

Abstract
Sturge-Weber syndrome (SWS), a rare sporadic neurocutaneous disease, is characterized by a congenital unilateral port-wine nevus affecting the area innervated by V1, ipsilateral leptomeningeal angiomatosis, and calcification in the occipital or frontoparietal region and glaucoma/vascular eye abnormality. Three types of SWS have been described in literature: Type I (classic) demonstrates facial and leptomeningeal angioma, often with glaucoma; type II has facial angioma and glaucoma, with no evidence of intracranial lesions; and type III (rarest) presents with only leptomeningeal angioma. Only a few cases of type III SWS have been reported. Here, we report a case of a seven-year-old boy with focal complex partial seizure, who was diagnosed with SWS without facial nevus. Recognition of this type of SWS is important, as our patient had been misdiagnosed and received inappropriate antiepileptic drugs for six years. We suggest that in the appropriate clinical scenario, the diagnosis of SWS without facial nevus should be considered before labelling idiopathic or cryptogenic localization-related epilepsy, and gadolinium-enhanced magnetic resonance imaging (MRI) should be done in clinically suspicious cases of SWS, without facial nevus.
Keywords: Facial nevus, gyral calcification, leptomeningeal angiomatosis, Sturge-Weber syndrome
Introduction
Sturge-Weber syndrome (SWS) is a rare sporadic neurocutaneous disease. It is characterized by the presence of a congenital unilateral port-wine nevus (flat capillary facial angioma) affecting the area innervated by the first sensory branch of the trigeminal nerve, ipsilateral leptomeningeal angiomatosis, and calcification in the occipital or frontoparietal region.[1] Some authors define SWS as a triad of the above two criteria and the third criteria as glaucoma and other vascular eye abnormalities.[2] SWS is classified into three different subtypes by Roach et al.[3] In type I (classic) SWS, the individual demonstrates facial angioma, leptomeningeal angioma, and glaucoma; in type II SWS, the patient has facial angioma and glaucoma, with no evidence of intracranial lesions; in type III SWS (rarest variant), the patient presents with only leptomeningeal angioma.[3] The incidence of type III SWS is not known, although few cases are reported.[4,5] In most SWS cases without facial angioma, epilepsy is the presenting symptom with leptomeningeal angiomatosis and calcification evident on initial imaging.[3]
Case Report
A seven-year-old boy with normal birth and development had presented with a focal complex partial seizure since nine months of age. His seizures were well controlled with Valproic acid and Clobazam for the initial three years. As his first MRI [Figure 1] was normal, the antiepileptic medicines were subsequently withdrawn. Since then, he has had breakthrough complex partial seizures despite being on medicine. For the last three years, he also had a hemicranial throbbing headache of moderate severity, lasting for few hours, associated with irritability and phonophobia, and intermittent vertiginous sensations, without imbalance when walking. His physical examination and neuropsychological assessment did not reveal any abnormalities; specifically, no facial port-wine stain was present. An ophthalmological examination did not reveal any abnormality, including glaucoma. For his breakthrough seizues, he was taking inadequate dosage of Oxcarbazepine and Lacosamide. His video electroencephalography (VEEG) revealed right posterior head region spike and wave discharges. A recent brain computed tomography (CT) [Figure 2] showed gyral calcification over the right parietal cortex. A recent MRI [Figure 3] showed focal cortical atrophy over the right parietal region and the gadolinium contrast revealed leptomeningeal angioma over the same region. The CT angiogram [Figure 4] did not show any venous or arterial malformation. We put the patient on an Oxcarbazepine and Clobazam combination, and on the last follow up six months later, the patient was seen to be seizure-free.
Figure 1.
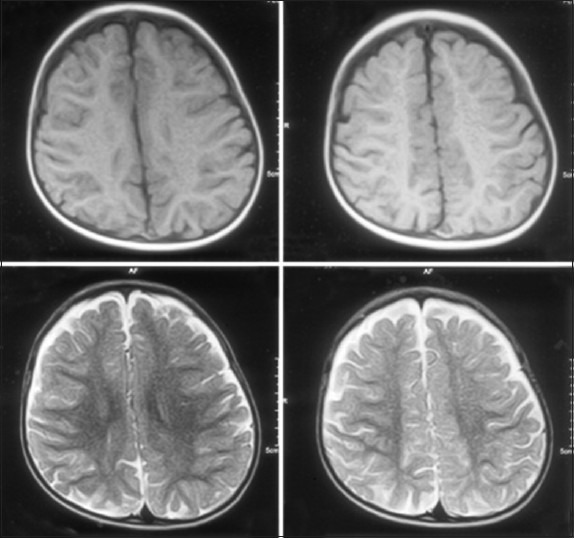
Axial T1, T2 images of an unenhanced MRI do not showany abnormality
Figure 2.
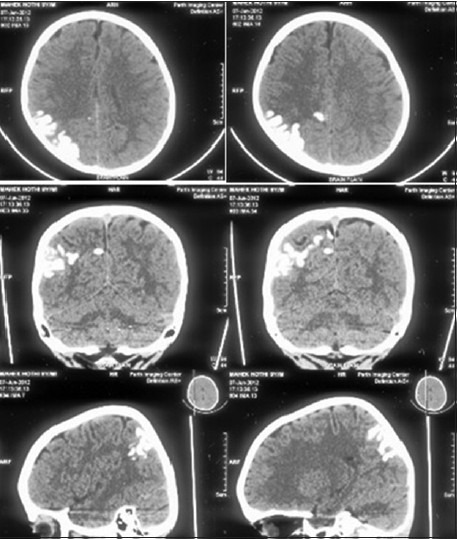
Axial, coronal, and sagittal images of a plain CT head show gyriform calcification in the right parietal cortex. Associated volume loss in the parietal lobe is noted
Figure 3.
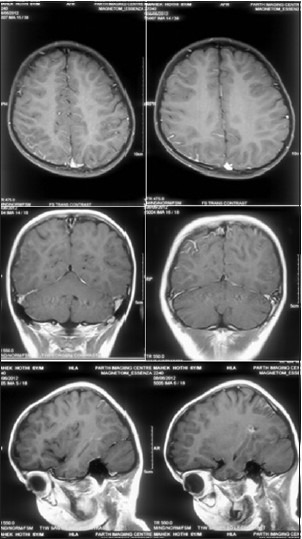
Axial, coronal, and sagittal images of a contrast-enhanced MRI reveal focal enhancement with the right parietal sulci
Figure 4.
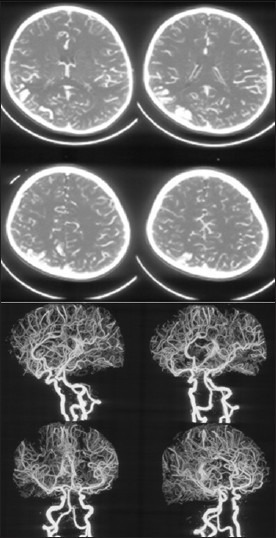
Axial CT angiogram source images and reconstructed CT angiogram images do not reveal any arteriovenous malformation
Discussion
Sturge-Weber syndrome typically presents at birth with facial angiomas. However, in SWS without facial nevus, the age of the onset is usually variable and marked by manifestation of other clinical features of the classic SWS, including epilepsy (75-90% of patients), mental retardation (50%) or hemiplegia/hemiatrophy (30%).[6] Glaucoma is present in almost 30% of classical SWS, but in SWS without facial nevus, glaucoma may be absent, because the nevus is not present in V1 distribution. Recurrent headaches and migraine-like attacks are other common manifestations in SWS patients, which were present in our patient.[7] Typical neuroradiological features of SWS include gyriform calcification in the posterior cerebral areas, focal cortical atrophy, and enlargement of the choroid plexus on the side of the pial angioma. These abnormalities are more evident on a CT scan, whereas, a brain MRI commonly reveals leptomeningeal enhancement after contrast infusion of gadolinium, and MR angiography detects abnormal veins draining into the deep venous circulation. In addition, functional neuroimaging investigations in SWS may show areas of hypometabolism and hypoperfusion around the vascular malformation.[8]
The pathogenesis of SWS is probably because of the abnormal persistence of an embryological venous plexus. The close proximity of the ectoderm, destined to form the facial skin, to the portion of the neural tube, destined to form the parieto-occipital area of the brain, may explain the association of a facial nevus and leptomeningeal angioma.[9] We propose that the abnormal persistence of the embryonic venous plexus only in the neural tube destined to form the parieto-occipital region, but not in the ectoderm, destined to form the facial skin, is responsible for this rarest (type III) form of SWS.
Our patient has only an imaging abnormality suggestive of SWS, with symptomatic epilepsy, without facial nevus, suggesting he has type III SWS, the rarest variant. The diagnosis of SWS in patients without facial nevus, who present with neurological manifestations typical of SWS, is made by the demonstration of a typical radiographic finding of calcification and leptomeningeal angioma.[4] Disorders known to produce cerebral calcifications, such as, celiac disease, encephalitis, purulent meningitis, ossifying meningoencephalopathy, and leukaemia, should be excluded in such cases. The necessity of recognizing this particular entity is to avoid misdiagnosis, which can often lead to incorrect management of epilepsy, including wrong choice of anti-epileptic medications.
We conclude that, in the appropriate clinical scenario, the diagnosis of SWS without facial nevus should be considered before labelling it as idiopathic or cryptogenic localization-related epilepsy of childhood, and a gadolinium-enhanced MRI should be done in clinically highly suspicious cases.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Bodensteiner JB, Roach ES, editors. SturgeWeber syndrome. Mt. Freedom, NJ: Sturge Weber Foundation; 1999. pp. 1–8. [Google Scholar]
- 2.Pascual-Castroviejo I, Díaz-Gonzalez C, García-Melian RM, Gonzalez-Casado I, Muñoz-Hiraldo E. Sturge-Weber syndrome: Study of 40 patients. PediatrNeurol. 1993;9:283–8. doi: 10.1016/0887-8994(93)90064-j. [DOI] [PubMed] [Google Scholar]
- 3.Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591–620. doi: 10.1016/s0031-3955(16)38367-5. [DOI] [PubMed] [Google Scholar]
- 4.Grururaj AK, Sztriha L, Johansen J, Nork M. Sturge-Weber syndrome without facial nevus: A case report and review of the literature. Acta Paediatr. 2000;89:740–3. doi: 10.1080/080352500750044133. [DOI] [PubMed] [Google Scholar]
- 5.Martínez-Bermejo A, Tendero A, López-Martín V, Arcas J, Royo A, Polanco I, et al. Occipital leptomeningealangiomatosis without facial angioma. Could it be considered a variant of Sturge-Weber syndrome? Rev Neurol. 2000;30:837–41. [PubMed] [Google Scholar]
- 6.Baselga E. Sturge-Weber syndrome. Semin Cutan Med Surg. 2004;23:87–98. doi: 10.1016/j.sder.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Sujansky E, Conradi S. Sturge-Weber syndrome: Age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49–58. doi: 10.1177/088307389501000113. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths PD. Sturge-Weber syndrome revisited: The role of neuroradiology. Neuropediatrics. 1996;27:284–94. doi: 10.1055/s-2007-973796. [DOI] [PubMed] [Google Scholar]
- 9.Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509–16. doi: 10.1177/08830738030180080701. [DOI] [PubMed] [Google Scholar]