

Abstract
The natural history of portal cavernoma cholangiopathy (PCC) is poorly defined and poorly understood. It develops early after acute portal vein thrombosis (PVT) if there is failure of recanalization. In PCC, the likelihood of progression of biliary abnormalities after 1 year is extremely low. The natural history of PCC is conveniently divided into asymptomatic and symptomatic stages. The majority of patients with PCC are asymptomatic and are detected incidentally on imaging. Limited data suggest that asymptomatic PCC is static or only slowly progressive in the initial stages. However, most workers agree that, overall, PCC is a slowly progressive disease. Symptomatic PCC represents a late stage in its natural history. Finding strictures with dilatation at cholangiography is associated with a higher risk of developing symptoms of PCC. Onset of symptoms is often precipitated by the development of biliary sludge or calculi and treating calculi usually relieves symptoms for prolonged periods of time. Clinical presentations include biliary pain, obstructive jaundice, acute cholangitis, acute cholecystitis, or other presentations of gallstone disease. Progressive liver dysfunction and secondary biliary cirrhosis can develop in a minority of patients.
Keywords: natural history, prognosis, extrahepatic portal venous obstruction, portal cavernoma cholangiopathy
Abbreviations: PCC, cavernoma cholangiopathy; PVT, portal vein thrombosis; PVCT, portal vein cavernomatous transformation; EHPVO, extra-hepatic portal venous obstruction; NCPH, non-cirrhotic portal hypertension; PH, portal hypertension; NCPF, non-cirrhotic portal fibrosis
Portal hypertension (PH) is usually due to liver cirrhosis but non-cirrhotic portal hypertension (NCPH) is a common entity in many parts of the developing world including India. Extra-hepatic portal venous obstruction (EHPVO) is the cause of portal hypertension in up to 40% of all adult patients1 and in 80–85% of children presenting with hematemesis in India.2 Thrombotic occlusion of the portal vein, whatever the cause is rapidly followed by compensatory responses aimed at re-establishing portal inflow to the liver, which includes partial or complete recanalization of the portal vein and the development of new collaterals around the occluded portal vein, which also engulf the bile ducts and gall bladder. The ensuing portal vein cavernomatous transformation (PVCT) results in the portal vein being eventually replaced by a “cavernoma”. Splenomegaly, esophageal and gastric varices, portal hypertensive gastropathy, and rarely ascites, may occur as complications of PHT due to PVCT. PVCT may also lead to changes in the biliary tree and pancreaticducts.3
Various terms have been used to describe the biliary changes in PVCT [See chapter Portal Cavernoma Cholangiopathy – History, Definition and Nomenclature, for detailed discussion]. The INASL working party agreed upon the consensus nomenclature “Portal Cavernoma Cholangiopathy”[PCC] as it emphasized the presence of a portal cavernoma with resultant abnormalities of the biliary tree including extra- and intra-hepatic bile ductal system, gall bladder and cystic duct.
Frequency and Clinical Presentation of Portal Cavernoma Cholangiopathy
Portal cavernoma cholangiopathy (PCC) has been reported mainly among patients with EHPVO. The frequency of PCC in patients with EHPVO is 81–100%.4–12 Although studies in the past have described PCC in patients with portal hypertension due to cirrhosis of liver (0–33%)8–15 and idiopathic portal hypertension or non-cirrhotic portal fibrosis (9–40%)8,15 this is misleading as the reported findings consisted of irregularities in bile ducts, mainly intrahepatic, which are secondary to parenchymal changes. An early report by Chandra et al16 described PCC in patients with liver cirrhosis and non-cirrhotic portal fibrosis (NCPF) as well as EHPVO. However, subsequent reports found that PCC occurred almost exclusively in patients with EHPVO with a portal cavernoma. Malkan et al8 reported cholangiographic changes suggestive of PCC in 9% patients with NCPF (dilated RHD) and 27% with liver cirrhosis (pruned intrahepatic branches only) while 85% of patients with EHPVO had typical PCC changes involving the extra-hepatic biliary tree with or without involvement of the intra-hepatic branches. They concluded that PCC changes occurred exclusively in patients with EHPVO with cavernoma. Kochhar et al17 reported changes on ERC in 12% of patients with liver cirrhosis and 8% with NCPF that were confined to intra-hepatic radicles, while changes were seen in 93% of patients with EHPVO, always involved the extra-hepatic biliary tree while the intrahepatic tree was involved in 60%. These observations prompt rethinking about whether changes solely involving intra-hepatic biliary radicles with a normal extrahepatic tree (Type II PB)14 are at all part of PCC and whether liver cirrhosis and NCPF can cause PCC in the absence of a portal cavernoma. In fact, recent series have not described type II PB in any patient.18–20 Careful review of previous reports of PCC with only intrahepatic changes in NCPF and liver cirrhosis and prospective studies documenting morphology and natural history in these patients are required before these patients are accepted as having PCC.
Most patients with these abnormalities are asymptomatic and are incidentally detected to have the presence of biliary abnormalities on cholangiography. Only a small percentage [5–38%] of patients present with symptoms of chronic cholestasis with or without biliary pain or acute cholangitis, related to the presence of biliary strictures or stones. Other than the age of the patient and duration of EHPVO, presence of gall stones and CBD stones are other risk factors for the causation of symptoms in patients with PCC [See chapter: Portal Cavernoma Cholangiopathy (PCC) – Clinical Characteristics, for detailed discussion].
Natural History Portal Cavernoma Cholangiopathy
The natural history of PCC is not known. The majority of patients (70%–95%) do not manifest with any symptoms of biliary obstruction. However, patients with symptomatic PCC are normally older than patients presenting with EHPVO by median of 8–14 years, which is suggestive of long-term obstruction12,13,15,18 and indicates that PCC is a progressive condition that develops late in the course of portal hypertension.
Stages in the Natural History of Portal Cavernoma Cholangiopathy
They are summarized in Table 1.
Table 1.
Stages in the Natural History of Portal Cavernoma Cholangiopathy.
| Stage | Portal cavernoma | Cholangiopathy | Liver biochemistry | Symptoms | Complications |
|---|---|---|---|---|---|
| Preclinical | Present | Absent | Normal | Absent | Absent |
| Asymptomatic | Present | Early changes | Normal or abnormal | Absent | Absent |
| Symptomatic | Present | Advanced changes | Abnormal | Present | Absent |
| Complicated | Present | Advanced changes | Abnormal | Present | Present |
Conventionally, the natural history of PCC has been described in two stages: asymptomatic and symptomatic PCC. Patients with asymptomatic PCC have no symptoms but have a portal cavernoma and ERC changes of early cholangiopathy (irregularity, serration, undulation, scalloping of ducts, smooth extrinsic nodular, spiral, or stenotic impressions and filling defects) with or without abnormal biochemical tests (elevated serum alkaline phosphatase level, minor elevation in serum bilirubin level). They do not require active management. Symptomatic PCC refers to patients with PCC who have ERC features of advanced cholangiopathy (ectasia, angulation, displacement, strictures and aneurysmal dilatation of the intrahepatic biliary tree), abnormal liver biochemistry and symptoms in the form of biliary pain, jaundice, cholestasis or cholangitis. It is noteworthy that in many patients, symptoms are triggered by the development of choledocholithiasis or sludge,19,20 which, in turn, is the result of biliary stasis from progressive choledochal compression or stenosis, and appropriate management of calculi often results in prolonged asymptomatic periods, till inexorable progression of biliary ductal changes results in the reappearance of symptoms.
Two other stages may usefully be added to this natural history. The portal cavernoma should be regarded as a preclinical stage of PCC, preceding the asymptomatic stage. This would help in identifying a high-risk group to be monitored for progression to PCC, for determining risk factors for progression and for studying possible interventions that may prevent or slow progression of PCC. At the other end of the spectrum, a complicated stage of PCC must be recognized to complete the clinical spectrum of this entity (Tables 1 and 2). This refers to patients with very advanced and extensive biliary ductal changes (e.g. long (>2 cm) or multifocal strictures, extrahepatic and/or intrahepatic strictures complicated by choledochal or intrahepatic calculi), biliopancreatic complications of PCC and associated biliary calculi (Table 2), patients where therapeutic options are limited due to extensive venous thromboses and patients with progressive liver dysfunction due to liver fibrosis, secondary biliary cirrhosis, and eventual progression to end-stage liver disease and, finally, those with significant co-morbidities. While symptoms and presentation are similar to those in the symptomatic stage, management is more difficult and challenging due to the complicated anatomy. The complicated PCC group is seen later in the natural history of PCC, in those who have had PCC for a decade or longer. These groups pose a formidable challenge since every intervention, whether endoscopic, surgical or percutaneous, is more morbid in them, complications are life-threatening and liver transplantation, if possible, may be the only salvage plan. Only a few patients have been successfully transplanted for PCC.21
Table 2.
Complicated Portal Cavernoma Cholangiopathy.
| PCC complications related to | ||
|---|---|---|
| Gallbladder stones | Bile duct stones | Biliary stricture |
| Acute cholecystitis | Multiple, large or impacted calculi | Long or multifocal strictures |
| Empyema | Mirizzi syndrome | Stones with strictures |
| Perforation | Intrahepatic lithiasis | Liver fibrosis |
| Gallbladder cancer | Bile duct injury during cholecystectomy | Secondary biliary cirrhosis |
Portal Cavernoma Cholangiopathy Does not Progress: ‘Early’ Natural History of Portal Cavernoma Cholangiopathy
The only full-length published study on natural history of PCC is from Spain.20 Sixty-seven consecutive patients with nontumoral, non-cirrhotic portal vein thrombosis (PVT) [22, acute PVT and 45 chronic PVT] were studied. Findings at magnetic resonance angiography and cholangiography (MRA/MRC) were classified into different degrees of severity: no abnormalities, grade I (irregularities or angulations of the biliary tree), grade II (indentations or strictures without dilation) and grade III (strictures with dilation; biliary dilation was considered when the intrahepatic duct was ≥4 mm or when the extrahepatic duct was ≥7 mm). After an initial episode of acute PVT, 73% patients (16 of 22 patients) developed PCC identified at MRA/MRC performed a median of 33 months (range 1–102) after acute PVT (4 grade I, 4 grade II and 8 grade III), whereas the remaining 27% (6 of 22) remained free of PCC after a median of 46 months (range 12–77) from the acute PVT episode. After an acute non-recanalized episode of PVT, PCC occurred mainly during the first year of follow-up and, once it occurred, progression of biliary abnormalities after 1 year was extremely low. Among the patients who had MRA/MRC performed during first year after acute PVT episode, 60% already had PCC. Of those patients who do not develop PCC during first year, 3/4th continued to be without PCC when MRA/MRC was performed a median of 36 months (range 14–53) after acute PVT episode. Eleven of 14 patients without grade III changes, had MRA/MRC repeated after a median follow-up of 43 months, and they showed no progression to grade III [Figure 1].
Figure 1.
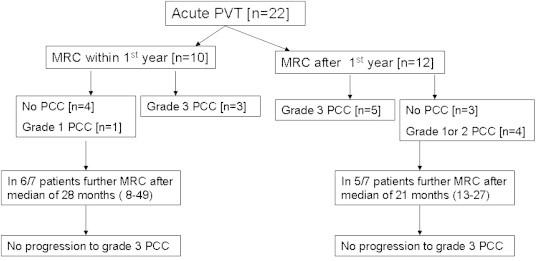
Magnetic resonance cholangiography (MRC) in patients followed after a diagnosis of acute portal vein thrombosis (PVT) [modified from Ref 16].
The rate of development of symptomatic PCC after an episode of acute PVT was low. Four of the 22 patients (18.2%) followed up after acute PVT developed symptoms of PCC a median of 42 months (range 11–120) after the diagnosis. The 5-year actuarial probability of developing symptoms of PCC after acute PVT was 19% and all patients had PCC grade III at the last MRA/MRC before developing symptoms.20
Among patients with chronic PVT, 80% (36 of 45 patients) had PCC at MRA/MRC [26, grade III, 19 less than grade III (9 no PCC; 3 grade 1 PCC and 7 grade 2PCC)]. Most patients without grade 3 PCC did not have progression of PCC lesions during median follow-up of 37 (11–77) months [Figure 2].
Figure 2.
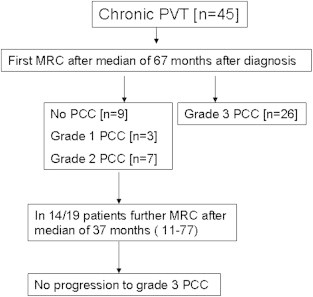
Magnetic resonance cholangiography (MRC) in patients followed after a diagnosis of chronic portal vein thrombosis (PVT) [modified from Ref 16].
The prevalence of symptomatic PCC in chronic PVT was 10/45 (22.2%). All patients with symptoms had grade III PCC. The 5-year and 10-year actuarial probability of symptomatic PCC after diagnosis of chronic PVT was 9% and 13%, respectively. Finding of grade III PCC at cholangiography was associated with a higher risk of developing symptoms of PCC.16
In this study, the first to follow a cohort of patients longitudinally from acute PVT to PCC, Llop et al19 have provided valuable insights into the natural history of PCC. They found that PCC occurred early during the course of acute PVT in Spain and symptomatic PCC was infrequent. They have argued that PCC is not a progressive disease and that MRC features that were noted in the first study did not progress over a median period of 36 months, with the longest period of observation being 8.5 years.
However, this study has a few limitations. Lesions were observed over a short period of time in relation to the known duration of natural history of PCC which may extend over 2–3 decades, so that the possibility of late progression is not ruled out in this study. It may be inappropriate to infer the full natural history of an entity spanning decades from observations over a shorter period. Further, although three grades of severity were described, even the most severe lesions were not as severe as reported in Indian series. Llop's cohort did not include any patients with advanced or complicated PCC i.e. those with very long or multifocal strictures, ‘fibrotic’ cavernomas, strictures with upstream calculi or marked ectasia. The shorter duration of follow-up and milder spectrum of disease in the Spanish study, when contrasted with the longer duration of follow-up and more severe disease reported in Indian studies, suggests that PCC is indeed a progressive disease with the Spanish series reflecting an earlier stage of disease that may progress to more advanced morphologic abnormalities over longer periods of time. Thus, this study may, at best, be termed ‘the early natural history of PCC’.
Portal Cavernoma Cholangiopathy is a Progressive Disease
The linkage between extent of anatomical abnormalities and duration of PVT was emphasized by Chevallier et al,22 who found stenoses, ductal narrowing or irregularities involving the CBD in three patients with extrahepatic PVT discovered a mean of 1.5 years earlier while changes involving both right and left intrahepatic bile ducts and CBD were present in six patients in whom extrahepatic PVT had been discovered a mean of 16.2 years earlier. Several studies from the Indian subcontinent support the view that PCC is indeed a progressive disease, albeit the rate of progression is slow, over a few decades rather than a few years. In an early series,13 median intervals between diagnosis of EHPVO and presentation with symptomatic PCC was 14.5 years (range, 0.8–23), supporting the notion that PCC is a slowly progressive condition. Another report,18 which had included patients with long strictures and strictures with upstream calculi, noted that the mean duration from diagnosis of EHPVO to onset of symptoms in PCC was 8 years (range 1–11 years), disregarding the period from acute PVT till detection of PVT. An unpublished study from GB Pant hospital, New Delhi, followed 14 asymptomatic patients with evidence of PCC. Biliary abnormalities were classified according to classification suggested by Chandra and Sarin as type I (involvement of extrahepatic bile duct), type II (involvement of intrahepatic bile ducts only), type IIIa (involvement of extrahepatic bile duct and unilateral intrahepatic bile duct) and type IIIb (involvement of extrahepatic bile duct and bilateral intrahepatic ducts).14 Repeat ERCP and LFTs were performed after a median of 29 months follow-up. During this interval, 5 patients (35%) became symptomatic for PCC. The median serum bilirubin and alkaline phosphatase levels were higher at the time of second ERCP. There was a significant progression of the ERCP features of portal biliopathy overtime compared to the baseline. More patients had type 3 changes on follow-up compared to baseline (45% vs. 70%, P°= 0.025). The percentage of patients with strictures also increased from 50% at baseline to 64% on follow-up (P = 0.010; unpublished data).
Reports of the occurrence of liver dysfunction and liver failure in patients with PCC provide another strand of evidence suggesting that PCC is a progressive condition. Overall, EHPVO is regarded as a progressive disease and hepatic dysfunction in the form of ascites, low serum albumin and prolonged prothrombin time have been reported in patients with EHPVO and, more so, in patients with prolonged portal hypertension.23,24 Development of ascites and derangements in liver function tests occurred with increasing age and increasing duration of disease. The deprivation of portal blood to the liver can influence the hepatic parenchymal functions.25–27 Lower MEGX clearance, comparable to that seen in the cirrhotic children, has been reported in EHPVO children.28 Among possible mechanisms that could contribute to hepatic dysfunction in EHPVO is the development and progression of ‘portal cavernoma cholangiopathy’. Whether histological changes progress concurrently with PCC remains to be seen in the long-term follow-up studies. Thus, it appears that PCC is progressive in nature and may lead to clinical and biochemical evidence of liver dysfunction.
Conclusion
The differences in natural history between the Spanish and Indian studies with respect to progression of PCC and development of symptoms [PCC occurred early during the course of acute PVT in Spanish study but had lower rate of symptom development, whereas Indian patients had higher incidence of symptomatic PCC] may be related to differences in age of onset [adult onset PVT in Spanish patients vs. childhood onset in Indian patients], etiological differences [secondary causes in Spanish patients vs. idiopathic in Indian patients] and prevalence of CBD stones [higher in Indian patients vs. Spanish patients]. However, more studies adhering to a uniform, sensitive, non-invasive monitoring protocol and with longer duration of follow-up are needed to uncover the natural history of PCC.
Conflicts of interest
All authors have none to declare.
References
- 1.Dilawari J.B., Chawla Y.K. Extrahepatic portal venous obstruction. Gut. 1988;29:554–555. doi: 10.1136/gut.29.4.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poddar U., Borkar V. Management of extra hepatic portal venous obstruction (EHPVO): current strategies. Trop Gastroenterol. 2011;32(2):94–102. [PubMed] [Google Scholar]
- 3.Bayraktar Y. Portal ductopathy: clinical importance and nomenclature. World J Gastroenterol. 2011 Mar 21;17(11):1410–1415. doi: 10.3748/wjg.v17.i11.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhiman R.K., Chawla Y., Vasishta R.K. Non-cirrhotic portal fibrosis (idiopathic portal hypertension): experience with 151 patients and a review of the literature. J Gastroenterol Hepatol. 2002;17:6–16. doi: 10.1046/j.1440-1746.2002.02596.x. [DOI] [PubMed] [Google Scholar]
- 5.Bayraktar Y., Balkanci F., Kayhan B., Ozenç A., Arslan S., Telatar H. Bile duct varices or “pseudo-cholangiocarcinoma sign” in portal hypertension due to cavernous transformation of the portal vein. Am J Gastroenterol. 1992;87:1801–1806. [PubMed] [Google Scholar]
- 6.Dilawari J.B., Chawla Y.K. Pseudosclerosing cholangitis in extrahepatic portal venous obstruction. Gut. 1992;33:272–276. doi: 10.1136/gut.33.2.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarin S.K., Bhatia V., Makwana U. Poratalbiliopathy in extrahepatic portal venous obstruction. Indian J Gastroenterol. 1992;11(suppl 1):A82. [abstract] [Google Scholar]
- 8.Malkan G.H., Bhatia S.J., Bashir K. Cholangiopathy associated with portal hypertension: diagnostic evaluation and clinical implications. Gastrointest Endosc. 1999;49:344–348. doi: 10.1016/s0016-5107(99)70011-8. [DOI] [PubMed] [Google Scholar]
- 9.Bayraktar Y., Balkanci F., Ozenc A. The ‘‘pseudo-cholangiocarcinoma sign’’ in patients with cavernous transformation of the portal vein and its effect on the serum alkaline phosphatase and bilirubin levels. Am J Gastroenterol. 1995;90:2015–2019. [PubMed] [Google Scholar]
- 10.Nagi B., Kochhar R., Bhasin D. Cholangiopathy in extrahepatic portal venous obstruction. Radiological appearances. Acta Radiol. 2000;41:612–615. doi: 10.1080/028418500127345992. [DOI] [PubMed] [Google Scholar]
- 11.Condat B., Vilgrain V., Asselah T. Portal cavernoma associated cholangiopathy: a clinical and MR cholangiography coupled with MR portography imaging study. Hepatology. 2003;37:1302–1308. doi: 10.1053/jhep.2003.50232. [DOI] [PubMed] [Google Scholar]
- 12.Sezgin O., Oguz D., Attintas E. Endoscopic management of biliary obstruction caused by cavernous transformation of the portal vein. Gastrointest Endosc. 2003;68:602–608. doi: 10.1067/s0016-5107(03)01975-8. [DOI] [PubMed] [Google Scholar]
- 13.Dhiman R.K., Chawla Y., Duseja A. Portal hypertensive biliopathy (PHB) in patients with extrahepatic portal venous obstruction (EHPVO) J Gastroenterol Hepatol. 2006;21:A504. [abstract] [Google Scholar]
- 14.Chandra R., Kapoor D., Tharakan A. Portal biliopathy. J Gastroenterol Hepatol. 2001;16:1086–1092. doi: 10.1046/j.1440-1746.2001.02562.x. [DOI] [PubMed] [Google Scholar]
- 15.Khuroo M.S., Yatoo G.N., Zargar S.A. Biliary abnormalities associated with extrahepatic portal venous obstruction. Hepatology. 1993;17:807–813. [PubMed] [Google Scholar]
- 16.Chandra R., Tharakan A., Kapoor D., Sarin S.K. Comparative study of portal biliopathy in patients with portal hypertension due to different etiologies. Indian J Gastroenterol. 1997;15(suppl 2):A59. [abstract] [Google Scholar]
- 17.Kochhar R., Nagi B., Singh K. Portal biliopathy in extrahepatic and intrahepatic portal hypertension. Gastrointest Endosc. 2001;53(3392):AB96. [Google Scholar]
- 18.Khare R., Sikora S.S., Srikanth G. Extrahepatic portal venous obstruction and obstructive jaundice: approach to management. J Gastroenterol Hepatol. 2005;20:56–61. doi: 10.1111/j.1440-1746.2004.03528.x. [DOI] [PubMed] [Google Scholar]
- 19.Oo Y.H., Olliff S., Haydon G., Thorburn D. Symptomatic portal biliopathy: a single centre experience from the UK. Eur J Gastroenterol Hepatol. 2009;21:206–213. doi: 10.1097/MEG.0b013e3283060ee8. [DOI] [PubMed] [Google Scholar]
- 20.Llop E., de Juan C., Seijo S. Portal cholangiopathy: radiological classification and natural history. Gut. 2011 Jun;60(6):853–860. doi: 10.1136/gut.2010.230201. [DOI] [PubMed] [Google Scholar]
- 21.Filipponi F., Urbani L., Catalano G. Portal biliopathy treated by liver transplantation. Transplantation. 2004;77:326–327. doi: 10.1097/01.TP.0000101795.29250.10. [DOI] [PubMed] [Google Scholar]
- 22.Chevallier P., Denys A., Novellas S., Schmidt S., Schnyder P., Bruneton J.N. Magnetic resonance cholangiography features of biliary abnormalities due to cavernous transformation of the portal vein. Clin Imaging. 2006;30:190–194. doi: 10.1016/j.clinimag.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 23.Rangari M., Gupta R., Jain M. Hepatic dysfunction in patients with extrahepatic portal vein obstruction. Liver Int. 2003;23:434–437. doi: 10.1111/j.1478-3231.2003.00879.x. [DOI] [PubMed] [Google Scholar]
- 24.Webb L.J., Sherlock S. The etiology, presentation and natural history of extrahepatic portal venous obstruction. Q J Med. 1979;48:627–639. [PubMed] [Google Scholar]
- 25.Fisher B., Lee S.U., Fisher S.R., Saffer E. Liver regeneration following portacaval shunt. Surgery. 1962;52:88–93. [PubMed] [Google Scholar]
- 26.Rozga J., Jeppsson B., Bergmark S. Hepatotrophic factors in liver growth and atrophy. Br J Exp Pathol. 1985;66:669–678. [PMC free article] [PubMed] [Google Scholar]
- 27.Mehrotra R.N., Bhatia V., Dabadghao P., Yaccha S.K. Extrahepatic portal vein obstruction in children: anthropometry, growth hormone and insulin-like growth factor I. J Paed Gastroenterol Nutr. 1997;25:520–523. doi: 10.1097/00005176-199711000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Misra A., Guptan R.C., Sarin S.K. Monoethylene glycine xylidide (MEGX). A sensitive non-invasive method to differentiate NCPF and child's a chronic liver disease. Ind J Gastroenterol. 1997;16(suppl 2):A-101. [Google Scholar]