

Abstract
Pharmacologic agents to enhance liver regeneration after injury would have wide therapeutic application. Based on previous work suggesting inhibition of bone morphogenetic protein (BMP) signaling stimulates liver regeneration, we tested known and novel BMP inhibitors for their ability to accelerate regeneration in a partial hepatectomy (PH) model. Compounds were produced based on the 3,6-disubstituted pyrazolo[1,5-a] pyrimidine core of the BMP antagonist dorsomorphin and evaluated for their ability to inhibit BMP signaling and enhance liver regeneration. Antagonists of the BMP receptor activin receptor–like kinase 3 (ALK3), including LDN-193189 (LDN; 4-[6-[4-(1-piperazinyl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]-quinoline), DMH2 (4-(2-(4-(3-(quinolin-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl)phenoxy)ethyl)morpholine; VU0364849), and the novel compound VU0465350 (7-(4-isopropoxyphenyl)-3-(1H-pyrazol-4-yl)imidazo[1,2-a]pyridine; VU5350), blocked SMAD phosphorylation in vitro and in vivo, and enhanced liver regeneration after PH. In contrast, an antagonist of the BMP receptor ALK2, VU0469381 (5-(6-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-3-yl)quinolone; 1LWY), did not affect liver regeneration. LDN did not affect liver synthetic or metabolic function. Mechanistically, LDN increased serum interleukin-6 levels and signal transducer and activator of transcription 3 phosphorylation in the liver, and modulated other factors known to be important for liver regeneration, including suppressor of cytokine signaling 3 and p53. These findings suggest that inhibition of ALK3 may be part of a therapeutic strategy for treating human liver disease.
Introduction
Despite important advances in understanding the response of the liver to injury, there are no pharmacologic treatments to enhance liver regeneration. Such therapies would find wide clinical use. Groups that could benefit include 1) patients with liver tumors deemed inoperable based on predicted insufficient residual mass after resection, 2) patients who develop small-for-size syndrome after liver resection or transplantation, and 3) patients with fulminant liver failure.
Our recent findings that bone morphogenetic protein (BMP) signaling constitutively inhibits liver regeneration suggested that pharmacologic agents that inhibit BMPs might have therapeutic utility (Do et al., 2012). BMP signaling is a subset of transforming growth factor β (TGF-β) signaling and involves multiple ligands, antagonists, and receptors. Similar to other TGF-β superfamily members, BMPs signal via two sets of serine-threonine kinase receptors: a type II receptor, which binds ligand, and a type I receptor, which is recruited to the activated type II receptor, cooperatively binds ligand, and transduces the signal (Nohe et al., 2004). Type I receptors expressed in the liver include activin receptor–like kinase 2 (ALK2) and ALK3 (Xia et al., 2008). These receptors mediate signaling via phosphorylation of SMAD1/5/8 (Moustakas et al., 2001).
Although direct inhibition of TGF-β signaling—for example, with follistatin—enhances liver regeneration (Russell et al., 1988; Schwall et al., 1993; Kogure et al., 1996), targeting the TGF-β type I receptor is complicated by cardiovascular toxicity (Anderton et al., 2011).
In contrast, pharmacologic antagonism of BMPs with a variety of compounds appears to be both achievable and tolerable in vivo. The first described selective small-molecule inhibitor of BMP type I receptor was dorsomorphin (DM), which is active in the liver and blocks iron-induced phosphorylation of SMADs (Yu et al., 2008). LDN-193189 (or LDN; 4-[6-[4-(1-piperazinyl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]-quinoline hydrochloride) is a structurally related compound that also antagonizes BMPs (Cuny et al., 2008). Long-term administration of LDN to low-density lipoprotein receptor–deficient mice ameliorates the development of vascular calcifications (Derwall et al., 2012).
In this report, we developed and tested known and novel pharmacologic agents active on BMP signaling, and tested their effect on liver regeneration after partial hepatectomy (PH). We find that inhibitors of ALK3—LDN, DMH2 [4-(2-(4-(3-(quinolin-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl)phenoxy)ethyl)morpholine], and the novel compound VU0465350 (7-(4-isopropoxyphenyl)-3-(1H-pyrazol-4-yl)imidazo[1,2-a]pyridine; VU5350)—enhance liver regeneration, whereas an inhibitor of ALK2, VU0469381 [5-(6-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-3-yl)quinolone], does not.
Materials and Methods
Animals.
Seven- or 8-week-old C57BL/6 male mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The animals were housed under standard conditions.
PH.
All surgeries were performed according to the National Institutes of Health guidelines for the humane treatment of laboratory animals according to the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health publication 86-23) and with approval of the Institutional Animal Care and Use Committee of Vanderbilt University Medical School or Harvard Medical School. Mice were anesthetized with 60 mg/kg ketamine (Hospira, Lake Forest, IL) and 7 mg/kg xylazine (Phoenix Pharmaceuticals, Burlingame, CA) and positioned supine. A transverse incision was made inferior to the xiphoid process, which was excised. The median and left lateral lobes of the liver were eviscerated and ligated. The result was removal of approximately two-thirds of the liver mass. At tissue recovery, mice were anesthetized and weighed. Livers were excised, rinsed, blotted, and weighed. Sections were snap-frozen in liquid nitrogen, fixed in 10% neutral buffered formalin, or preserved in RNAlater (Qiagen, Gaithersburg, MD).
Synthesis of BMP Analogs.
BMP analogs were synthesized as previously described (Hao et al., 2010). Briefly, we concentrated on the 3,6-disubstituted pyrazolo[1,5-a] pyrimidine core of DM with particular attention to the R1 and R2 groups (C-3 and the 4-phenyl group on C-6, respectively). Compounds were produced according to methods previously described (Hao et al., 2010) and chosen for study based on their inhibitor constant (Ki) for different receptors.
Kinase Assays.
All kinase assays were conducted by Reaction Biology Corp. (Malvern, PA) as previously described (Hao et al., 2010). Briefly, compounds were tested at 10 concentrations by 3-fold serial dilutions starting at 100 μM and using the nonspecific kinase inhibitor staurosporine as control. In vitro kinase reactions were carried out in the presence of 10 μM [33P]γATP. The kinases tested included the human BMP type-1 receptors ALK2, ALK3, ALK4, ALK5, and ALK6; the human BMP type-2 receptor; the human TGF-β type-2 receptor; the human AMP-activated protein kinase; and the human vascular endothelial growth factor type-2 receptor.
LDN, DMH2, VU5350, and 1LWY Injection.
LDN, DMH2, VU5350, or 1LWY was dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO) + 50 mM Tris at 4 mg/ml and adjusted to a final pH of 7.0. Solutions were administered by intraperitoneal injection at indicated doses twice a day for 2 days prior to PH, and for 2 days after PH for all experiments, unless otherwise indicated.
In Vitro Smad Phosphorylation.
C2C12 cells were maintained in standard culture conditions. Cells were pretreated for 30 minutes with either VU5350 or 1LWY at the indicated concentration (nanograms per milliliter). Cells were then stimulated with either BMP4 (+) or nothing (−) and incubated for 45 minutes. Cells were then lysed. Western blot was performed by loading 30 μg of protein/lane and using antibodies to phosphorylated SMAD (p-SMAD) 1/5/8. Results were normalized to α-tubulin.
Proliferation Assay with 5-Bromo-2-Deoxyuridine and Immunohistochemistry.
All proliferation data were obtained by dividing the number of hepatocytes staining positive for 5-bromo-2-deoxyuridine (BrdU) by those not stained. For these assays, mice received 1 mg of intraperitoneal injection of BrdU (BD Pharmingen, Brea, CA) 2 hours before sacrifice. Staining for BrdU was performed on fixed paraffin-embedded liver sections according to the manufacturer’s instructions (Vectastain ABC kit; Vector Laboratories, Burlingame, CA). Primary antibody was rat anti–mouse BrdU (Santa Cruz Biotechnology, Inc., Dallas, TX). At least three high-power fields (400×) were counted to quantify the percentage of labeled hepatocytes to determine the proliferation index.
mRNA Isolation and Reverse-Transcription Polymerase Chain Reaction.
Total mRNA was purified from 30 mg of liver tissue preserved in RNAlater using an RNeasy Mini kit (Qiagen). One microgram of mRNA was reverse transcribed to cDNA using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA). StepOnePlus (Applied Biosystems) was used for all real-time polymerase chain reactions (PCRs). The cDNA template was diluted 1:5 and amplified using inventoried TaqMan gene expression assays (Applied Biosystems) under standard conditions. Gene expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase using the comparative Ct method. Data were analyzed using StepOne Software v2.1 (Applied Biosystems). TaqMan probe and primer sets (Applied Biosystems) were as follows: inhibitor of DNA binding 1 (ID1) (Mm00775963_g1), c-JUN (Mm00495062_s1), CCAAT-enhancer binding protein-α (C/EBP-α; Mm01265914_s1), interleukin-6 (IL-6; Mm00446191_m1), p21 (Mm01303209_m1), p27 (Mm00438168_m1), p53 (Mm01731287_m1), suppressor of cytokine signaling 3 (SOCS3) (Mm00545913_s1), BMP4 (Mm00432087_m1), p38 (Mm00442497_m1), and glyceraldehyde-3-phosphate dehydrogenase (Mm99999915_g1).
Protein Sample Preparation and Western Blot Analysis.
Whole-cell liver lysates were prepared by homogenizing 50 mg of frozen tissue in lysis buffer (50 mM HEPES, 10% glycerol, 1 mM EDTA, 50 mM NaF, 1 mM dithiothreitol, and 0.1% NP40) containing phosphatase and protease inhibitors (Sigma-Aldrich). Samples were sonicated for 10 seconds and clarified by centrifugation. Protein concentration was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA). Protein was denatured and separated by SDS-PAGE in 4–12% or 12% NuPAGE Bis-Tris precast gels (Life Technologies, Carlsbad, CA) and transferred to a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA). All antibody dilutions were done in 5% nonfat dry milk in Tris-buffered saline buffer. Primary antibodies used were as follows: mouse anti–β-actin (Abcam, Cambridge, MA), mouse anti-STAT3 (Abcam), mouse anti-Smad1/5/8 (Santa Cruz Biotechnology), mouse anti–p-Smad (Santa Cruz Biotechnology), mouse anti–cyclin A2 (Santa Cruz Biotechnology), mouse anti–p-signal transducer and activator of transcription 3 (STAT3; Abcam), mouse anti-p38 (Cell Signaling Technology, Danvers, MA), and mouse anti-p21 (Abcam). Secondary antibody was anti horseradish peroxidase–conjugated species-specific secondary antibodies (Promega, Madison, WI; or Santa Cruz Biotechnology). Western blot films were scanned and analyzed quantitatively by ImageJ software (NIH, Bethesda, MD). In all cases, at least three mice in each group were analyzed.
IL-6, Retinol-Binding Protein 4 and Bilirubin Serum Analysis.
Serum levels of IL-6 and retinol-binding protein 4 (RBP4) were measured using a commercial enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Serum bilirubin levels were determined using a QuantiChrom bilirubin assay kit (BioAssay Systems, Hayward, CA) according to the manufacturer’s instructions.
Data Analysis.
Comparisons were performed using a two-tailed unpaired Student’s t test. All statistical tests used at least three different samples for each time point. In the figures, statistical significance is shown as follows: *P < 0.05; **P < 0.01; and ***P < 0.001. All error bars in the figures are the S.E.
Results
In Vitro Activity and Structure of BMP Receptor Antagonists.
Our previous results using conditional inactivation of ALK3 in hepatocytes suggested that inhibiting ALK3 signaling could be a therapeutic strategy to enhance liver regeneration (Do et al., 2012). We therefore investigated selective inhibitors of ALK3, and compared them to selective inhibitors of ALK2 for similar effects. Table 1 lists the compounds tested along with their Ki for the indicated kinase. Data for LDN and VU0469381 have been published (Engers et al., 2013) and are included here for comparison. LDN is a selective inhibitor of ALK3 and a relatively weak inhibitor of the other receptors tested, with the exception of ALK6, which is not expressed on hepatocytes (Xia et al., 2008). DMH2 has a very similar profile to LDN. VU5350, a novel compound, is qualitatively similar to LDN and DMH2 in that it has a higher affinity for ALK3 compared with ALK2, although it does have an off-target effect on vascular endothelial growth factor type-2 receptor. In contrast and as reported (Engers et al., 2013), 1LWY is a very specific inhibitor of ALK2, more than 300 times more potent compared with its Ki for ALK3. The structure of the novel compound VU0465350 (VU5350) is shown in Fig. 1A. Other previously reported structures are included for comparison (Engers et al., 2013).
TABLE 1.
Ki in nanomolar concentrations for each experimental compound versus the specific receptor indicated
| ALK2 | ALK3 | ALK4 | ALK5 | ALK6 | BMPR2 | TGFBR2 | AMPK | VEGFR2 | |
|---|---|---|---|---|---|---|---|---|---|
| LDN | 40.72 | <5 | 1825 | 565 | <1 | 3845 | 140.4 | 1122 | 214.7 |
| DMH2 | 42.77 | 5.4 | 1407 | 2418 | <1 | 3845 | 86.48 | 3527 | 2418 |
| VU5350 | 1970 | 92.3 | Not done | Inactive | 895 | 1660 | 85.6 | 150 | 14.9 |
| 1LWY | 32 | 10,800 | Inactive | Inactive | 9830 | Inactive | Inactive | Inactive | 19,700 |
BMPR2, bone morphogenetic protein receptor 2; TGFBR2, transforming growth factor β receptor 2; AMPK, adenosine monophosphate–activated protein kinase; VEGFR2, vascular endothelial growth factor receptor 2.
Fig. 1.
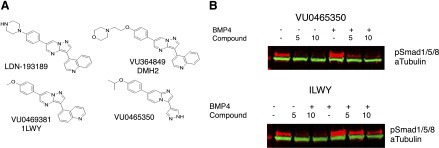
Structures and activity of specific BMP inhibitors. (A) Previously reported structures for LDN, DMH2, and VU0469381 are included for comparison with VU0465350. (B) Representative Western blot demonstrating VU0465350 (VU5350) and 1LWY inhibit phosphorylation of SMAD1/5/8 in C2C12 cells. Concentrations of experimental compounds are provided above each lane in micromolar concentrations. Both compounds strongly inhibited SMAD phosphorylation in a dose-dependent fashion. Each experiment was performed in triplicate with P <0.01 except for 2 μM 1LWY (P = 0.018).
We next examined the activity of our novel compound VU0465350 (VU5350) and 1LWY in cell culture experiments to determine whether they could inhibit phosphorylation of SMAD1/5/8 in C2C12 cells. Both with and without BMP stimulation, both compounds strongly inhibited SMAD phosphorylation in a dose-dependent fashion as measured by Western blot for p-SMAD1/5/8 (Fig. 1B). Each experiment was performed in triplicate and quantitated using ImageJ software [all P values were <0.01 except for 2 μM 1LWY (P = 0.018)]. Representative Western blots are shown.
BMP Antagonists Are Active In Vivo.
To determine whether the in vitro data showing LDN, DMH2, VU5350, and VU0469381 inhibited BMP signaling could be reproduced in vivo, we examined how each compound affected phosphorylation of SMAD1/5/8, a critical event in BMP signaling, in adult mouse liver. Western blot analysis for p-SMAD1/5/8 was performed in the liver after two doses of 6 mg/kg LDN, 2 mg/kg DMH2, 20 mg/kg VU5350, or 5 mg/kg VU0469381. Doses were chosen to correlate with our preliminary data demonstrating in vivo effects. Administration of each compound resulted in a decrease in p-SMAD (Fig. 2A). Compared with control, LDN led to a 24% decrease in p-SMAD (P = 0.04), VU5350 a 30% decrease in p-SMAD (P = 0.02), 1LWY a 28% decrease in p-SMAD (P = 0.04), and DMH2 a 39% decrease in p-SMAD (P = 0.01). Western blot was then used to determine the relative amounts of p-SMAD to SMAD (Fig. 2B) after PH. At baseline, 6 hours, and 48 hours after PH, LDN inhibited p-SMAD relative to SMAD compared with control animals (t = 0, P = 0.017; t = 6, P = 0.048; t = 48, P = 0.0053). Curiously, at 24 hours after PH, this result was reversed (P = 0.0041). Taken together, these results establish that the agents tested inhibit BMP signaling both in vitro and, in the liver, in vivo. The result at 24 hours may represent a physiologic rebound.
Fig. 2.

Specific inhibitors of ALK3 and ALK2 decrease SMAD phosphorylation in the liver and are therefore active in vivo. (A) Compared with control, LDN led to a 24% decrease in p-SMAD (P = 0.04), VU5350 a 30% decrease in p-SMAD (P = 0.02), 1LWY a 28% decrease in p-SMAD (P = 0.04), and DMH2 a 39% decrease in p-SMAD (P = 0.01) after two doses of the drug. (B) Western blot demonstrates that at baseline, 6 hours, and 48 hours after PH, LDN inhibits p-SMAD relative to SMAD compared with control animals (t = 0, P = 0.017; t = 6, P = 0.048; t = 48, P = 0.0053). At 24 hours after PH, this result is reversed (P = 0.004). *P < 0.05; **P < 0.01.
ALK3, but Not ALK2, Inhibitors Enhance Liver Regeneration.
Next it was necessary to determine whether these agents produced the predicted effect and enhanced liver regeneration in vivo. To test the effect of ALK3 and ALK2 inhibition on liver regeneration, mice received LDN, DMH2, VU5350, or VU0469381 in DMSO or pH-matched DMSO only as a control prior to PH. Hepatocyte proliferation rates were determined 48 hours after surgery (Fig. 3). Using LDN, mice receiving 0.6 mg/kg b.i.d. demonstrated no difference in hepatocyte proliferation at 48 hours compared with control mice (Fig. 3A). At 2 mg/kg, proliferation rates increased from the control rate of 13.7 to 22% (P = 0.014), and at 6 mg/kg, the rate rose to 31% (P = 0.001).
Fig. 3.
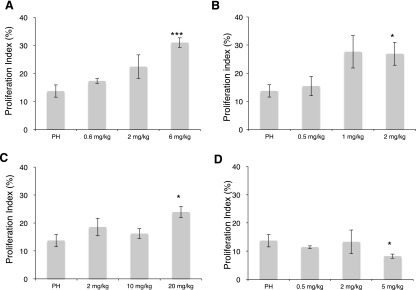
Inhibitors of ALK3 [(A) LDN, (B) DMH2, (C) VU5350] enhance hepatocyte proliferation 48 hours after PH, whereas an inhibitor of ALK2 [(D) VU0469381] does not. Low-dose LDN (0.6 mg/kg b.i.d.) did not affect hepatocyte proliferation compared with control mice (A). Increasing the dose to 2 mg/kg increased proliferation rates from the control level of 13.7 to 22% (P = 0.104). At 6 mg/kg, the rate rose to 31% (P = 0.001). Low-dose DMH2 (B) (0.5 mg/kg b.i.d.) did not increase hepatocyte proliferation, but higher dose (2 mg/kg) doubled hepatocyte proliferation from 13.7 to 26.9% (P = 0.027). Increasing VU5350 (C) from 2 to 20 mg/kg also increased hepatocyte proliferation to nearly double with the larger dose (P = 0.01 at 20 mg/kg). In contrast, VU0469381 (D) led to a decrease in hepatocyte proliferation at the highest dose (13.7 versus 8.2%, P = 0.035). *P < 0.05; ***P < 0.001.
We next performed similar experiments using DMH2 (Fig. 3B). Mice receiving 0.5 mg/kg b.i.d. exhibited absolutely, but not significantly, higher hepatocyte proliferation at 48 hours compared with control animals. Mice receiving 1 mg/kg DMH2 demonstrated a trend toward increased hepatocyte proliferation, from 13.7 to 27.6% (P = 0.056). Mice receiving 2 mg/kg DMH2 demonstrated doubling of hepatocyte proliferation, 13.7 versus 26.9% (P = 0.027).
Similarly, doses of VU5350 from 2 to 20 mg/kg increased hepatocyte proliferation 48 hours after PH to nearly double with the larger dose (Fig. 3C; P = 0.01 at 20 mg/kg).
To determine whether enhancement of liver regeneration was specific to ALK3 antagonism, we administered the ALK2-specific antagonist VU0469381 under the same schedule to mice prior to PH (Fig. 3D). In contrast to the results with ALK3 inhibitors, with escalating doses of 0.5, 2, and 5 mg/kg, there was not an increase in hepatocyte proliferation, and in fact, at the highest dose there was a statistically significant decrease in hepatocyte proliferation (13.7 versus 8.2%, P = 0.035).
Analyzed together, these results suggest that BMP inhibition of liver regeneration is mediated by ALK3 and not ALK2. This is consistent with our previous results (Do et al., 2012) and with other reports that BMP signaling is precisely modulated by different agonists and receptors that can have opposite effects (Piscione et al., 1997).
LDN Accelerates Restoration of Liver Mass over Time.
Based on these results, we chose to focus on LDN for a more detailed analysis of the effects of ALK3 inhibition on liver regeneration over a 1-week period after PH. BrdU incorporation as a measure of hepatocyte proliferation was measured in mice receiving LDN compared with control. At 48 hours after PH, treated mice demonstrated a >2-fold increase in hepatocyte proliferation compared with control mice (Fig. 4A; n = 4, P = 0.001), with a trend toward higher numbers at other time points. Representative immunohistochemistry for BrdU demonstrated higher proportions of BrdU+ hepatocyte nuclei in mice receiving LDN compared with control (Fig. 4B).
Fig. 4.

LDN enhances liver regeneration 48 hours after PH. (A) Administration of LDN around the time of PH increased hepatocyte proliferation over time. Treated mice demonstrated a more than 2-fold increase in hepatocyte proliferation compared with control mice (A) (P = 0.001), with a trend toward higher numbers at other time points. (B) Immunohistochemistry for BrdU injected 2 hours prior to sacrifices reveals increased nuclear staining (brown color) 48 hours after PH. (C) Administration of LDN around the time of PH leads to accelerated restoration of liver mass after PH. Higher proliferation rates in LDN-treated mice compared with control mice translated into more rapid restoration of liver mass after PH. Mice treated with LDN demonstrated a trend toward more rapid restoration of liver mass by 72 hours after PH, approximately 24 hours after peak BrdU incorporation. By 96 and continuing to 168 hours after PH, liver mass in the LDN-treated group was significantly higher compared with the control group (C) (*P = 0.011; **P = 0.001). (D) Western blot demonstrates LDN increased cyclin A2 protein levels compared with controls 36 hours after PH (t = 36 hours, P = 0.005). *P < 0.05; **P < 0.01.
Higher proliferation rates in LDN-treated mice compared with control mice translated into more rapid restoration of liver mass after PH. Mice treated with LDN demonstrated a trend toward more rapid restoration of liver mass by 72 hours after PH, approximately 24 hours after peak BrdU incorporation. By 96 and continuing to 168 hours after PH, liver mass in the LDN-treated group was significantly higher compared with the control group (Fig. 4C; P = 0.011; P = 0.001). Histologic sections were similar in both groups and did not show evidence of hepatocyte injury (data not shown).
To begin to identify the mechanism of this effect, we performed Western blot for cyclin A2, which plays an important role in the transition from mitosis to S phase (Malumbres and Barbacid, 2009). As expected, LDN produced an early rise in cyclin A2 at 36 hours after PH compared with control (Fig. 4D; P = 0.005).
Taken together these results suggest that after PH, LDN accelerates restoration of liver mass by increasing hepatocyte proliferation. Furthermore, these results suggest that we have translated an improved understanding of the basic mechanisms of liver regeneration into a therapy that has an effect in an in vivo system.
LDN Inhibits BMP Signaling In Vivo.
To begin to explore the mechanism of action of LDN, we focused on examining known in vitro effects of the compound in vivo. Previous studies demonstrated that in cultured Hep3B cells, IL-6 induces ID1 and hepcidin in a manner dependent on BMPs (Nemeth et al., 2004). Furthermore, in an in vivo model, PH induces ID1 expression in sinusoidal endothelial cells (Ding et al., 2010). We therefore tested whether LDN inhibits induction of ID1 and hepcidin in vivo (Fig. 5, A and B). Administration of LDN prior to PH inhibits the induction of ID1. Preoperative LDN has no effect on baseline ID1 mRNA levels. In contrast, 6 hours after PH, when control animals mice demonstrate a >3-fold increase in ID1, mice treated with LDN show only a modest increase (Fig. 5A; P = 0.037). After early induction in control animals, ID1 mRNA returns to baseline by 24 hours, then increases again at 36 and 48 hours in a manner not inhibited despite continued LDN administration.
Fig. 5.

LDN effects on ID1 and hepcidin induction and liver metabolic and synthetic function after PH as measured by real-time PCR. (A) Pretreating with LDN did not affect baseline ID1 mRNA levels. Six hours after PH, mice treated with LDN show a blunted increase compared with control mice (P = 0.037). (B) ID1 mRNA returns to baseline by 24 hours, then increases again at 36 and 48 hours despite continued LDN administration. Six hours after PH, control mice demonstrated a 4-fold induction of hepcidin mRNA, which was blocked completely by LDN (B) (P = 0.003). (C) LDN has no effect on bilirubin levels compared with controls after PH. (D) LDN has no effect on plasma RBP compared with controls after PH. *P < 0.05.
In a similar manner, 6 hours after PH, control mice demonstrated a 4-fold induction of hepcidin mRNA which was blocked completely by LDN (Fig. 5B; P = 0.003).
These results confirm specific in vivo activity of LDN as an inhibitor of BMP signaling and BMP-dependent events known to be important for liver regeneration.
LDN Does Not Compromise Liver Function or Drive Hepatocyte Proliferation in the Absence of PH.
To determine the effect of LDN on liver function and hepatocyte proliferation, we administered LDN for 5 days and measured plasma direct bilirubin and RBP4 compared with control mice. We chose RBP4 due to its short half-life, which makes it a sensitive indicator of liver synthetic function. We found no difference in either, suggesting that LDN does not impact bilirubin metabolism or synthetic function in this model (Fig. 5, C and D). Similarly, LDN did not increase hepatocyte proliferation in the absence of PH (data not shown).
LDN Affects Signaling Pathways Known to Be Important for Liver Regeneration.
Liver regeneration is controlled by a wide variety of molecules. To discover likely targets of LDN, we performed a broad survey of molecules known to play an important role during liver regeneration to determine how they are affected by LDN.
IL-6 is a critical mediator of liver regeneration. IL-6 signals through STAT3 phosphorylation, and mice lacking IL-6 demonstrate impaired liver regeneration (Cressman et al., 1995, 1996). To determine the effect of LDN on IL-6, we performed real-time PCR measurements of IL-6 mRNA after PH in mice administered LDN versus controls. Administration of LDN to unoperated mice for 2 days led to a nearly 4-fold increase in baseline IL-6 mRNA levels compared with controls not receiving LDN (P = 0.04) (Fig. 6A). To determine the effect of LDN on serum IL-6 levels, we performed direct measurement of IL-6 in serum (Fig. 6b). LDN not only increased the baseline level of IL-6, but also augmented the rise in IL-6 in response to hepatectomy (t = 0, P = 0.04; t = 6, P = 0.009; t = 24, P = 0.0038). We next examined STAT3 phosphorylation over time after PH (Fig. 6C). Twenty-four hours after hepatectomy, LDN increased p-STAT3 compared with mice not receiving LDN (P = 0.01). These results are consistent with a mechanism of action for LDN to increase levels of IL-6, resulting in increased STAT3 phosphorylation and enhanced liver regeneration.
Fig. 6.

LDN affects expression of genes in the IL-6/STAT3 pathway. (A) LDN increased baseline IL-6 mRNA levels 4-fold compared with control mice not receiving the drug at baseline (P = 0.004). (B) LDN increased plasma IL-6 levels compared with control and augmented the rise in IL-6 in response to hepatectomy (t = 0, P = 0.04; t = 6, P = 0.009; t = 24, P = 0.0038). (C) LDN increased p-STAT3 compared with controls 24 hours after hepatectomy (P = 0.01). *P < 0.05; **P < 0.01.
SOCS3 is highly induced after PH and limits cytokine activation during early liver regeneration. Liver-specific SOCS3-null mice demonstrate increased liver regeneration (Riehle et al., 2008). An inhibitor of SOCS3 induction, therefore, would be expected to enhance liver regeneration. To determine whether LDN inhibits SOCS3 transcription, real-time PCR was performed (Fig. 7A). Although SOCS3 mRNA is higher at baseline after LDN treatment in unoperated mice (P = 0.031), LDN significantly blunts the rise in SOCS3 seen after PH. Whereas control mice exhibited nearly 20-fold increase in the expression of SOCS3, LDN-treated mice exhibited <3-fold induction (P = 0.039) at 6 hours.
Fig. 7.

(A) LDN increases SOCS3 mRNA at baseline compared with controls (P = 0.031), but significantly blunts the post-PH rise in SOCS3 at 6 hours after PH (P = 0.039). (B) LDN decreases p53 expression 6 hours after PH to less than one-third the level of controls (P = 0.004). (C) LDN does not affect expression of BMP4 at baseline, but mildly decreases expression 36 hours after PH (P < 0.05). *P < 0.05; **P < 0.01.
Normal hepatocyte proliferation after PH requires repression of p53 (Stepniak et al., 2006). To determine if LDN affected this pathway, we examined transcription of p53 after PH (Fig. 7B). Since p53 promotes growth arrest, we predict that a proliferative stimulus provided by LDN would tend to repress p53 transcription. In the presence of LDN, p53 expression, in fact, is repressed 6 hours to less than one-third the level of controls after PH (n = 3, P = 0.004). At other times, p53 expression is unaffected by LDN. These results suggest a further explanation for the effects of LDN.
Next, we investigated whether LDN affected the expression of BMP4, which we have shown to be important for liver regeneration (Do et al., 2012). LDN did not affect BMP4 expression in the initial stages after injury, although by 48 hours after PH, LDN mildly decreased BMP4 expression (Fig. 7C; P < 0.05).
We next examined mRNA and protein levels of p38 and p21, important modulators of liver regeneration (Albrecht et al., 1998; Hayashi et al., 2003; Stepniak et al., 2006). mRNA analysis demonstrated LDN did not alter transcription of either. There was a modest decrease in p38 protein 48 hours after PH (P = 0.003), and a decrease in p21 protein (P = 0.04) 6 hours after PH with LDN (Fig. 8). There was no change in mRNA for p27, C/EBP-α, or c-JUN, other proteins that play important roles in liver regeneration (Flodby et al., 1996; Huang et al., 2006) (Fig. 9).
Fig. 8.

LDN did not affect mRNA levels of p38 or p21 compared with controls after PH. p38 was mildly decreased 48 hours after PH (P = 0.003), and p21 was decreased 6 hours after PH (P = 0.04). *P < 0.05; **P < 0.01.
Fig. 9.

LDN did not affect p27, C/ebp-α, or c-Jun mRNA compared with controls after PH.
Discussion
Our findings support the hypothesis that BMPs play an inhibitory role in liver regeneration, and suggest a target for clinical therapies. These results are consistent with and predicted by our previous findings that hepatocyte-specific loss of ALK3 enhances liver regeneration, viral-mediated expression of BMP4 inhibits regeneration, and viral-mediated expression of the BMP antagonist Noggin enhances liver regeneration in an ALK3-dependent manner (Do et al., 2012).
These results validate a methodology for producing ALK antagonists and provide evidence that these compounds are active in vivo. Given the overlap of ligands for ALK, BMP, AMP-activated protein kinase, and VEGF receptors, off-target effects may be common. The ability to generate a large variety of biologically active inhibitors is critical to generate pharmaceuticals with specific affinity for this pathway and minimal off-target effects.
Although possible off-target effects potentially limit the clinical utility of these compounds, a number of findings strengthen the attractiveness of these compounds for clinical development. First, the fact that a variety of ALK3 inhibitors have a similar effect in vivo validates the robustness of the discovery pipeline. Next, LDN does not seem to inhibit liver function as measured by bilirubin and RBP4, suggesting that ALK3 inhibition may not be toxic to the liver. In more severe models of injury, it will be interesting to determine if ALK3 inhibitors might improve liver function. The enhancement of hepatocyte proliferation with normal bilirubin levels suggests that the effect of LDN is not dependent on bilirubin levels.
LDN inhibition of ID1 increases is consistent with LDN as an inhibitor of ID1 transcription. Other reports demonstrate that LDN decreases levels of ID1 mRNA 2 hours after administration (Steinbicker et al., 2011). In the experiments described earlier, ID1 mRNA levels were measured 6 hours after administration, and this likely accounts for the discrepancy in the findings at this time point. Since ID1 is thought to be an inductive angiocrine signal from sinusoidal epithelial cells (Ding et al., 2010), our data showing that, at later times, LDN has no effect on ID1 transcription suggest that ID1 may play a signaling role that is required only for a short time.
In a PH model, ALK3 inhibition using LDN is associated with changes in transcription and protein expression of other molecules critical for liver regeneration. IL-6 is a cytokine necessary for liver regeneration and signals through STAT3; mice lacking IL-6 have impaired liver regeneration, and administration of IL-6 leads to massive liver growth (Zimmers et al., 2003). Our finding that LDN leads to an increase in baseline IL-6 mRNA followed by increased serum levels of IL-6 and increased STAT phosphorylation after PH suggests a mechanism of action for LDN. Taken together, our results suggest that LDN acts on ALK3, resulting in increased IL-6 levels leading to increased hepatocyte proliferation. The precise connection between ALK3 and IL-6 levels, however, remains unknown.
Loss of SOCS3 accelerates liver regeneration, and SOCS3 is thought to limit cytokine activation (Riehle et al., 2008). Our finding that LDN prevents an early rise in SOCS3 suggests an additional possible mechanism of action for LDN, although the link between LDN, ALK3, and SOCS3 remains unclear. Since the signaling pathways for SOCS3 and BMP are not known to overlap, it is likely that LDN affects SOCS3 via an indirect mechanism. Repression of p53 activity by c-Jun is essential for proper liver regeneration (Stepniak et al., 2006). Administration of LDN represses p53 transcription after PH, suggesting a further mechanism for the action of LDN.
Previous laboratory studies suggest a variety of compounds, including bile acids, hepatocyte growth factor, and TGF-β pathway members, can enhance liver regeneration (Michalopoulos et al., 1984; Gohda et al., 1988; Huang et al., 2006). In contrast to these findings that have not been translated into the clinic (Cataldegirmen et al., 2005; Rehman et al., 2011), the compounds discussed here may be more easily translated to human use. Recombinant BMP ligands are already approved for clinical applications relating to growth and repair (Brandoff et al., 2008), and animal studies have demonstrated tolerability of chronic pharmacologic BMP suppression (Derwall et al., 2012). In certain clinical settings relevant to the liver, short-term drug administration may be sufficient, and will minimize risks associated with long-term treatment.
There is a compelling clinical need for pharmacologic therapies to enhance liver repair. In this report, we suggest pharmacologic inhibition of BMP signaling may have therapeutic utility.
Acknowledgments
The authors dedicate this work to the memory of Dr. Kenneth Bloch, a brilliant friend and colleague.
Abbreviations
- ALK
activin receptor–like kinase
- BMP
bone morphogenetic protein
- BrdU
5-bromo-2-deoxyuridine
- C/EBP-α
CCAAT-enhancer binding protein-α
- DM
dorsomorphin
- DMH2
4-(2-(4-(3-(quinolin-4-yl)pyrazolo[1,5-a]pyrimidin-6-yl)phenoxy)ethyl)morpholine
- DMSO
dimethylsulfoxide
- ID1
inhibitor of DNA binding 1
- IL-6
interleukin-6
- LDN-193189/LDN
4-[6-[4-(1-piperazinyl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]-quinoline
- PCR
polymerase chain reaction
- PH
partial hepatectomy
- RBP4
retinol binding protein 4
- SOCS3
suppressor of cytokine signaling 3
- STAT3
signal transducer and activator of transcription 3
- TGF-β
transforming growth factor β
- VU0465350
7-(4-isopropoxyphenyl)-3-(1H-pyrazol-4-yl)imidazo[1,2-a]pyridine
- VU0469381
5-(6-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-3-yl)quinolone
Authorship Contributions
Participated in research design: Tsugawa, Oya, Masuzaki, Dib, Do, Yu, Bloch, Karp.
Conducted experiments: Tsugawa, Oya, Masuzaki, Ray, Kuramitsu, Frist, Karp.
Contributed new reagents or analytic tools: Engers, Yu, Bloch, Lindsley, Hopkins, Hong.
Wrote or contributed to the writing of the manuscript: Tsugawa, Oya, Masuzaki, Ray, Engers, Dib, Do, Kuramitsu, Ho, Yu, Bloch, Lindsley, Hopkins, Hong, Karp.
Footnotes
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grants 7R01-DK081387 and R01-DK0829710]; Vanderbilt University Medical Center; and the Foundation LeDucq.
Kenneth D. Bloch died during the preparation of this manuscript.
References
- Albrecht JH, Poon RY, Ahonen CL, Rieland BM, Deng C, Crary GS. (1998) Involvement of p21 and p27 in the regulation of CDK activity and cell cycle progression in the regenerating liver. Oncogene 16:2141–2150. [DOI] [PubMed] [Google Scholar]
- Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, Steele SJ, Roberts RR, Heier A. (2011) Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol 39:916–924. [DOI] [PubMed] [Google Scholar]
- Brandoff JF, Silber JS, Vaccaro AR. (2008) Contemporary alternatives to synthetic bone grafts for spine surgery. Am J Orthop 37:410–414. [PubMed] [Google Scholar]
- Cataldegirmen G, Zeng S, Feirt N, Ippagunta N, Dun H, Qu W, Lu Y, Rong LL, Hofmann MA, Kislinger T, et al. (2005) RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J Exp Med 201:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressman DE, Diamond RH, Taub R. (1995) Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology 21:1443–1449. [PubMed] [Google Scholar]
- Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. (1996) Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 274:1379–1383. [DOI] [PubMed] [Google Scholar]
- Cuny GD, Yu PB, Laha JK, Xing X, Liu JF, Lai CS, Deng DY, Sachidanandan C, Bloch KD, Peterson RT. (2008) Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg Med Chem Lett 18:4388–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS, Zapol WM, Bloch KD, Yu PB. (2012) Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler Thromb Vasc Biol 32:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B-S, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, et al. (2010) Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 468:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do N, Zhao R, Ray K, Ho K, Dib M, Ren X, Kuzontkoski P, Terwilliger E, Karp SJ. (2012) BMP4 is a novel paracrine inhibitor of liver regeneration. Am J Physiol Gastrointest Liver Physiol 303:G1220–G1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers DW, Frist AY, Lindsley CW, Hong CC, Hopkins CR. (2013) Synthesis and structure-activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of dorsomorphin: the discovery of ML347 as an ALK2 versus ALK3 selective MLPCN probe. Bioorg Med Chem Lett 23:3248–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flodby P, Barlow C, Kylefjord H, Ahrlund-Richter L, Xanthopoulos KG. (1996) Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein alpha. J Biol Chem 271:24753–24760. [DOI] [PubMed] [Google Scholar]
- Gohda E, Tsubouchi H, Nakayama H, Hirono S, Sakiyama O, Takahashi K, Miyazaki H, Hashimoto S, Daikuhara Y. (1988) Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J Clin Invest 81:414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, Hopkins CR, Lindsley CW, Hong CC. (2010) In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem Biol 5:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi E, Yasui A, Oda K, Nagino M, Nimura Y, Nakanishi M, Motoyama N, Ikeda K, Matsuura A. (2003) Loss of p27(Kip1) accelerates DNA replication after partial hepatectomy in mice. J Surg Res 111:196–202. [DOI] [PubMed] [Google Scholar]
- Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, Dong B, Huang X, Moore DD. (2006) Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 312:233–236. [DOI] [PubMed] [Google Scholar]
- Kogure K, Zhang YQ, Kanzaki M, Omata W, Mine T, Kojima I. (1996) Intravenous administration of follistatin: delivery to the liver and effect on liver regeneration after partial hepatectomy. Hepatology 24:361–366. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9:153–166. [DOI] [PubMed] [Google Scholar]
- Michalopoulos G, Houck KA, Dolan ML, Leutteke NC. (1984) Control of hepatocyte replication by two serum factors. Cancer Res 44:4414–4419. [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. (2001) Smad regulation in TGF-beta signal transduction. J Cell Sci 114:4359–4369. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. (2004) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 113:1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohe A, Keating E, Knaus P, Petersen NO. (2004) Signal transduction of bone morphogenetic protein receptors. Cell Signal 16:291–299. [DOI] [PubMed] [Google Scholar]
- Piscione TD, Yager TD, Gupta IR, Grinfeld B, Pei Y, Attisano L, Wrana JL, Rosenblum ND. (1997) BMP-2 and OP-1 exert direct and opposite effects on renal branching morphogenesis. Am J Physiol 273:F961–F975. [DOI] [PubMed] [Google Scholar]
- Rehman H, Sun J, Shi Y, Ramshesh VK, Liu Q, Currin RT, Lemasters JJ, Zhong Z. (2011) NIM811 prevents mitochondrial dysfunction, attenuates liver injury, and stimulates liver regeneration after massive hepatectomy. Transplantation 91:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, Bammler TK, Fausto N. (2008) Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med 205:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell WE, Coffey RJ, Jr, Ouellette AJ, Moses HL. (1988) Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci USA 85:5126–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwall RH, Robbins K, Jardieu P, Chang L, Lai C, Terrell TG. (1993) Activin induces cell death in hepatocytes in vivo and in vitro. Hepatology 18:347–356. [DOI] [PubMed] [Google Scholar]
- Steinbicker AU, Bartnikas TB, Lohmeyer LK, Leyton P, Mayeur C, Kao SM, Pappas AE, Peterson RT, Bloch DB, Yu PB, et al. (2011) Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 118:4224–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, Wagner EF. (2006) c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev 20:2306–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Babitt JL, Sidis Y, Chung RT, Lin HY. (2008) Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 111:5195–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 4:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmers TA, McKillop IH, Pierce RH, Yoo JY, Koniaris LG. (2003) Massive liver growth in mice induced by systemic interleukin 6 administration. Hepatology 38:326–334. [DOI] [PubMed] [Google Scholar]