

Abstract
A female patient was found to have meningioma when she was 3 years and 11 months old and subtotal excision was performed. The residual tumour recurred 3 months after the first excision, and again 11 months after the second one. She was also found to have subcutaneous neurofibroma. However, her clinical features did not fulfil the diagnostic criteria for neurofibromatosis type 2 (NF2), and her family history was unremarkable. Considering that primary meningioma is extremely rare in the paediatric population, the diagnosis of NF2 was considered. It was thought that this might have an impact on her subsequent management. Genetic testing on blood DNA for NF2 was arranged, and the results confirmed that she had mosaic deletion of the promoter to exon 16 of NF2. With uncertainty of whether NF2 mutations are also present in other tissues, vigilant follow-up for other NF2-related complications would be required in the future.
Background
The incidence of meningioma increases with age.1 Only 1.5–2% of all meningiomata occur in children and adolescents.2 Most paediatric meningiomata occur in the second decade of life and are mainly secondary to cranial irradiation.3 4 Several genetic tumour predisposition syndromes are associated with early onset meningioma, including neurofibromatosis type 2 (NF2),5 Gorlin syndrome,6 Li-Fraumeni syndrome,7 von Hippel-Lindau syndrome8 and Rubinstein-Taybi syndrome.9
NF2 has an incidence of 1 in 60 000–80 000.10 Although NF2 is traditionally thought to present in young adults, Manchester Children's Tumour Registry showed that only 18% of newly diagnosed patients with NF2 are in paediatric age (0–15 years old) and 9% are diagnosed before 10 years of age.11 More importantly, more than half of these paediatric patients had no family history to alert clinicians to NF2 susceptibility.
Accurate diagnoses of underlying genetic disorders are important as they have significant implications on treatment and prognosis. Here we describe a case of NF2 in childhood presenting as recurrent meningioma.
Case presentation
A previously healthy girl presented with insidious onset of unsteady gait at 3 years and 11 months. CT and MRI showed a large intraventricular tumour with hydrocephalus (figure 1). First, subtotal excision of the tumour was performed and pathology confirmed atypical meningioma. The residual meningioma enlarged rapidly and a second resection was performed 3 months later. However, the meningioma recurred again (figure 2) and the patient was referred to us for further management. Resection was repeated the third time at 11 months after the second operation. As the tumour was encasing the internal cerebral veins, the meningioma could not be completely resected. Pathology was again found to be atypical meningioma. After multiple recurrences with incomplete resection, the tumour was treated with 54 Gy external beam irradiation. The latest MRI showed a small residual tumour static in size.
Figure 1.
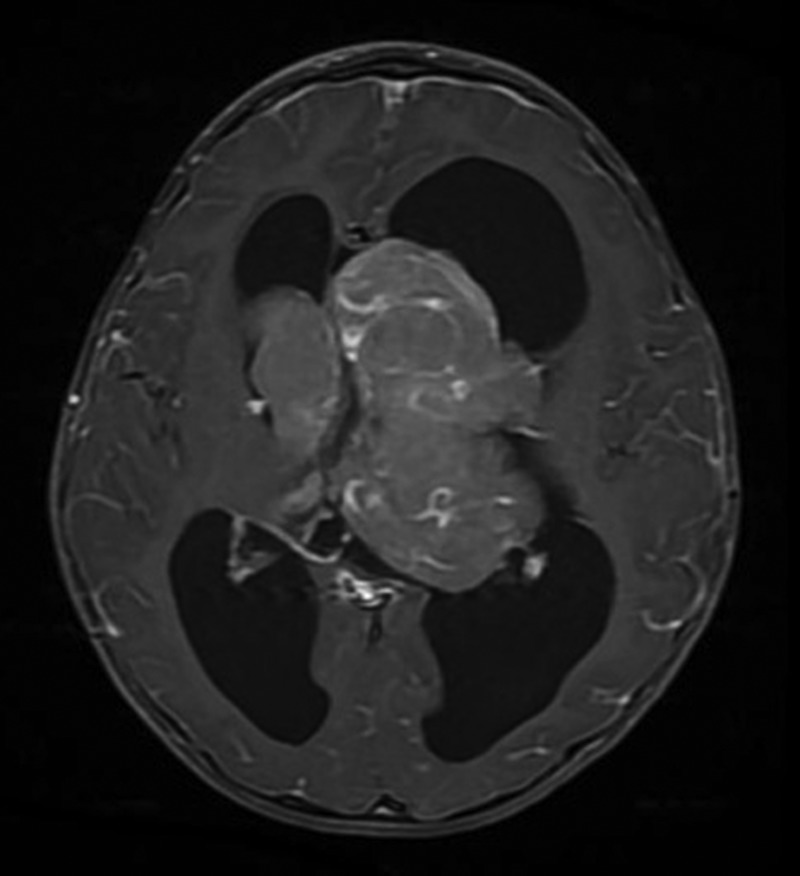
Gadolinium-enhanced T1-weighted MRI of the brain at initial diagnosis. Axial scan shows contrast-enhanced tumour mass measuring 6.9×7.9 cm in the third ventricle extending to bilateral lateral ventricles.
Figure 2.
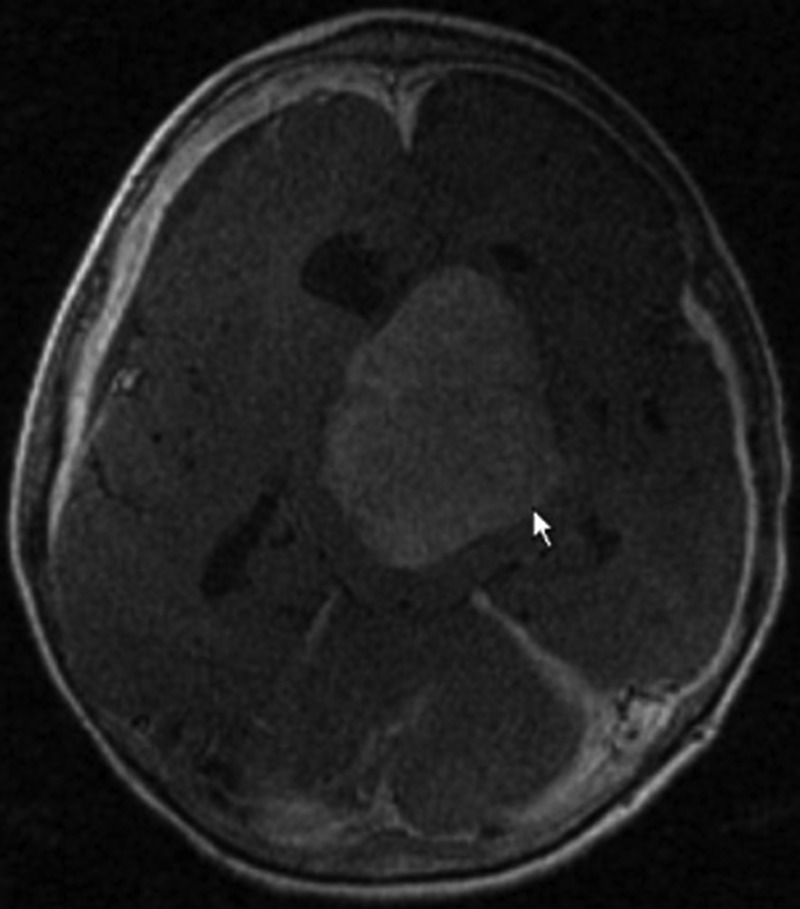
Gadolinium-enhanced T1-weighted MRI of the brain at second recurrence. Axial scan shows contrast-enhanced intraventricular meningioma measuring 5.1×6.5 cm recurring in the same location as initial diagnosis.
Detailed history revealed that the patient's parents were non-consanguinous. There was no family history of tumour or syndromal/genetic disorders. Neither was there a family history of visual or hearing loss. Antenatal history was unremarkable and the patient's development was normal. On physical examination, the patient showed no dysmorphic feature apart from macrocephaly. Two non-tender, firm subcutaneous nodules were noted over her right forearm and right thigh respectively. Biopsy of the right thigh nodule confirmed neurofibroma. Ophthalmological assessment showed no cataract or retinal lesion and hearing assessment was normal.
Investigations
Blood was sent for genetic testing of NF2 gene defects. NF2 gene mutations were screened using bidirectional fluorescent Sanger sequencing of all 17 coding exons, which included the alternatively spliced exons 16 and 17. The whole coding sequence, including the immediate splice donor and acceptor sites, was screened. Multiplex ligation-dependent probe amplification (MLPA) dosage test was also used to measure copy number of all 17 exons of the NF2 gene and promoter (P044-B1 NF2 MLPA kit from MRC-Holland). The result showed a mosaic deletion of the promoter to exon 16 of the NF2 gene, meaning some but not all blood cells harbour one copy of NF2 with the deletion (figure 3). The result was confirmed by independent MLPA analysis of DNA extracted from second blood sample.
Figure 3.
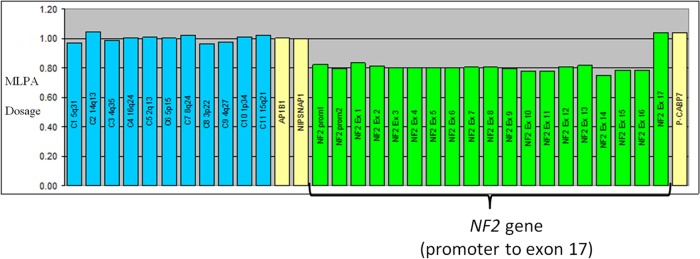
Multiplex ligation-dependent probe amplification (MLPA) dosage test for NF2 gene. The green histogram bars represent MLPA probes in the promoter and each of the 17 exons of the NF2 gene. For a normal sample the bars should be at a dosage quotient of 1.0, which represents two copies of each MLPA probe target. For a sample with a heterozygous deletion the histogram bars should be at dosage quotient of 0.5, which represents one copy of each MLPA probe target. For the patient's samples the histogram bars are at around a dosage quotient of 0.8 from the NF2 promoter to exon 16, which indicates a mosaic deletion of these MLPA probe targets.
Differential diagnosis
While the diagnosis of atypical meningioma was unequivocal, underlying tumour predisposition syndrome was considered when the patient was referred to us, in view of the young age of onset of meningioma. NF2 was highest on the list of differential diagnoses in view of the presence of subcutaneous neurofibromata. Other tumour predisposing conditions associated with meningioma such as Gorlin syndrome, Li-Fraumeni syndrome, von Hippel-Lindau syndrome and Rubinstein-Taybi syndrome were also considered but their unique clinical picture make them less likely.
Treatment
Before the confirmation of mosaic NF2, the patient was treated as a case of sporadic meningioma by surgical resections. In adults with atypical meningioma, adjuvant radiotherapy is usually recommended but this was not initially given in this child owing to her young age. However, because of multiple recurrences with unresectable tumour residual, full-dose radiotherapy was eventually given and the child tolerated it well without significant acute adverse effects.
Outcome and follow-up
The patient remained well at the latest 6 months follow-up after radiotherapy. The residual tumour remained static in size. Comprehensive genetic counselling was given to parents about the diagnosis and implications of mosaic NF2. We shall continue to follow-up for progress of the meningioma and complications of NF2 according to current recommendations.
Discussion
Meningiomas occur in approximately 50% of NF2 cases, and intracranial meningiomas are the second most common type of tumour found in patients with NF2 (the most common being schwannomas).10 Hence NF2 should be suspected in children presenting with meningioma or schwannoma, especially when they also have subtle findings such as skin lesions (eg, neurofibromas/schwannomas) or eye changes (eg, cataracts/retinal hamatomas).12 Data from the Manchester Children's Tumour Registry showed that 3 of 22 children presenting with meningioma have gone on to develop classic features of NF2 later in life.10 11
NF2 is a condition with evolving presentation, and patients with NF2 do not always fulfil the diagnostic criterion at their first assessment. Our patient presented with subcutaneous neurofibromata, but lacked vestibular schwannoma, hearing or visual problems. Hence she did not fulfil the common diagnostic criteria such as the Manchester criteria, NIH criteria or National Neurofibromatosis Foundation (NNFF) criteria.13 Without genetic testing, it would have been difficult to exclude the important differential diagnosis of NF2.
The genetics of NF2 have been studied extensively. Standard mutation techniques are able to detect 35–66% of pathogenic mutations, which include missense mutations and large deletions.10 There are also studies of genotype–phenotype correlations in NF2, showing the types of mutation that correlate with prognosis of the disease severity. In general, constitutional nonsense and frameshift NF2 mutation is associated with a more severe prognosis, compared with missense mutations, in-frame deletions and large deletions.10 14 As well as mutation type correlating with prognosis, early presentation of disease and high number of meningiomas are also associated with a poorer prognosis.10
The clinical presentation of mosaic disorders is different among affected individuals with variable degrees of severity. The genetic test for mosaicism should not be limited to either the observable features or the known genetic aetiology of the disorder.15 Sometimes it requires genetic testing in different tissues for the confirmation of mosaicism. It is estimated that a minimum of 20–30% of sporadic classical NF2 cases are mosaic, with the mutation detectable only in tumour but not in blood DNA.10
The assessment of disease recurrence risk of any mosaic condition is challenging. As the patient is confirmed to have mosaic NF2, the mosaicism occurs postzygotically and the possibility of one parent carrying NF2 mutation can be excluded. The risk for the parents to have another affected child will be the same as that of the general population. However, for the patient herself, the risk of passing the NF2 mutation to her offspring depends on whether or not the mosaic mutation is present in her germ line, and the proportion of her germ cells that carry the mutation.15 The proportion of affected lymphocytes cannot give an accurate risk assessment. There has been a case of an affected child with NF2. The parent had mosaic NF2, but less than 3% of lymphocytes were affected and risk of transmission was below the 5% level.14 In conclusion, the risk of our patient having an affected offspring is therefore uncertain.
Learning points.
Meningiomas are seen uncommonly in childhood and the commonest association is NF2. Children with NF2 seldom fulfil the typically used clinical diagnostic criteria. They may present as central nervous tumours tumours with subtle skin lesions or eye findings.
For clinicians, understanding the genotype–phenotype correlation of NF2 (the effect of mutation types and mosaicism) is helpful to predict the prognosis.
Acknowledgments
The authors would like to thank the patient and her family for their contribution.
Footnotes
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Wiemels J, Wrensch M, Claus E. Epidemiology and etiology of meningioma. J Neurooncol 2010;99:307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao X, Zhang R, Mao Y et al. Childhood and juvenile meningiomas. Childs Nerv Syst 2009;25:1571–80. [DOI] [PubMed] [Google Scholar]
- 3.Menon G, Nair S, Sudhir J et al. Childhood and adolescent meningiomas: a report of 38 cases and review of literature. Acta Neurochir (Wien) 2009;151:239–44; discussion 44. [DOI] [PubMed] [Google Scholar]
- 4.Kotecha RS, Junckerstorff RC, Lee S et al. Pediatric meningioma: current approaches and future direction. J Neurooncol 2011;104:1–10. [DOI] [PubMed] [Google Scholar]
- 5.Blakeley JO, Evans DG, Adler J et al. Consensus recommendations for current treatments and accelerating clinical trials for patients with neurofibromatosis type 2. Am J Med Genet A 2012;158A:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rushing EJ, Olsen C, Mena H, et al. Central nervous system meningiomas in the first two decades of life: a clinicopathological analysis of 87 patients. J Neurosurg 2005;103(6 Suppl):489–95. [DOI] [PubMed] [Google Scholar]
- 7.Rieske P, Zakrzewska M, Biernat W et al. Atypical molecular background of glioblastoma and meningioma developed in a patient with Li-Fraumeni syndrome. J Neurooncol 2005;71:27–30. [DOI] [PubMed] [Google Scholar]
- 8.Kanno H, Yamamoto I, Yoshida M et al. Meningioma showing VHL gene inactivation in a patient with von Hippel-Lindau disease. Neurology 2003;60:1197–9. [DOI] [PubMed] [Google Scholar]
- 9.Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet 1995;56:112–15. [DOI] [PubMed] [Google Scholar]
- 10.Evans D. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 2009;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans D, Birch J, Ramsden R. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child 1999;81:496–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruggieri M, Iannetti P, Polizzi A et al. Earliest clinical manifestations and natural history of neurofibromatosis type 2 (NF2) in childhood: a study of 24 patients. Neuropediatrics 2005;36:21–34. [DOI] [PubMed] [Google Scholar]
- 13.Baser ME, Friedman JM, Wallace AJ et al. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology 2002;59:1759–65. [DOI] [PubMed] [Google Scholar]
- 14.Baser ME, Friedman JM, Aeschliman D et al. Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet 2002;71:715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biesecker L, Spinner N. A genomic view of mosaicism and human disease. Nat Rev Genet 2013;14:307–21. [DOI] [PubMed] [Google Scholar]