

Abstract
Complement component 3 glomerulopathy (C3G) is a recently defined entity comprising of dense deposit disease and C3 glomerulonephritis. The key histological feature is the presence of isolated C3 deposits without immunoglobulins. Often masqueradng as some of the common glomerulonephritides this is a prototype disorder occurring from dysregulated alternate complement pathway with recently identified genetic defects and autoantibodies. We review the pathophysiology, clinical features, and diagnostic and treatment strategies.
Keywords: Alternate complement pathway, complement component 3 glomerulonephritis, complement component 3 glomerulopathy, complements, dense deposit disease
Introduction
Complement component 3 glomerulopathy (C3G) is an entity with glomerular deposits made solely of complement C3 and no immunoglobulin (Ig). It consists of dense deposit disease (DDD) and C3 glomerulonephritis (C3GN)[1] and was previously classified under membranoproliferative glomerulonephritis (MPGN).[2] C3G usually manifests with an MPGN pattern of injury predicated upon the severity of injury, the phase of the disease process (acute or chronic) and prior treatment. Other histological patterns include mesangial proliferative GN, diffuse proliferative GN and crescentic GN.[1] Currently all renal biopsies showing dominant C3 staining with little or no Ig are reclassified as C3G. The hallmark of this disorder is the dysregulation of the alternative complement pathway (AP) through inherited or acquired defects.
The Evolution of Complement Component 3 Glomerulopathy
Membranoproliferative glomerulonephritis is a pattern seen on light microscopy where there is enlargement and lobular accentuation of the glomerular tufts along with mesangial and endocapillary proliferation and, capillary wall thickening and double contouring. Immunofluorescence (IF) technique is used to identify deposits which may contain both Ig and complements or C3 alone. A new proposed definition of C3 dominance of at least two orders of magnitude more intensity on IF than any other immune reactant, is more inclusive and practical than ‘C3 only’ definition[3] leading to investigation of alternate complement pathway dysregulation in those patients. Electron microscopy (EM) locates these deposits in the sub endothelial (type 1), intramembranous (type 2 or DDD) or subendothelial, subepithelial with occasional intramembranous (type 3) regions. In DDD, the glomerular basement membrane (GBM) is transformed by extremely dark, ribbon-like electron-dense deposits located within the lamina densa. These deposits are also seen within the mesangium, tubular basement membrane and Bowman's capsule.[4]
Historical perspective
The association between GN and low serum levels of complement proteins was recognized a century ago when Gunn reported a markedly reduced serum hemolytic activity in two children with nephritis complicating scarlet fever.[5] This was followed by a revolution in complement biology in the 1960s. The ability to detect C3 in serum[6] and early reports of low serum C3 in patients with lupus nephritis[7] and MPGN[8,9] coincided with the development of an IF technique for identifying C3 deposits in renal sections.[10] The existence of a C3 nephritic “factor” (C3NeF) was inferred from the accelerated breakdown of C3 in normal human serum upon adding serum from a patient with persistent hypocomplementemic GN.[11] A rare glomerular lesion characterized by dense intramembranous deposits was recognized with transmission EM.[12] In the 1970s, DDD was described in conjunction with MPGN,[13] where predominant C3 glomerular deposition and low levels of serum C3 were attributed to the activation of the AP.[14] In the 1980s, a defect in the control of amplification of C3 convertase was found in five members of a family spanning three generations, implying a genetic basis for some cases of DDD.[15]
The entity of C3GN was first described in 2007 when Servais et al., identified a group of patients with a glomerular lesion they termed “primary GN with isolated C3 deposits” without electron-dense intramembranous deposits.[16]
Pathogenesis of complement component 3 glomerulopathy
The pathogenesis of C3G appears to be heterogeneous with insights mainly from familial and single case studies.[17,18,19] The presence of unaffected relatives with genetic abnormalities implies that a single hit may not be sufficient to cause disease. As a second hit, an inciting event like an infection, or an accumulation of mutations of AP causing uncontrolled “C3 tick over” activity influence the pathogenesis.[20] The reports by Licht and later by Habbig show that the specific sites of AP dysregulation determine the severity of disease and place both DDD and C3GN in a spectrum of the same disease process.[17,18] Our knowledge of the complement system is central to the understanding of the pathogenesis of C3G.
Complement system
The complement system is an integral part of innate immunity which “complements” the ability of antibody and phagocytic cells to clear pathogens from an organism. There is a fine balance between inhibition and activation of the complement system which if altered, can result in injury. Uncontrolled activation occurs with a gain of function of activators or with a loss of function of regulators. These changes are further modulated by mutations, polymorphisms and environmental triggers. (https://www.youtube.com/watch?v = ton3Npq1g-8)
Complement activation pathways
Complement activation occurs via proteolytic cleavage in three pathways: The classical, lectin and AP - all leading to the deposition of complement fragment C3b on the target. A major breakthrough in our understanding of the pathogenesis of C3G was the discovery of genetic mutations and deficiencies in complement proteins leading to dysregulation of the AP and isolated C3 deposits.[21] The activation of the classical and lectin pathways is triggered by antibodies binding to antigens or lectin attaching to sugar residues on the surface of pathogens. In contrast AP activation occurs spontaneously at a low level in the circulation due to hydrolysis of the internal thioester bond of the C3 molecule which is called the “C3 tick over”. This mechanism enables the complement system to monitor and probe the environment. C3 activation generates fragments called “anaphylatoxins” like C3a and C3b. These fragments deposit in large numbers on microbes and immune aggregates or necrotic tissue, leading to opsonization by phagocytes. In humans, complement factor B (CFB) protein is encoded by the CFB gene. Upon activation of the AP, it is cleaved by CF D yielding inactive chain, Ba and the active subunit Bb. The active subunit Bb is a serine protease that binds with C3b to form “AP C3 convertase” that amplifies C3 activation as a positive feedback [Figure 1].
Figure 1.
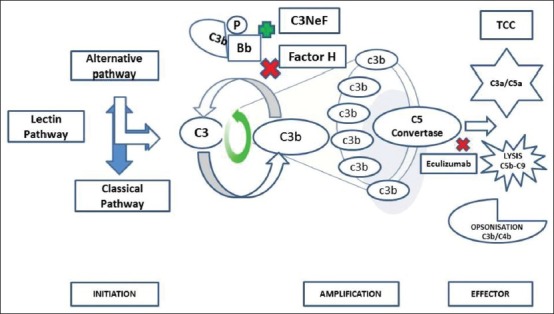
Complement activation cascade (C3NeF: Complement component 3 nephritic factor, TCC: Terminal complement cascade)
Amplification loop
The C3b amplification loop is a powerful means through which millions of C3b molecules are generated following initial activation of C3 [Figure 1]. The binding of additional C3b molecules to AP C3 convertase generates C5 convertase that activates C5, yielding anaphylatoxins C5a and C5b. C5b initiates terminal complement cascade (TCC) resulting in the formation of the membrane attack complex (MAC, C5b-9) [Figure 1]. The dense deposits of DDD contain components of the AP, including C3b, and the breakdown products inactive C3b (iC3b), C3dg and C3c, as well as components of the TCC.[22]
Regulators of the complement system
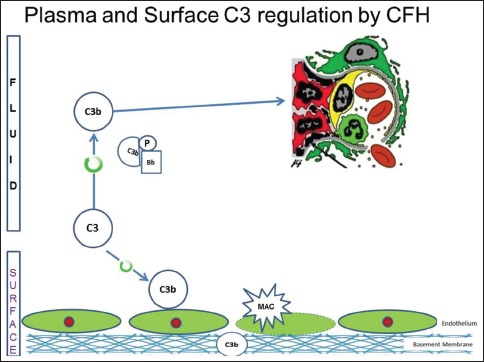
There are several regulatory proteins present both in the circulation (fluid phase) and on cell surfaces (surface phase) that inhibit AP. CF H (CFH), CF I (CFI), membrane cofactor protein (MCP), five CF H-related proteins (CFHR1-5) are some of the well-known regulators involved in the pathogenesis of C3G. CFH mainly competes with CFB for C3b binding, impeding the formation of the AP C3 convertase [Figure 2a]. CFH accelerates the decay of AP C3 convertase. It is also a cofactor for CF I (CFI)-mediated proteolysis of C3b. CFI protein was first isolated in 1966 in guinea pig serum. It regulates complement activation by cleaving cell-bound or fluid phase C3b generating iC3b and subsequently C3dg. Unlike C3b, iC3b cannot associate with factor B thereby preventing activation of C3b amplification loop. CFH has two terminals, a C terminal, the surface regulatory end and an N terminal, the C3 regulatory end. The mutation or deficiency of the C terminal end leads to atypical hemolytic uremic syndrome while that of the N terminal end leads to C3G. Animal studies have shown that different fragments of C3 attach to specific glomerular regions;[23] the inactive component iC3b deposits on the basement membrane as in DDD while C3b deposits in the mesangium as in C3GN [Figures 2a–c]. A defective uncontrolled C3 convertase activity would lead to DDD while that in C5 convertase activity would cause C3GN [Figure 2d].
Figure 2a.
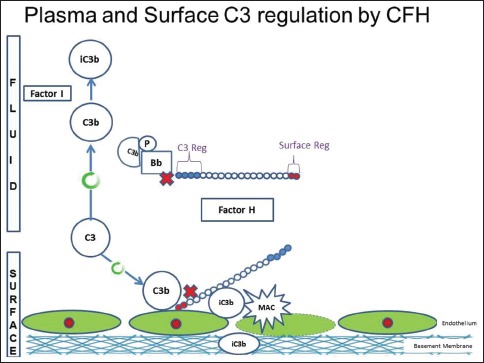
Complement factor H with its C terminal which is surface regulatory end and N terminal which is complement component 3 (C3) regulatory end. Defects in C terminal leads to atypical hemolytic uremic syndrome while defects in N terminal leads to C3glomerulonephritis. (Factor I and factor H - Regulators of complement system, C3b - Fragments from breakdown of C3, iC3b - Inactive fragment from breakdown of C3b, factor P-Regulator of complement cascade stabilizes C3 convertase, Bb - Active subunit of CFB, FLUID - Fluid or circulatory phase, SURFACE - Cell surface phase, MAC - Membrane attack complex, C3 Reg - C3 regulatory or N terminal end of factor H, surface reg - Surface regulatory or C terminal end of factor H)
Figure 2c.
Animal model with complement factor I deficiency with accumulation of C3b, with a predilection for mesangium. (Factor I and factor H - Regulators of complement system, C3b - Fragments from breakdown of C3, iC3b-Inactive fragment from breakdown of C3b, factor P - Regulator of complement cascade stabilizes C3 convertase, Bb-Active subunit of CFB, FLUID-Fluid or circulatory phase, SURFACE-Cell surface phase, MAC - Membrane attack complex, C3 reg-C3 regulatory or N terminal end of factor H, surface reg-Surface regulatory or C terminal end of factor H)
Figure 2d.
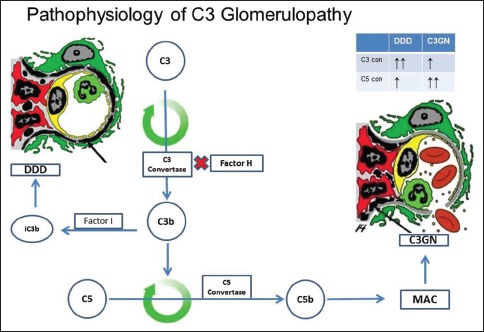
Uncontrolled complement component 3 convertase activity leads predominantly to dense deposit disease while that of C5 convertase activity causes complement component 3 glomerulonephritis (C3GN). (DDD - Dense deposit disease, C3GN-C3 glomerulonephritis, factor I and factor H - Regulators of complement system, C3b - Fragments from breakdown of C3, iC3b - Inactive fragment from breakdown of C3b, factor P-Regulator of complement cascade stabilizes C3 convertase, Bb-Active subunit of CFB, FLUID - Fluid or circulatory phase, SURFACE-Cell surface phase, MAC - Membrane attack complex, C3 reg-C3 regulatory or N terminal end of factor H, surface reg - Surface regulatory or C terminal end of factor H)
Figure 2b.
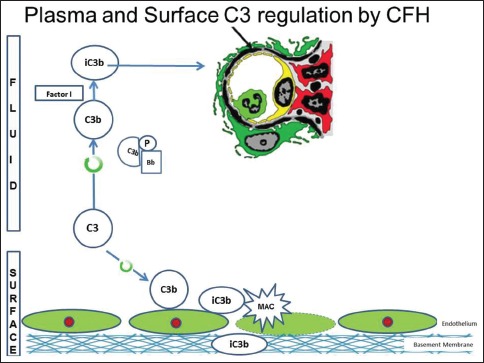
Animal model with complement factor H deficiency leads to accelerated complement component 3 (C3) convertase activity and formation of inactive C3b, which has a predilection for the glomerular basement membrane. (Factor I and factor H-Regulators of complement system, C3b - Fragments from breakdown of C3, iC3b-Inactive fragment from breakdown of C3b, factor P-Regulator of complement cascade stabilizes C3 convertase, Bb - Active subunit of CFB, FLUID - Fluid or circulatory phase, SURFACE - Cell surface phase, MAC - Membrane attack complex, C3 Reg - C3 regulatory or N terminal end of factor H, surface reg - Surface regulatory or C terminal end of factor H)
Role of complement factor H-related proteins
Gale et al., first reported CFHR mutation on 26 patients with a familial glomerulopathy called Cypriot Nephropathy.[19] This cohort has subsequently been expanded to include 91 patients in 16 families.[19,24] The family of CFHRs includes five plasma proteins, CFHR1-CFHR5, with concentrations ranging 5-50 μg/ml that are structurally and functionally related to factor H.[25] The genes encoding these CFHRs, CFHR1-CFHR5, likely originated by tandem duplication events from FH gene (CFH) leading to the formation of dimers and tetramers within CFHR genes. CFHR have analogous domains with factor H at the surface regulatory end and compete with factor H for binding with C3B, leading to familial C3GN. The internal duplication of CFHR5, leads to Cypriot Nephropathy,[19] while a unique hybrid CFHR3-1 protein, in excess leads to familial C3GN, reported from Ireland.[26] Interestingly, deletion of CFHR1 and CFHR3 genes, a common polymorphism in humans, is associated with a lower risk of age-related macular degeneration[27] and IgA nephropathy,[28] whereas generation of mutant FHRs by gene fusion or internal duplication events is associated with an increased risk for kidney diseases, like atypical hemolytic uremic syndrome and C3GN.[29,30,31] This epigenetic information provides a good insight of the pathophysiology, useful in the management of C3GN.
Autoimmune abnormalities
Autoantibodies targeting the activator or regulator components of C3 and/or C5 convertases of the AP have been identified. The first such autoantibodies is C3NeF,[32,33,34] which directly stabilizes the C3 activating complex of the AP and prevents the inhibitory actions of CFH. By blocking the normal inhibitory actions of CFH, C3NeFs prolong the half-life of C3 convertase from a few seconds to up to 60 min.[35] The massive C3 consumption leads to very low serum levels of C3 and an increased generation of C3 convertase and C5 convertase.
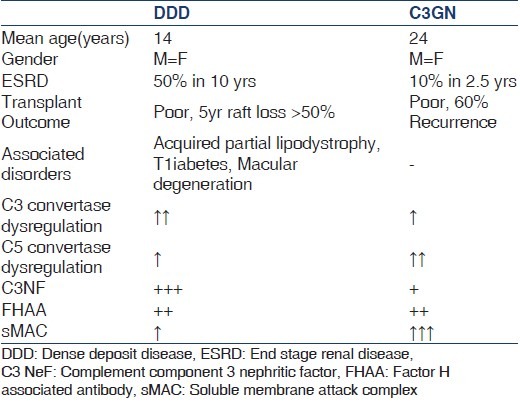
Complement component 3 nephritic factor are detected in the serum of approximately 80% of patients with DDD and 40-50% of patients with C3GN [Table 1]. As C3NeFs have been detected in healthy individuals[36] as well as in patients with other glomerular[37] and nonglomerular diseases,[38] the exact degree to which C3NeFs contribute to C3G remains undefined. In a report from Servais et al. on a large French cohort of patients with C3G, a fluctuation of C3NeF activity was seen in a third of the patients during follow-up and a normal range of serum C3 levels in approximately 40% of C3NeF-positive patients.[39] Furthermore, more than half of patients with C3G had mutations in the genes encoding CFH, CFI, or MCP identified in the complement pathway in addition to detectable C3NeF. This could explain why treatment directed solely at reducing or removing the C3NeF antibody have not shown consistent results[40,41,42] and also highlights the possibility of a two-hit hypothesis in the pathogenesis of C3G.
Table 1.
The main differences between DDD and complement component 3 glomerulonephritis
Prevalence of complement component 3 glomerulopathy
Dense deposit disease has an estimated prevalence of 2-3/million population and traditionally is viewed as a diagnosis of childhood and young adulthood.[43,44] The ratio of C3GN: DDD was 3:1. In the French cohort of 134 patients, 29 patients had DDD and 56 had C3GN of whom 71% were MPGN, and 29% were mesangial proliferative GN.[39]
Clinical features
Complement component 3 glomerulopathy is a rare renal entity that typically presents with proteinuria and hematuria in the face of low serum C3 levels (59% in DDD and 40% in C3 GN). The main differences between DDD and C3GN are shown in Table 1.
Dense deposit disease
Dense deposit disease usually presents in children between the ages of 5 and 15, although a recent series included more patients diagnosed in adulthood with a female preponderance.[45] It usually manifests with a nephritic-nephrotic syndrome, often preceded by an infection.[39,45] Biochemical evaluation reveals low serum C3 levels with normal levels of other complement components. C3NeF is found in 80% of these patients. They are occasionally accompanied by extra renal abnormalities like drusen in Bruch's membrane of retina[46,47] that develops at a much younger age and infrequently leads to loss of vision. Secondly, an association with Acquired Partial lipodystrophy, (Barraquer-Simons' or Dunnigan-Koeberling syndrome) which is characterized by loss of subcutaneous fat in the upper half of the body often predating renal clinical manifestations has been long described.[48] Monoclonal gammopathy causing complement dysregulation resulting in C3GN is seen in the older population without monoclonal deposits in kidney tissue.[49,50] There are reports of an increased risk of type 1 diabetes mellitus in families with DDD.[51] Progression to end-stage renal disease (ESRD) occurs in 50% patients already diagnosed with DDD in a decade especially in young women.[51] Spontaneous clinical remission is rare in DDD.[52] In 5 years after kidney transplantation 45% of allografts are lost to recurrence.[40,51,53]
Complement component 3 glomerulonephritis
Complement component 3 glomerulonephritis manifests as a membranoproliferative or mesangioproliferative pattern with isolated C3 deposits [Figure 3]. Many cases of MPGN type 3 and the so called resolving or persistent post streptococcal GN may actually be C3GN.[54,55] They are a pathogenetically heterogeneous group. There is a range of mutations in these patients as in Cypriot Nephropathy described below[19] and in familial C3GN from Ireland.[26] The common clinical manifestations are proteinuria, occasionally in the nephrotic range,[45] hematuria, and variable degrees of hypertension and azotemia. C3 levels were low in 40% and 45% had C3NeF. ESRD occurred in 10% of these patients by 2.5 years with 60% recurrence after renal transplantation.[45]
Figure 3.
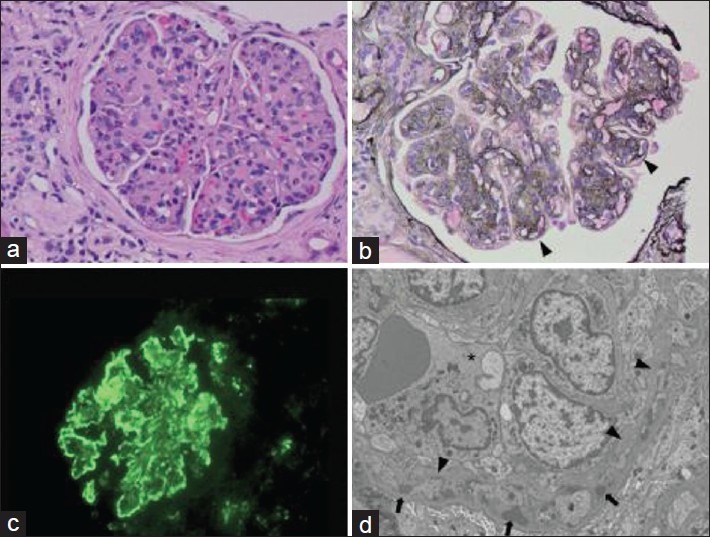
Pathological findings from a 53-year-old man with complement component 3 glomerulonephritis who presented with nephrotic range proteinuria, microscopic hematuria and renal failure. (a) Light microscopy shows accentuated lobularity and global glomerular hypercellularity (H and E) (b) Increased mesangial matrix with tram tracks in peripheral capillary loops (arrowheads) (c) Immunofluorescence showing strong granular capillary loop reactivity for complement component 3. (d) Electron microscopy with an endocapillary inflammatory cell (*), glomerular basement membrane duplication (arrowheads) and subendothelial electron-dense deposits (arrows)
Complement component 3 glomerulonephritis
Cypriot cohort
Cypriot (CFHR5) nephropathy is characterized by microscopic hematuria, turning macroscopic with intercurrent illness simulating IgA nephropathy, occurring in half of the affected individuals.[19] Proteinuria is reported in 38% of the original cohort, with a high rate of progression to chronic renal failure.[19] Serum C3 levels are usually normal, suggesting that excessive C3 activation occurs not in the circulation (as in DDD) but within the glomerulus.[56] Interestingly, among mutation carriers, men are by far more likely to progress to chronic renal failure and ESRD than women (80% vs. 21%). Ten patients with CFHR5 nephropathy had a successful transplantation,[24] with recurrence reported in one.[57]
Complement component 3 glomerulonephritis French cohort
From the French cohort (85 C3G patients; 29 with DDD and 56 with C3GN), we are afforded a view of the phenotypic differences within the C3GN disease family, this time from a broader geographical population and with an increased genetic diversity.[39] Greater than 60% of patients presented with microscopic hematuria (range 64.3-75.8%) and the urine protein was less in C3GN than in DDD (3.6 ± 3.3 vs. 5.6 ± 4.5 g). In a relatively young afflicted population, the gender distribution was equal between the two groups and the age at onset was statistically higher in C3GN than with DDD (30.3 ± 19.3 vs. 18.9 ± 17.7).[39] Low C3 level was seen in 62%. C3NeFs were more likely to be abnormal in DDD than in C3GN (86.4 vs. 45.3%), and importantly, mutations in the CFH and CFI genes were identified in 28%. The CFH H402 variant was significantly more common in DDD, and an additional “at-risk” MCP haplotype was found in a patient with C3GN. Two patients with recurrence of C3GN have also been described.[58]
Investigations
Complement component 3 glomerulonephritis typically would be diagnosed in a young patient with hematuria, proteinuria, a low C3 and normal C4 where renal histology shows an MPGN pattern. High C3NeF levels would suggest DDD, however their correlation with the course of disease is still unclear.[1] An algorithm for the investigation and diagnosis is shown in Figures 4a and b. It is presently recommended all such patients should have measurements of serum C3, C4, and factor H levels; screening for C3NeF, paraproteins and CFHR5 mutation.[1] The whole panel is however, available in just a few centers around the world.
Figure 4.
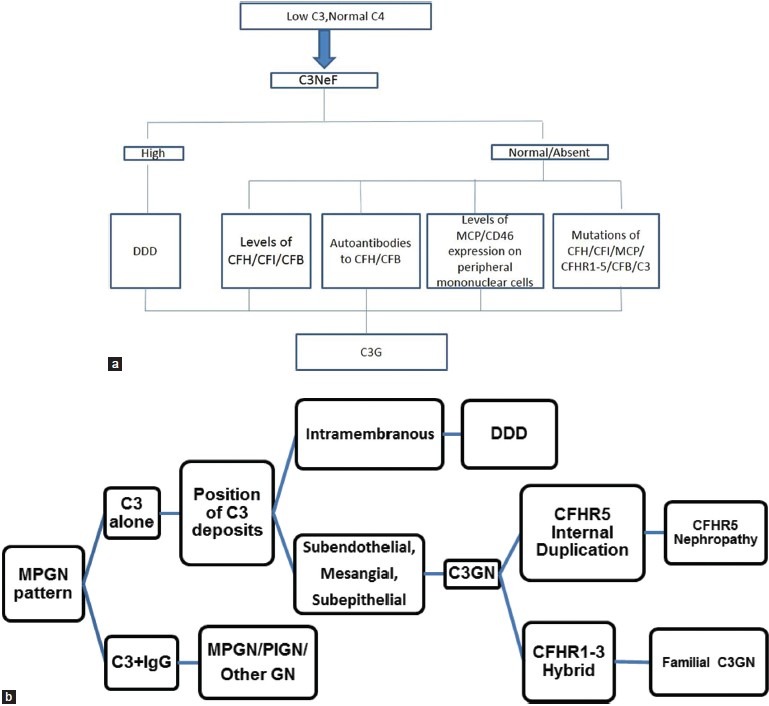
(a) Stepwise serological investigation for complement component 3 glomerulonephritis, (b) Stepwise biopsy diagnosis for complement component 3 glomerulonephritis
Prognosis of complement component 3 glomerulopathy
Servais et al., have reported a 10-year renal survival of 63.5% in a cohort of 134 patients.[39] In adults, the cumulative renal survival was lower in patients with DDD than in those with MPGN I and C3GN.
Treatment
There is no effective disease-specific therapy for C3GNs, and consequently treatment has included supportive measures, plasmatherapy and cellular immune suppression. Supportive measures with angiotensin converting enzyme inhibitors/angiotensin receptor blockers, and lipid lowering agents are recommended extrapolating their usefulness from other proteinuric renal diseases and also from the French cohort.[39,59,60]
Plasma exchange is the first-line therapy for factor H defects and elevated C3NeF. Treatment with Fresh frozen plasma/recombinant factor H is recommended for factor H deficiency. C3GN-due to autoantibodies to a complement protein may benefit from immunosuppressive therapy with steroids/Rituximab®. C3GN has been treated with Soliris® eculizumab which is a recombinant humanized monoclonal antibody marketed by Alexion, that inhibits the cleavage of C5 to C5a and C5b by C5 convertase [Figure 1]. In a series of six patients, which included three patients with DDD (including one patient with recurrent DDD in allograft) and three patients with C3GN (including two patients with recurrent C3GN in allograft). Genetic and complement function testing revealed a mutation in CFH and MCP in one each, C3 NeF and elevated levels of serum MAC in three subjects each.[61] After 12 months, subjects with elevated serum MAC levels responded to eculizumab more than those with normal serum MAC levels. Currently, a multicenter open label trial is underway for a new drug-soluble CR1 (CDX1135). CR1 is a regulator of complement activity along with factor H and MCP. It also mediates factor I dependent cleavage of iC3B to C3C and C3D and regulates C3 and C5 convertase activity. In mice made deficient in CF H and transgenic for human CR1, soluble CR1 therapy stopped AP activation, resulting in normalization of serum C3 levels and clearance of iC3b from GBMs.[62] Short-term data in a pediatric patient with ESRD demonstrated its safety and ability in normalizing activity of the terminal complement pathway.[62]
Renal transplantation in patients with C3G have high rates of recurrence in the allograft.[39] Because C3GN is a relatively new diagnostic category, long-term data on transplantation are lacking, but recurrences are likely to be at least as high as those reported with “idiopathic” MPGN type I up to 65% in some series.[63,64] Allograft survival may be similar to the 50% at 5 years rate reported for DDD.[40] Plasma exchange has been reported to be useful in the treatment of recurrent disease in some,[65] with the most success likely in cases of DDD or C3GN mediated primarily through defective factor H function. Eculizumab has been used in a few patients with recurrence of C3G in allograft.[61]
Conclusions
Complement component 3 glomerulopathy is an increasingly recognized entity arising from dysregulation of the complement system, usually manifesting as a nephritic nephrotic syndrome. It can mimick persistent postinfectious GN or present with synpharyngitic hematuria. The glomeruli show isolated deposits of C3 without Ig and varying patterns of proliferation. C3 is the prime target for further advances in therapy.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Pickering MC, D'Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: Consensus report. Kidney Int. 2013;84:1079–89. doi: 10.1038/ki.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Habib R, Gubler MC, Loirat C, Mäiz HB, Levy M. Dense deposit disease: A variant of membranoproliferative glomerulonephritis. Kidney Int. 1975;7:204–15. doi: 10.1038/ki.1975.32. [DOI] [PubMed] [Google Scholar]
- 3.Hou J, Markowitz GS, Bomback AS, Appel GB, Herlitz LC, Barry Stokes M, et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int. 2014;85:450–6. doi: 10.1038/ki.2013.340. [DOI] [PubMed] [Google Scholar]
- 4.Walker PD, Ferrario F, Joh K, Bonsib SM. Dense deposit disease is not a membranoproliferative glomerulonephritis. Mod Pathol. 2007;20:605–16. doi: 10.1038/modpathol.3800773. [DOI] [PubMed] [Google Scholar]
- 5.Gunn WC. The variation in the amount of complement in the blood in some acute infectious diseases and its relation to the clinical features. J Pathol Bacteriol. 1915;19:155–81. [Google Scholar]
- 6.Muller-Eberhard HJ, Nilsson U, Aronsson T. Isolation and characterization of two beta1-glycoproteins of human serum. J Exp Med. 1960;111:201–15. doi: 10.1084/jem.111.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seligmann M, Hanau C. Immuno-electrophoretic study of the blood of disseminated lupus erythematosus patients. Rev Hematol. 1958;13:239–48. [PubMed] [Google Scholar]
- 8.West CD, Northway JD, Davis NC. Serum levels of beta-1C globulin, a complement component, in the nephritides, lipoid nephrosis, and other conditions. J Clin Invest. 1964;43:1507–17. doi: 10.1172/JCI105027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West CD, McAdams AJ, McConville JM, Davis NC, Holland NH. Hypocomplementaemic and normocomplementaemic persistent (chronic) glomerulonephritis: Clinical and pathologic characteristics. J Pediatr. 1965;67:1089–9112. [Google Scholar]
- 10.Lachmann PJ, Muller-Eberhard HJ, Kunkel HG, Paronetto F. The localization of in vivo bound complement in tissue section. J Exp Med. 1962;115:63–82. doi: 10.1084/jem.115.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spitzer RE, Vallota EH, Forristal J, Sudora E, Stitzel A, Davis NC, et al. Serum C'3 lytic system in patients with glomerulonephritis. Science. 1969;164:436–7. doi: 10.1126/science.164.3878.436. [DOI] [PubMed] [Google Scholar]
- 12.Berger J, Galle P. Unusual change of the basal membranes of the kidney. J Urol Nephrol (Paris) 1962;68:116–22. [PubMed] [Google Scholar]
- 13.Mathew TH, Kincaid-Smith P. Membranoproliferative glomerulonephritis (MPGN) with dense deposits in basement membrane. ASN Abstr. 1971;5:51. [Google Scholar]
- 14.Marder HK, Coleman TH, Forristal J, Beischel L, West CD. An inherited defect in the C3 convertase, C3b, Bb, associated with glomerulonephritis. Kidney Int. 1983;23:749–58. doi: 10.1038/ki.1983.89. [DOI] [PubMed] [Google Scholar]
- 15.Levy M, Halbwachs-Mecarelli L, Gubler MC, Kohout G, Bensenouci A, Niaudet P, et al. H deficiency in two brothers with atypical dense intramembranous deposit disease. Kidney Int. 1986;30:949–56. doi: 10.1038/ki.1986.278. [DOI] [PubMed] [Google Scholar]
- 16.Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, et al. Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44:193–9. doi: 10.1136/jmg.2006.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Licht C, Heinen S, Józsi M, Löschmann I, Saunders RE, Perkins SJ, et al. Deletion of Lys224 in regulatory domain 4 of Factor H reveals a novel pathomechanism for dense deposit disease (MPGN II) Kidney Int. 2006;70:42–50. doi: 10.1038/sj.ki.5000269. [DOI] [PubMed] [Google Scholar]
- 18.Habbig S, Mihatsch MJ, Heinen S, Beck B, Emmel M, Skerka C, et al. C3 deposition glomerulopathy due to a functional factor H defect. Kidney Int. 2009;75:1230–4. doi: 10.1038/ki.2008.354. [DOI] [PubMed] [Google Scholar]
- 19.Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, McLean AG, et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794–801. doi: 10.1016/S0140-6736(10)60670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. 2012;8:634–42. doi: 10.1038/nrneph.2012.213. [DOI] [PubMed] [Google Scholar]
- 21.Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, et al. Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol. 2012;23:1229–37. doi: 10.1681/ASN.2011121186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sethi S, Gamez JD, Vrana JA, Theis JD, Bergen HR, 3rd, Zipfel PF, et al. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney Int. 2009;75:952–60. doi: 10.1038/ki.2008.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, et al. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest. 2008;118:608–18. doi: 10.1172/JCI32525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, Zavros M, et al. Familial C3 glomerulopathy associated with CFHR5 mutations: Clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol. 2011;6:1436–46. doi: 10.2215/CJN.09541010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Józsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol. 2008;29:380–7. doi: 10.1016/j.it.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 26.Malik TH, Lavin PJ, Goicoechea de Jorge E, Vernon KA, Rose KL, Patel MP, et al. A hybrid CFHR3-1 gene causes familial C3 glomerulopathy. J Am Soc Nephrol. 2012;23:1155–60. doi: 10.1681/ASN.2012020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–7. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 28.Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43:321–7. doi: 10.1038/ng.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sánchez-Corral P, González-Rubio C, Rodríguez de Córdoba S, López-Trascasa M. Functional analysis in serum from atypical Hemolytic Uremic Syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol. 2004;41:81–4. doi: 10.1016/j.molimm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Goicoechea de Jorge E, Caeser J, Malik TH, Patel MM, Colledge M, Johnson S, et al. Structural findings in complement factor H-related proteins unravel the pathogenesis of C3 glomerulopathy. Immunobiology. 2012;217:1205. [Google Scholar]
- 31.Neary J, Dorman A, Campbell E, Keogan M, Conlon P. Familial membranoproliferative glomerulonephritis type III. Am J Kidney Dis. 2002;40:E1. doi: 10.1053/ajkd.2002.33932. [DOI] [PubMed] [Google Scholar]
- 32.Ruley EJ, Forristal J, Davis NC, Andres C, West CD. Hypocomplementemia of membranoproliferative nephritis. Dependence of the nephritic factor reaction on properdin factor B. J Clin Invest. 1973;52:896–904. doi: 10.1172/JCI107254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallota EH, Götze O, Spiegelberg HL, Forristal J, West CD, Müller-Eberhard HJ. A serum factor in chronic hypocomplementemic hephritis distinct from immunoglobulins and activating the alternate pathway of complement. J Exp Med. 1974;139:1249–61. doi: 10.1084/jem.139.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West CD. Nephritic factors predispose to chronic glomerulonephritis. Am J Kidney Dis. 1994;24:956–63. doi: 10.1016/s0272-6386(12)81068-7. [DOI] [PubMed] [Google Scholar]
- 35.West CD, Witte DP, McAdams AJ. Composition of nephritic factor-generated glomerular deposits in membranoproliferative glomerulonephritis type 2. Am J Kidney Dis. 2001;37:1120–30. doi: 10.1053/ajkd.2001.24511. [DOI] [PubMed] [Google Scholar]
- 36.Gewurz AT, Imherr SM, Strauss S, Gewurz H, Mold C. C3 nephritic factor and hypocomplementaemia in a clinically healthy individual. Clin Exp Immunol. 1983;54:253–8. [PMC free article] [PubMed] [Google Scholar]
- 37.Frémeaux-Bacchi V, Weiss L, Demouchy C, May A, Palomera S, Kazatchkine MD. Hypocomplementaemia of poststreptococcal acute glomerulonephritis is associated with C3 nephritic factor (C3NeF) IgG autoantibody activity. Nephrol Dial Transplant. 1994;9:1747–50. [PubMed] [Google Scholar]
- 38.Sissons JG, West RJ, Fallows J, Williams DG, Boucher BJ, Amos N, et al. The complement abnormalities of lipodystrophy. N Engl J Med. 1976;294:461–5. doi: 10.1056/NEJM197602262940902. [DOI] [PubMed] [Google Scholar]
- 39.Servais A, Noël LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–64. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 40.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): An update. J Am Soc Nephrol. 2005;16:1392–403. doi: 10.1681/ASN.2005010078. [DOI] [PubMed] [Google Scholar]
- 41.Radhakrishnan S, Lunn A, Kirschfink M, Thorner P, Hebert D, Langlois V, et al. Eculizumab and refractory membranoproliferative glomerulonephritis. N Engl J Med. 2012;366:1165–6. doi: 10.1056/NEJMc1106619. [DOI] [PubMed] [Google Scholar]
- 42.Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, et al. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol. 2007;18:2447–56. doi: 10.1681/ASN.2007030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith RJ, Harris CL, Pickering MC. Dense deposit disease. Mol Immunol. 2011;48:1604–10. doi: 10.1016/j.molimm.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galle P, Mahieu P. Electron dense alteration of kidney basement membranes. A renal lesion specific of a systemic disease. Am J Med. 1975;58:749–64. doi: 10.1016/0002-9343(75)90631-2. [DOI] [PubMed] [Google Scholar]
- 45.Nasr SH, Valeri AM, Appel GB, Sherwinter J, Stokes MB, Said SM, et al. Dense deposit disease: Clinicopathologic study of 32 pediatric and adult patients. Clin J Am Soc Nephrol. 2009;4:22–32. doi: 10.2215/CJN.03480708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duvall-Young J, MacDonald MK, McKechnie NM. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: A histopathological report. Br J Ophthalmol. 1989;73:297–302. doi: 10.1136/bjo.73.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: Implications for the role of complement activation in drusen biogenesis. Eye (Lond) 2001;15:390–5. doi: 10.1038/eye.2001.142. [DOI] [PubMed] [Google Scholar]
- 48.Williams DG, Scopes JW, Peters DK. Hypocomplementaemic membranoproliferative glomerulonephritis and nephrotic syndrome associated with partial lipodystrophy of the face and trunk. Proc R Soc Med. 1972;65:591. [PMC free article] [PubMed] [Google Scholar]
- 49.Sepandj F, Trillo A. Dense deposit disease in association with monoclonal gammopathy of unknown significance. Nephrol Dial Transplant. 1996;11:2309–12. doi: 10.1093/oxfordjournals.ndt.a027156. [DOI] [PubMed] [Google Scholar]
- 50.Sethi S, Sukov WR, Zhang Y, Fervenza FC, Lager DJ, Miller DV, et al. Dense deposit disease associated with monoclonal gammopathy of undetermined significance. Am J Kidney Dis. 2010;56:977–82. doi: 10.1053/j.ajkd.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu DF, Moon M, Lanning LD, McCarthy AM, Smith RJ. Clinical features and outcomes of 98 children and adults with dense deposit disease. Pediatr Nephrol. 2012;27:773–81. doi: 10.1007/s00467-011-2059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marks SD, Rees L. Spontaneous clinical improvement in dense deposit disease. Pediatr Nephrol. 2000;14:322–4. doi: 10.1007/s004670050768. [DOI] [PubMed] [Google Scholar]
- 53.Angelo JR, Bell CS, Braun MC. Allograft failure in kidney transplant recipients with membranoproliferative glomerulonephritis. Am J Kidney Dis. 2011;57:291–9. doi: 10.1053/j.ajkd.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 54.Sethi S, Fervenza FC, Zhang Y, Zand L, Meyer NC, Borsa N, et al. Atypical postinfectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013;83:293–9. doi: 10.1038/ki.2012.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sandhu G, Bansal A, Ranade A, Jones J, Cortell S, Markowitz GS. C3 glomerulopathy masquerading as acute postinfectious glomerulonephritis. Am J Kidney Dis. 2012;60:1039–43. doi: 10.1053/j.ajkd.2012.04.032. [DOI] [PubMed] [Google Scholar]
- 56.Gale DP, Pickering MC. Regulating complement in the kidney: Insights from CFHR5 nephropathy. Dis Model Mech. 2011;4:721–6. doi: 10.1242/dmm.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vernon KA, Gale DP, de Jorge EG, McLean AG, Galliford J, Pierides A, et al. Recurrence of complement factor H-related protein 5 nephropathy in a renal transplant. Am J Transplant. 2011;11:152–5. doi: 10.1111/j.1600-6143.2010.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sethi S, Fervenza FC, Zhang Y, Zand L, Vrana JA, Nasr SH, et al. C3 glomerulonephritis: Clinicopathological findings, complement abnormalities, glomerular proteomic profile, treatment, and follow-up. Kidney Int. 2012;82:465–73. doi: 10.1038/ki.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brenner BM, Cooper ME, de Zeeuw D, Grunfeld JP, Keane WF, Kurokawa K, et al. The losartan renal protection study - rationale, study design and baseline characteristics of RENAAL (Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan) J Renin Angiotensin Aldosterone Syst. 2000;1:328–35. doi: 10.3317/jraas.2000.062. [DOI] [PubMed] [Google Scholar]
- 60.Ruggenenti P, Perna A, Gherardi G, Garini G, Zoccali C, Salvadori M, et al. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet. 1999;354:359–64. doi: 10.1016/S0140-6736(98)10363-X. [DOI] [PubMed] [Google Scholar]
- 61.Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7:748–56. doi: 10.2215/CJN.12901211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, et al. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–9. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lorenz EC, Sethi S, Leung N, Dispenzieri A, Fervenza FC, Cosio FG. Recurrent membranoproliferative glomerulonephritis after kidney transplantation. Kidney Int. 2010;77:721–8. doi: 10.1038/ki.2010.1. [DOI] [PubMed] [Google Scholar]
- 64.Karakayali FY, Ozdemir H, Kivrakdal S, Colak T, Emiroğlu R, Haberal M. Recurrent glomerular diseases after renal transplantation. Transplant Proc. 2006;38:470–2. doi: 10.1016/j.transproceed.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 65.Saxena R, Frankel WL, Sedmak DD, Falkenhain ME, Cosio FG. Recurrent type I membranoproliferative glomerulonephritis in a renal allograft: Successful treatment with plasmapheresis. Am J Kidney Dis. 2000;35:749–52. doi: 10.1016/s0272-6386(00)70025-4. [DOI] [PubMed] [Google Scholar]