. rtcb-1 mutant animals were grown until day 4 after hatching and exposed to tunicamycin (for 6 h) along with N2 control worms. Based on qPCR results, xbp-1s and xbp-1t mRNA expression was significantly reduced in rtcb-1 mutant worms compared with control animals (Fig. 2C). It should be noted that xbp-1 mutant animals, containing a nonsense mutation in the XBP-1 predicted protein, are unable to mount a UPR (Bischof et al., 2008; Chen et al., 2010). These combined RNAi and mutant analyses support the hypothesis that RTCB-1 regulates xbp-1 splicing.
We next wanted to determine whether RTCB-1-mediated splicing of xbp-1 mRNA was extended to DA neurons. To examine a neuron-specific effect of xbp-1 splicing in C. elegans, we used a selectively DA neuronal-sensitive strain to knockdown rtcb-1 and measured xbp-1 mRNA levels. Using rtcb-1-specific primers, we first determined whether the knockdown of rtcb-1 in the eight DA neurons of C. elegans hermaphrodites could be assessed successfully via qPCR. The results indicated sufficient sensitivity to detect a significant decrease in the expression levels of rtcb-1 mRNA in RNAi-treated worms when compared with EV control (Fig. 2D). Furthermore, expression of xbp-1s was substantially lowered in rtcb-1 RNAi worms compared with the control. Therefore, it is clear that RTCB-1 also functions in vivo to mediate xbp-1 splicing in DA neurons.
RNAi knockdown of rtcb-1 lowers ER stress response in vivo
In PD, as well as other neurodegenerative diseases, the presence of misfolded proteins disrupts the balance between the synthesis of new proteins and the ability of the ER to process these proteins. As a result, ER stress in turn provokes the UPR, which may protect cells against the toxic accumulation of misfolded proteins (Harding et al., 2002). To examine the function of RTCB-1 during ER stress-induced UPR, an in vivo quantitative readout assay for the ER stress response was performed. Levels of ER stress was monitored in transgenic animals using a fluorescent reporter, GFP, under the control of hsp-4 promoter (Phsp-4::GFP). HSP-4 (worm homolog of BiP) is one of the ER chaperones regulated by an active xbp-1 (Calfon et al., 2002). During tunicamycin-initiated ER stress, transcription of hsp-4 is induced and GFP is highly expressed, thereby providing a direct and quantitative measure of the in vivo stress response. A previous study has shown that hsp-4::GFP worms treated with xbp-1 RNAi (in the presence of tunicamycin) exhibit reduced GFP intensity compared with controls (Chen et al., 2010). To determine the role of RTCB-1 in ER stress response, worms fed with rtcb-1 RNAi (or xbp-1 RNAi as a positive control) were treated using a gradient of tunicamycin concentrations and examined by fluorescent intensity changes. As expected, a significant decrease in hsp-4::GFP expression was observed for both xpb-1 RNAi and rtbc-1 RNAi compared with the EV RNAi control at all concentrations tested (Fig. 3A–C). We also performed qPCR to measure the mRNA expression of hsp-4 in these RNAi-treated worms that were exposed to 10 μg/ml tunicamycin. Similar to the in vivo results, the levels of hsp-4 expression in rtcb-1 and xbp-1 RNAi worms were lower than that of the EV RNAi treated animals (Fig. 3D). These data further strengthen the genetic link between RTCB-1 activity and xbp-1 in regulating ER stress response.
Figure 3.

Knockdown of RTCB-1 lowers ER stress response. A, Bar charts of GFP intensity from the stress reporter hsp-4::GFP alone (with EV RNAi) or after RNAi knockdown of rtcb-1 or xbp-1 measured consistently in the regions shown in B and C. Strains were treated with increasing concentrations of tunicamycin for 6 h (or DMSO as control). Data presented have been normalized to EV RNAi, untreated, hsp-4::GFP samples and is calculated as the mean ± SD of three experiments, in which 30 animals were analyzed per replicate (p < 0.05, one-way ANOVA). RNAi knockdown of both rtcb-1 and xbp-1 (positive control) caused significant downregulation of hsp-4::GFP expression. B, C, Representative C. elegans images for RNAi treatments described in A. Pixel intensities were measured in a 100 × 100 μm region of the intestine proximal to the pharynx. The region highlighted in the white box shows GFP expression driven by the hsp-4 gene promoter that was measured in all animals. Scale bar, 50 μm. D, RNAi depletion of rtcb-1 and xbp-1 (positive control) in N2 worms caused a decrease in hsp-4 mRNA expression when compared with EV control, as shown by qPCR (*p < 0.05, one-way ANOVA). Values are the mean ± SD of three independent experiments and were normalized to the control.
RNAi depletion of xbp-1 blocks the neuroprotective function of RTCB-1
Having revealed a role for RTCB-1 in xbp-1 mRNA splicing, we sought to determine whether its neuroprotective function is mediated by XBP-1. In vitro studies in SH-SY5Y cells and in vivo studies in mice have shown that overexpression of xbp-1s protects DA neurons from dying from MPTP (Sado et al., 2009). Here we used a pan-neuronal-sensitive RNAi strain expressing α-syn and GFP in DA neurons to perform RNAi knockdown of xbp-1 and rtcb-1 separately. Strikingly, we observed a significant change in neurodegeneration in each of them compared with the α-syn control (Fig. 4A). To examine the genetic interaction between RTCB-1 and xbp-1 in regulating neuroprotection, we crossed our pan-neuronal-specific RNAi strain (expressing α-syn and GFP in DA neurons) with worms overexpressing RTCB-1 in DA neurons (Pdat-1::rtcb-1) to generate a pan-neuronal-selective RNAi-sensitive strain expressing α-syn, RTCB-1, and GFP in DA neurons. Analysis of this new strain showed that RTCB-1 overexpression attenuated the DA neuron loss induced by α-syn, consistent with our previous results using a non-RNAi-specific strain (Fig. 4A). By using this strain for RNAi analysis, we further discerned that xbp-1 knockdown suppressed the neuroprotective phenotype attained through RTCB-1 overexpression (Fig. 4A). Thus, RTCB-1-mediated neuroprotection requires XBP-1.
To further support this conclusion, we overexpressed XBP-1 in DA neurons of worms expressing α-syn and observed significant neuroprotection with the endogenous level of rtcb-1 (Fig. 4B). Alternately, knocking down rtcb-1 in this XBP-1 overexpression background caused a reversal of the neuroprotective phenotype (Fig. 4B); this was anticipated, because loss of rtcb-1 would block the splicing of xbp-1 mRNA (as already shown in Fig. 2). Together, these results suggest that RTCB-1-induced neuroprotection is mediated through XBP-1 signaling. Whereas loss of xbp-1 negates the neuroprotective phenotype of RTCB-1, loss of rtcb-1 blocks the neuroprotective phenotype of XBP-1 on α-syn-induced degeneration.
Mutation of a conserved residue abolishes neuroprotective activity of RTCB-1
Previously, it has been shown that mutation of a conserved residue (C122A) in the human RTCB-1 homolog HSPC117 abolished its RNA ligase activity (Popow et al., 2011). To investigate whether putative RNA ligase activity of RTCB-1 is required for its neuroprotection, we created a ligase-dead version of C. elegans RTCB-1 by mutating the same site in the worm homolog, which contains a conserved residue at the same amino acid position (Cys122 to Ala122). The rtcb-1 (C122A) variant was overexpressed in DA neurons of worms also expressing α-syn. Unlike WT rtcb-1, this mutated version was unable to provide significant neuroprotection when compared with α-syn alone as worms aged (compare Figs. 5 and 1).
Discussion
Humans, bacteria, and archaea all have RTCB-1 homologs, thus showing the evolutionary conservation of this protein. These proteins are characterized to be novel RNA ligases implicated in tRNA splicing and repair (Englert et al., 2011; Popow et al., 2011; Tanaka et al., 2011; Chakravarty et al., 2012). However, with the exception of the C. elegans ortholog, these proteins have not been correlated directly with neuronal function or survival to date. In the study by Kanai et al. (2004), the human homolog HSPC117 was identified to be a component of RNA transporting granule, thereby suggesting an overall role in RNA processing or RNA metabolism. The high degree of conservation among RtcB proteins suggests a shared function for C. elegans RTCB-1 with other family members. In this context, we determined that RTCB-1, a previously uncharacterized protein in C. elegans, is a neuroprotective gene product that regulates xbp-1 mRNA splicing in vivo. Likewise, it is evident that the underlying mechanism associated with RTCB-1-mediated neuroprotection is based on XBP-1-mediated UPR signaling.
Conventional splicing is catalyzed by the spliceosome and takes place exclusively in the nucleus (Tarn and Steitz, 1997). In contrast, the unconventional splicing of HAC1 and XBP1 is mechanistically distinct and is completely independent of the spliceosome in that it is catalyzed by IRE1 endonuclease and an RNA ligase (Sidrauski et al., 1996; Kawahara et al., 1998; Gonzalez et al., 1999; Yoshida et al., 2001). Furthermore, unconventional splicing of yeast HAC1 and XBP1 mRNAs occurs in the cytoplasm during the UPR (Ruegsegger et al., 2001; Uemura et al., 2009). Notably, there are no RtcB homologs in yeast or plants, but bacterial RtcB can complement the function of yeast tRNA ligase in HAC1 mRNA splicing (Tanaka et al., 2011).
In mammals, along with the conserved IRE1 signaling pathway, two additional UPR pathways have evolved to maintain proteostasis. These two pathways are respectively mediated by pancreatic-enriched ER kinase (PERK), which facilitates translational inhibition to reduce protein production, and activating transcription factor 6 (ATF6), which mediates transcription to increase the levels of chaperone proteins (Haze et al., 1999; Sood et al., 2000). The coordination of these three pathways provides protection against ER stress in mammals. C. elegans has homologs of the vertebrate UPR components IRE1, ATF6, and PERK (Shen et al., 2001; Urano et al., 2002). It also retains the essential function of these components in upregulating the expression of UPR target genes during ER stress. Worms treated with ER stress inducers, such as tunicamycin, thapsigargin, or DTT, exhibit an increase of spliced or stressed xbp-1 (xbp-1s), followed by activation of UPR genes involved in ER biogenesis (Shen et al., 2001; Calfon et al., 2002). The requirement for the UPR pathway in C. elegans is cell type and age dependent, and expression of XBP-1 is elevated in neurons during development (Hetz et al., 2008). Studies have shown that C. elegans XBP-1 functions in the nervous system and loss of xbp-1 blocks UPR signaling (Calfon et al., 2002). Therefore, our data showing the neuroprotective function of RTCB-1 and its role in regulation of UPR xbp-1 splicing further strengthens our conclusions. For additional studies, it will be instructive to determine a putative neuronal role of RTCB-1 in mitochondrial stress response, because both the ER and mitochondria coordinate the UPR.
Misfolded proteins and associated ER stress are common features of neurodegenerative diseases. Overexpression of mutant forms of α-syn in cultured neuronal cells leads to proteasomal dysfunction and DA neurodegeneration (Stefanis et al., 2001; Tanaka et al., 2001). We reported previously that α-syn aggregation impairs vesicle trafficking and degradation of selective ERAD substrates and causes ER stress (Cooper et al., 2006). Neurotoxins, such as 6-OHDA and MPTP, also cause dopaminergic neuronal death and trigger the activation of genes involved in ER stress and the UPR (Ryu et al., 2002; Holtz and O'Malley, 2003). Inactivation of the UPR leads to organelle dysfunction and cell death. For example, deletion of PERK impairs cell survival, whereas increased PERK activity is protective (Harding et al., 2000; Scheuner et al., 2001; Lu et al., 2004). Similarly, IRE1–XBP1 signaling has a protective role during ER stress. In vitro ectopic expression of XBP1s activated various secretory pathway genes, increased cell size, and elevated total protein synthesis (Shaffer et al., 2004). XBP-1 also plays a role in controlling the autophagic clearance of aggregated proteins (Hetz et al., 2009). A recent study reported that silencing of XBP1 in mice led to ER stress and DA neurodegeneration, indicating the functional requirement of the UPR in maintaining neuronal proteostasis (Valdés et al., 2014). In addition, overexpression of XBP1 has been shown to be protective against proteasome inhibition- or ischemic-induced cell death in neuronal cells (Sado et al., 2009; Ibuki et al., 2012). Likewise, in our C. elegans model for α-syn-induced DA neurodegeneration, we demonstrated that overexpression of XBP-1 is neuroprotective, whereas neuron-specific RNAi knockdown of xbp-1 exacerbated the neurodegeneration.
These previous discoveries highlight the importance of identifying mediators of xbp-1 activity, which take on added significance when considering neurodegeneration. Here we have shown that overexpression of RTCB-1 in DA neurons of worms expressing α-syn attenuated degenerating neurons from α-syn-induced toxicity, whereas neuron-specific depletion of xbp-1 loses the protective function of RTCB-1 in the same strain of worms coexpressing RTCB-1 and α-syn in DA neurons. Similarly, we also showed that neuron-selective knockdown of rtcb-1 attenuates the neuroprotective phenotype of XBP-1 in worms expressing α-syn. These results uncover a functional relationship between RTCB-1 and XBP-1 in regulating neuroprotection against proteostatic stress caused by α-syn (Fig. 6). For the road ahead, an important goal is to explore the transcriptional status of RTCB-1 and XBP-1 in α-syn transgenic worms. Using qPCR and transgenic worms overexpressing α-syn in the nematode body wall muscles (Hamamichi et al., 2008), we have discerned that rtcb-1 is not regulated by α-syn (data not shown). However, it is yet to be determined whether α-syn induces transcriptional activation of xbp-1. In vitro studies showed that α-syn oligomers induce xbp-1 splicing (Castillo-Carranza et al., 2012), indicating that α-syn is an ER stressor. In vivo studies have also shown that XBP-1 significantly reduces α-syn levels (Si et al., 2012). Pathologically, our results support the idea that RTCB-1-induced activation of XBP-1 promotes clearance of abnormally accumulated α-syn and thus plays a neuroprotective role. Thus, identification of the signals modulating this cellular response may contribute to the treatment of PD.
Figure 6.

Proposed model for RTCB-1-mediated DA neuroprotection in C. elegans. This tentative model describes our current understanding of the underlying mechanism involved in RTCB-1-induced neuroprotection against DA neuron cell death caused by various stressors. We have determined previously that RTCB-1 protects cells from α-syn protein misfolding or α-syn-induced neuronal toxicity. It is possible that, during ER stress caused by accumulation of such toxic misfolded or unwanted proteins, RTCB-1 acts as an RNA modifier that regulates the splicing of the XBP-1 transcription factor in UPR. This in turn activates downstream factors, such as ER chaperones and the hexosamine biosynthetic pathway, and stimulates cytoprotective mechanisms to reestablish protein homeostasis (Denzel et al., 2014). In our C. elegans PD model, cells depleted with rtcb-1 might block the UPR pathway because of a failure in xbp-1 splicing, leading to cell death or DA neuronal death caused by ER overload. Alternatively, overexpression of RTCB-1 might lead to prolonged xbp-1 splicing, thereby enhancing proteostasis activity, maintaining ER stress response, and thus protecting against neuronal damage caused by α-syn. Based on our neurodegeneration and splicing assay, the association between RTCB-1 and XBP-1 could be direct or indirect in the regulation of the UPR pathway.
Previously, an ER chaperone, Hsp70, was shown to inhibit α-syn-induced toxicity in a Drosophila PD model (Auluck et al., 2002). According to another study, mammalian Hsp70 interacts with IRE1, and this interaction prolongs the splicing of XBP1, thereby activating UPR target genes and protecting cells from cell death (Gupta et al., 2010). Likewise, we propose a model in which overexpression of RTCB-1 in C. elegans DA neurons might prolong the splicing of xbp-1, thus activating ER chaperone proteins and rescuing α-syn-induced neurodegeneration (Fig. 6), whereas cells depleted of RTCB-1 would hinder xbp-1 splicing, thereby blocking UPR signaling and promoting cell death caused by α-syn (Figs. 2, 4). Recently, the mammalian RtcB has been identified as a UPR RNA ligase regulating XBP1 splicing in vitro (Lu et al., 2014). Moreover, cells expressing the ligase-dead RtcB (C122A) exhibited XBP-1 splicing defects. Our findings are consistent with these results in that C. elegans RTCB-1 also regulates XBP-1 splicing in the UPR pathway. We also determined that the ligase-active site of worm RTCB-1 (Cys122) is necessary for its neuroprotective function. Our findings further extend the understanding of RTCB-1 function in the context of neurodegeneration and as a previously uncharacterized neuroprotective gene product with potential as a therapeutic target.
Growing evidence suggests that proteins functioning in RNA synthesis, processing, splicing, or degradation are mechanistically significant for neurodegenerative disorders (Cooper et al., 2009). Mutations in genes that are involved in RNA metabolism, such as TDP-43 (TAR DNA-binding protein) and FUS (fused in sarcoma, translocated in liposarcoma), have been found to cause motor neuron diseases (Gitcho et al., 2008; Vance et al., 2009). Both of these proteins are implicated in mRNA splicing (Belly et al., 2005; Buratti and Baralle, 2008). Additionally, mutations in tRNA splicing endonuclease (TSEN54) and tRNA synthetase have also been shown to cause motor neurodegeneration (Wan et al., 2012). The neuronal phenotypes caused by mutations in these genes could be a result of RNA processing defects as observed with RTCB-1 studies or could be attributable to alternate biochemical and cellular activities distinct from RNA processing (Antonellis et al., 2003; Jordanova et al., 2006; Latour et al., 2010). Interestingly, a recent C. elegans RNAi screen identified RTCB-1 (F16A11.2) as a modifier for GABAergic axonal regeneration (Nix et al., 2014). These and other studies suggest that defects in RNA metabolism (regulated at many different levels) can be pathogenically related to various neurodegenerative disorders. In this regard, an in-depth understanding of the pathways involving RTCB-1, or its potential functional effectors, might yield promising and unexploited targets for therapeutic development to prevent neurodegeneration.
Footnotes
We thank Laura Berkowitz, Xiaohui Yan, and Susan DeLeon for their assistance and expert advice in the progress of this project. Some C. elegans strains were provided by the Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office Research Infrastructure Programs (Grant P40 OD010440). This research was funded by National Institutes of Health Grants R15 NS075684 (G.A.C.) and R15 NS078728 (K.A.C.). Other support came from a Howard Hughes Medical Institute Undergraduate Science Program Grant to The University of Alabama (C.R.) and the Parkinson's Support Group of Huntsville.
The authors declare no competing financial interests.
References
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VMY, Bonini NM. Chaperone suppression of α-Synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Belly A, Moreau-Gachelin F, Sadoul R, Goldberg Y. Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurons: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci Lett. 2005;379:152–157. doi: 10.1016/j.neulet.2004.12.071. [DOI] [PubMed] [Google Scholar]
- Bischof LJ, Kao CY, Los FCO, Gonzalez MR, Shen Z, Briggs SP, van der Goot FG, Aroian RV. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008;4:e1000176. doi: 10.1371/journal.ppat.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci. 2005;25:3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Zhang Y, Guerrero-Muñoz MJ, Kayed R, Rincon-Limas DE, Fernandez-Funez P. Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem Res. 2012;37:1707–1717. doi: 10.1007/s11064-012-0780-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty AK, Subbotin R, Chait BT, Shumana S. RNA ligase RtcB splices 3′-phosphate and 5′-OH ends via covalent RtcB-(histidinyl)-GMP and polynucleotide-(3′)pp(5′)G intermediates. Proc Natl Acad Sci U S A. 2012;109:6072–6077. doi: 10.1073/pnas.1201207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Burdette AJ, Porter JC, Ricketts JC, Fox SA, Nery FC, Hewett JW, Berkowitz LA, Breakefield XO, Caldwell KA, Caldwell GA. The early-onset torsion dystonia-associated protein, torsinA, is a homeostatic regulator of endoplasmic reticulum stress response. Hum Mol Genet. 2010;19:3502–3515. doi: 10.1093/hmg/ddq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzel MS, Storm NJ, Gutschmidt A, Baddi R, Hinze Y, Jarosch E, Sommer T, Hoppe T, Antebi A. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell. 2014;156:1167–1178. doi: 10.1016/j.cell.2014.01.061. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Tredici DK, Wszolek ZK, Litvan I. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurology. 2009;8:1150–1157. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- Englert M, Sheppard K, Aslanian A, Yates JR, 3rd, Söll D. Archaeal 3′-phosphate RNA splicing ligase characterization identifies the missing component in tRNA maturation. Proc Natl Acad Sci U S A. 2011;108:1290–1295. doi: 10.1073/pnas.1018307108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez TN, Sidrauski C, Dörfler S, Walter P. Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 1999;18:3119–3132. doi: 10.1093/emboj/18.11.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8:e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson's disease model. Proc Natl Acad Sci U S A. 2008;105:728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/S1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Harrington AJ, Yacoubian TA, Slone SR, Caldwell KA, Caldwell GA. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson's disease. J Neurosci. 2012;32:2142–2153. doi: 10.1523/JNEUROSCI.2606-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Lee AH, Gonzalez-Romero D, Thielen P, Castilla J, Soto C, Glimcher LH. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc Natl Acad Sci U S A. 2008;105:757–762. doi: 10.1073/pnas.0711094105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hoogewijs D, Houthoofd K, Matthijssens F, Vandesompele J, Vanfleteren JR. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Mol Biol. 2008;9:9. doi: 10.1186/1471-2199-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks KB, Griffiths-Jones S. Conserved RNA structures in the non-canonical Hac1/Xbp1 intron. RNA Biol. 2011;8:552–556. doi: 10.4161/rna.8.4.15396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibuki T, Yamasaki Y, Mizuguchi H, Sokabe M. Protective effects of XBP1 against oxygen and glucose deprivation/reoxygenation injury in rat primary hippocampal neurons. Neurosci Lett. 2012;518:45–48. doi: 10.1016/j.neulet.2012.04.053. [DOI] [PubMed] [Google Scholar]
- Inoue T, Thomas JH. Targets of TGF-β Signaling in Caenorhabditis elegans dauer formation. Dev Biol. 2000;217:192–204. doi: 10.1006/dbio.1999.9545. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Hosoda A, Okuda T, Kamigori Y, Nomura-Furuwatari C, Kimata Y, Tsuru A, Kohno K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat Cell Biol. 2001;3:158–164. doi: 10.1038/35055065. [DOI] [PubMed] [Google Scholar]
- Jordanova A, Irobi J, Thomas FP, Dijck PV, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D'Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, Hirokawa N. Isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Yanagi H, Yura T, Mori K. Unconventional splicing of HAC1/ERN4 mRNA required for the unfolded protein response. Sequence-specific and non-sequential cleavage of the splice sites. J Biol Chem. 1998;273:1802–1807. doi: 10.1074/jbc.273.3.1802. [DOI] [PubMed] [Google Scholar]
- Latour P, Thauvin-Robinet C, Baudelet-Méry C, Soichot P, Cusin V, Faivre L, Locatelli MC, Mayençon M, Sarcey A, Broussolle E, Camu W, David A, Rousson R. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Liang FX, Wang X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell. 2014;55:758–770. doi: 10.1016/j.molcel.2014.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix P, Hammarlund M, Hauth L, Lachnit M, Jorgensen EM, Bastiani M. Axon regeneration genes identified by RNAi screening in C.elegans. J Neurosci. 2014;34:629–645. doi: 10.1523/JNEUROSCI.3859-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lührmann R, Söll D, Martinez J. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science. 2011;331:760–764. doi: 10.1126/science.1197847. [DOI] [PubMed] [Google Scholar]
- Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463:1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 2002;21:6505–6514. doi: 10.1093/emboj/cdf639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüegsegger U, Leber JH, Walter P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell. 2001;107:103–114. doi: 10.1016/S0092-8674(01)00505-0. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, Wada D, Kitagawa H, Watanabe TK. Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. 2009;1257:16–24. doi: 10.1016/j.brainres.2008.11.104. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/S1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/S0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Si L, Xu T, Wang F, Liu Q, Cui M. X-box-binding protein 1-modified neural stem cells for treatment of Parkinson's disease. Neural Regen Res. 2012;7:736–740. doi: 10.3969/j.issn.1673-5374.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Cox JS, Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87:405–413. doi: 10.1016/S0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Sood R, Porter AC, Ma K, Quilliam LA, Wek RC. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. Biochem J. 2000;346:281–293. doi: 10.1042/0264-6021:3460281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Meineke B, Shuman S. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J Biol Chem. 2011;286:30253–30257. doi: 10.1074/jbc.C111.274597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson L, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. Pre-mRNA splicing: the discovery of a new spliceosome doubles the challenge. Trends Biochem Sci. 1997;22:132–137. doi: 10.1016/S0968-0004(97)01018-9. [DOI] [PubMed] [Google Scholar]
- Thompson ML, Chen P, Yan X, Kim H, Borom AR, Roberts NB, Caldwell KA, Caldwell GA. TorsinA rescues ER-associated stress and locomotive defects in C. elegans models of ALS. Dis Model Mech. 2014;7:233–243. doi: 10.1242/dmm.013615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura A, Oku M, Mori K, Yoshida H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J Cell Sci. 2009;122:2877–2886. doi: 10.1242/jcs.040584. [DOI] [PubMed] [Google Scholar]
- Urano F, Calfon M, Yoneda T, Yun C, Kiraly M, Clark SG, Ron D. A survival pathway for Caenorhabditis elegans with a blocked unfolded protein response. J Cell Biol. 2002;158:639–646. doi: 10.1083/jcb.200203086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- Valdés P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A. 2014;111:6804–6809. doi: 10.1073/pnas.1321845111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, Vos KJD, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Yourshaw M, Mamsa H, Rudnik-Schöneborn S, Menezes MP, Hong JE, Leong DW, Senderek J, Salman MS, Chitayat D, Seeman P, von Moers A, Graul-Neumann L, Kornberg AJ, Castro-Gago M, Sobrido MJ, Sanefuji M, Shieh PB, Salamon N, Kim RC, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Shuman S. Structure-function analysis of yeast tRNA ligase. RNA. 2005;11:966–975. doi: 10.1261/rna.2170305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian TA, Standaert DG. Targets for neuroprotection in Parkinson's disease. Biochim Biophys Acta. 2009;1792:676–687. doi: 10.1016/j.bbadis.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]