

Abstract
Ribonucleotide reductases (RNRs) catalyze the conversion of nucleotides to deoxynucleotides in all organisms. Active E. coli class Ia RNR is an α2β2 complex that undergoes reversible, long-range proton-coupled electron transfer (PCET) over a pathway of redox active amino acids (β-Y122 → [β-W48] → β-Y356 → α-Y731 → α-Y730 → α-C439) that spans ∼35 Å. To unmask PCET kinetics from rate-limiting conformational changes, we prepared a photochemical RNR containing a [ReI] photooxidant site-specifically incorporated at position 355 ([Re]-β2), adjacent to PCET pathway residue Y356 in β. [Re]-β2 was further modified by replacing Y356 with 2,3,5-trifluorotyrosine to enable photochemical generation and spectroscopic observation of chemically competent tyrosyl radical(s). Using transient absorption spectroscopy, we compare the kinetics of Y· decay in the presence of substrate and wt-α2, Y731F-α2 ,or C439S-α2, as well as with 3′-[2H]-substrate and wt-α2. We find that only in the presence of wt-α2 and the unlabeled substrate do we observe an enhanced rate of radical decay indicative of forward radical propagation. This observation reveals that cleavage of the 3′-C–H bond of substrate by the transiently formed C439· thiyl radical is rate-limiting in forward PCET through α and has allowed calculation of a lower bound for the rate constant associated with this step of (1.4 ± 0.4) × 104 s–1. Prompting radical propagation with light has enabled observation of PCET events heretofore inaccessible, revealing active site chemistry at the heart of RNR catalysis.
Introduction
Managing the coupled translocation of protons and electrons is the keystone to energy storage and conversion.1−5 Biological systems have evolved to capitalize on proton-coupled electron transfer (PCET) to execute energy conversions efficiently and with exquisite control.6,7E. coli class Ia ribonucleotide reductase (RNR) maintains reversible8,9 PCET over ∼35 Å10−13 via a multistep, proton-coupled hopping mechanism and thus serves as a paradigm for the study of PCET in biology.14,15
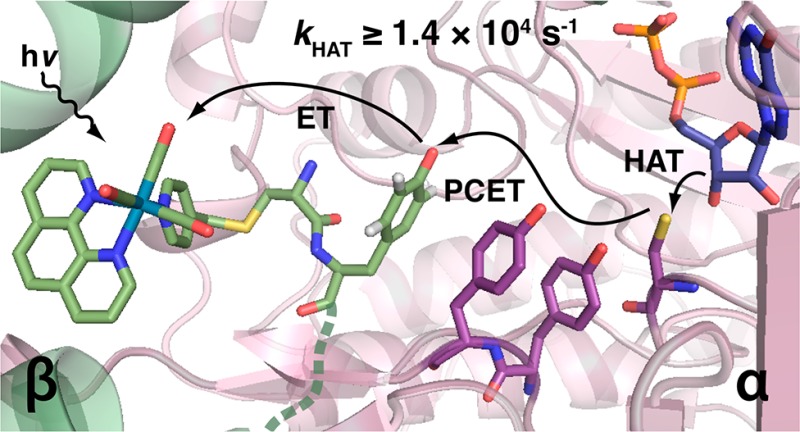
RNR catalyzes the conversion of nucleotides to deoxynucleotides, the bottleneck in de novo production of monomeric DNA building blocks.16 The active form of E. coli class Ia RNR is composed of two homodimeric subunits, α2 and β2 (Figure 1a).13,17 The active site is located in α, while the diferric-Y122· cofactor required to initiate active site chemistry is buried deep within β (Figure 1a). The rate-determining step in turnover consists of a conformational change triggered by substrate binding.8 This process initiates radical translocation by way of bidirectional PCET to β-Y122· in which a proton is transferred from a specific water molecule ligated to the diferric cluster,18 while the electron transfer (ET) results in oxidation of β-Y356.19 β-Y356· then oxidizes α-Y731 across the α/β subunit interface which subsequently oxidizes α-Y730 and, in turn, α-C439 in sequential PCET steps (Figure 1b).15,20,21 The C439· thiyl radical initiates active site chemistry by abstracting H· from the C3′-position of substrate.22,23 Multistep active site radical chemistry follows,16,24 resulting in reoxidation of C439 and reverse PCET along the same pathway of redox active amino acid residues to restore the radical resting state at β-Y122·.8,9
Figure 1.

(a) Pseudoatomic model of the active E. coli class Ia RNR α2β2 complex10,13,32 using PDB files 1MXR(33) and 4R1R.34 α2 (purple and violet) binds substrate (blue), and effector (slate), and crystallizes with the 20-mer peptide corresponding to the C-terminal tail of β. β2 (forest and light green) contains the μ-O-FeIII2/Y122· cofactor (Fe, brown; μ-O, red). (b) Illustration of photoinitiated radical transport over the PCET pathway in α (purple), and β (green) via excitation of [ReI] appended to β-S355C and with 2,3,5-F3Y replacing β-Y356. Residues 340–360 are disordered in the β2 structure, and the [ReI]-C355–F3Y356 fragment (constructed from the [ReI]-Br crystal structure)29 is placed in a hypothetical position (dashed green line) intended for illustrative purposes only.
Despite the fact that this multistep radical transport pathway presents RNR as an ideal system in which to examine biological PCET kinetics, rate-determining conformational changes have largely precluded such studies. In order to disentangle conformational gating from PCET kinetics, we have developed photochemical RNRs.25 Bypassing conformationally triggered reduction of the Y122· cofactor, we instead initiate PCET events midway through the RNR mechanism by photooxidation of Y356 (Figure 1b). Synchronization in this way has enabled detailed studies of photoinitiated substrate turnover,26 spectroscopic observation of photogenerated radicals,27 and direct measurement of radical injection rates into α2.28 For initial constructs, the β2 subunit was replaced by a short peptide encompassing the 20 C-terminal residues of the β2 protein. More recently, we have developed a β2 in which three mutations (C268S, C305S, and S355C) render a single cysteine residue surface-exposed, facilitating site-specific conjugation of a bromomethylpyridyl rhenium(I) tricarbonyl phenanthroline complex to position 355 ([ReI], Figure 1b).29 By measuring the [ReI]* excited-state lifetimes of [Re]-β2 and [Re]-Y356F-β2 under different conditions we have shown that this photochemical β2 is capable of reporting on Y356 oxidation.30 However, attempts to measure radical propagation kinetics directly were prevented by fast charge recombination and thus a low yield of photochemically produced radical.
We have now achieved direct observation of Y· propagation by circumventing the requirement for concomitant proton transfer during the generation of Y356·. Installation of an unnatural 2,3,5-trifluorotyrosine31 in place of Y356 ([Re]-F3Y356-β2, Figure 1b) has successfully boosted the yield of the photochemically generated radical, allowing spectroscopic resolution of downstream radical propagation kinetics by transient absorption (TA) spectroscopy. We have shown that this construct is competent for photoinitiated enzymatic turnover and present the first direct measure of Y· propagation kinetics through the active α2β2 RNR complex. By comparing the kinetics of Y· decay in the presence of substrate and wt-α2, Y731F-α2, or C439S-α2, as well as with [3′-2H]-substrate and wt-α2, we find that only in the presence of wt-α2 and substrate with natural isotopic abundance are radical decay kinetics enhanced. These data support that cleavage of the 3′-C–H bond of substrate by the transiently formed C439· thiyl radical is rate-limiting in forward PCET through α. We report a lower limit for the rate constant associated with this step of (1.4 ± 0.4) × 104 s–1. Unmasking PCET events in the active α2β2 RNR has provided a first direct measure of active site kinetics in the class Ia enzyme, yielding new evidence for a long-standing model and shedding light on the mechanism by which RNR maintains control and specificity during long-range PCET.
Materials and Methods
Materials
Wt-α2 (2000 nmol/mg/min) was expressed from pET28a-nrdA and purified as previously described.35 Glycerol stocks of Y731F-α2 and C439S-α2 were available from a previous study28 and were expressed and purified as wt-α2. All α2 proteins were prereduced prior to use.21 [5-3H]-cytidine 5′-diphosphate sodium salt hydrate ([5-3H]-CDP) was purchased from ViTrax (Placentia, CA). 3′-Deuterated cytidine 5′-diphosphate ([3′-2H]-CDP) was available from a previous study,36 in which it was synthesized as reported.22,23 Tricarbonyl(1,10-phenanthroline)(4-bromomethyl-pyridine)rhenium(I) hexafluorophosphate ([ReI]-Br) was available from a previous study.29E. coli thioredoxin (TR, 40 μmol/min/mg) and thioredoxin reductase (TRR, 1800 μmol/min/mg) were prepared as previously described.37,38 2,3,5-Trifluorotyrosine was synthesized enzymatically from pyruvate, ammonia, and 2,3,6-trifluorophenol with tyrosine phenol lyase as the catalyst.39 Assay buffer consists of 50 mM HEPES, 15 mM MgSO4, and 1 mM EDTA adjusted to the specified pH.
Preparation of [Re]-F3Y356-β2
Construction of C268S/C305S/S355C/Y356Z-pBAD-nrdB was achieved by site-directed mutagenesis using pBAD-nrdB as a template, and primers listed in the Supporting Information. E. coli TOP10 cells were cotransformed with the newly constructed nrdB plasmid and pEVOL-FnY-aaRS obtained from a previous study,31 plated on LB-agar plates supplemented with 100 μg/mL ampicillin and 35 μg/mL chloramphenicol and incubated at 37 °C overnight. A 1 mL culture containing the same antibiotics was inoculated with a single colony, incubated at 37 °C for 10 h, and then used to inoculate a small culture grown overnight at 37 °C. This starter-culture was used to inoculate 4 × 2L cultures of 2xYT at a 100-fold dilution. The cells were grown in the presence of 1.5 mM 2,3,5-F3Y until reaching an OD600 of 0.5, at which point the FnY-aaRS and nrdB genes were induced with arabinose (0.05% w/v). The cells were grown for an additional 4 h to a final OD600 of ∼1.5 and then harvested by centrifugation (3000 × g, 10 min, 4 °C). Yields of ∼2 g/L were obtained. Success of expression was assessed by 10% SDS-PAGE. The protein was purified by anion-exchange chromatography following a previously reported protocol,40 to give 10–15 mg per g of cell paste. Holo-S355C/2,3,5-F3Y356-β2 contained 0.6 Y·/β2 and exhibited no enzymatic activity. This variant is inactive due to the presence of a thiol/thiolate in the tricysteine mutant, which may be oxidized by F3Y·. This quenching process is not a concern in the photoRNR experiments because conjugation to [ReI] results in a thioether, which is difficult to oxidize. Purified material contained ≤5% ββ′ heterodimer resulting from the presence of protein truncated at position 356 (a consequence of the method used for unnatural amino acid incorporation). Treatment with hydroxyurea to quantitatively reduce Y122·, and labeling with [ReI]-Br were achieved as reported previously,29 to yield met-[Re]-F3Y356-β2 exhibiting >95% labeling efficiency.
Steady-State Emission pKa Titration
The steady-state emission intensity of 5 μM [Re]-F3Y-β2 in the presence of 1 mM CDP, 3 mM ATP, and 20 μM wt-α2 was measured in buffer containing 50 mM of either MES (pH 5.2–6.8) or HEPES (pH 7.0–7.6), 15 mM MgSO4, and 1 mM EDTA. Excitation at 315 nm using a 420 nm long-pass cutoff filter allowed spectra to be recorded over 450–650 nm, scanning 3 times per sample at a rate of 0.1 nm/s and detecting in 0.5 nm steps. Samples were held at 25 °C for 2 min prior to scan and throughout the duration of the measurement. Integrated emission intensity was plotted versus pH and fit to eq 1 (Figure S1). Here, I corresponds to integrated emission intensity and Imax and I0 correspond to I at pH 7.6 and 5.2, respectively.
| 1 |
Photochemical Turnover
Single turnover experiments under photochemical conditions were performed by mixing 10 μM each of met-[Re]-F3Y-β2 with wt-α2, Y731F-α2, or C439S-α2 in the presence of 0.2 mM [5-3H]-CDP (specific activity 26,700 cpm/nmol), 1 mM ATP, and 10 mM Ru(NH3)6Cl3 in assay buffer at pH 7.6. Samples were placed in a 4 mm × 4 mm quartz cuvette and held at 25 °C under illumination for 10 min with white light powered at 800 W (35 V and 24 A DC) in conjunction with a 313 nm long-pass cutoff filter. Quantitation of radioactive products by scintillation counting and confirmation of product identity (Figure S2) were performed as previously described.30 Equivalents of dCDP per α2 produced in the presence of [Re]-F3Y-β2 were normalized to dCDP production in the presence of wt-β2 under the photochemical conditions, which produces 1.2 equiv/α2 of a theoretical maximum of 4 equiv/α2. Photochemical activity for 3–6 independent samples was determined, and the standard deviation associated with both wt-β2 and [Re]-F3Y-β2 dCDP production was propagated during normalization.
Nanosecond Laser Flash Photolysis
Samples were prepared in a total volume of 750 μL and recirculated through a 1 cm path length flow cell to reduce sample decomposition. An inline filter (Acrodisc 13 mm 0.2 μM Supor Membrane, Pall Corporation) was used to collect solid photoproducts. Optical long-pass cutoff filters (λ > 375 nm) were used to filter probe light before detection to remove scattered 355 nm pump light. Samples contained 50 μM [Re]-F3Y-β2; 75 μM of wt-α2, Y731F-α2, or C439S-α2; 3 mM ATP; and either 1 or 0.5 mM CDP or [3′-2H]-CDP.
Laser experiments were performed using a system that has previously been described.28 Single wavelength kinetics data were collected at 412.5 nm using slit widths corresponding to 0.7 nm resolution and recorded over 1000 laser shots for each sample. TA spectra were collected over 500 four-spectrum sequences where two of four conditions result in exposure to the pump beam.
Lifetimes were obtained by averaging three sets of decay traces from three unique samples of a single protein preparation (both wt-α2 and [Re]-F3Y-β2; expression, purification, and [Re]-labeling) (Trial 1 in Table S1), according to eq 2:
| 2 |
Lifetimes from the decay traces for another three sets of unique samples were then obtained using a second protein preparation (Trial 2 in Table S1). Table 1 lists the propagated error for the six measurements across the two trials with weighted averages derived from error associated with the fit for each data set within a trial, compounded with the standard deviation between the two trials. An exemplary data set, along with fits to eq 2, and associated residuals analysis are included in Figure S3.
Table 1. Y· Lifetimes for α2-Variant/Substrate Combinations.
| α2-variant | substrate | τ/μsa |
|---|---|---|
| wt | CDP | 18 (1) |
| Y731F | CDP | 24 (1) |
| C439S | CDP | 25 (2) |
| wt | [3′-2H]-CDP | 26 (1) |
Photogenerated Y· lifetimes monitored by TA spectroscopy at 412.5 nm and fit to monoexponential decay from 3 to 76.5 μs (Figure S3). Weighted averages represent duplicate sets of three measurements each, on two separate protein preparations of [Re]-F3Y-β2 and wt-α2. Samples contain 50 μM met-[Re]-F3Y-β2, 75 μM α2 variant, 1 mM or 0.5 mM CDP or [3′-2H]-CDP, 3 mM ATP, 10 mM Ru(NH3)6Cl3, and assay buffer at pH 8.2.
Results
pKa of 2,3,5-F3Y356 within the α2β2 Complex
Photochemical generation of an observable population of F3Y356· is enhanced when the amino acid resides in its deprotonated state. This enhanced radical generation is a direct consequence of the ability to generate the radical by removal of only an electron as opposed to removal of an electron and proton (i.e., PCET). To determine the optimum pH for photochemical radical generation, we measured the pKa of F3Y356 within the α2/[Re]-β2 complex. This measurement was accomplished by monitoring the steady-state emission from the rhenium complex excited state ([ReI]*), which is quenched much more effectively when the adjacent F3Y is deprotonated.41 Plotting emission intensity as a function of pH revealed a pKa of 6.2 ± 0.1 (Figure S1). This value is in line with the pKa of 6.4 measured for the free amino acid derivative,42 particularly in light of the fact that a positively charged [ReI] complex is present. The pKa measured here is also in line with the value of 6.8 predicted by titrations using 3-NO2Y356-β2, in which, by comparison with free NO2Y, the ΔpKa due to perturbations arising from the protein environment at position 356 was inferred.40 All subsequent spectroscopy was conducted at pH 8.2 such that ∼99% of F3Y356 is deprotonated.
Observation of Transient Y·
In order to observe photogenerated Y·’s, charge recombination within the initially formed charge-separated state ([Re0]-F3Y356·-β2) must be prevented. Therefore, we applied flash-quench methodology by including an excess of Ru(NH3)6Cl3 in reaction mixtures. Bimolecular quenching of the [ReI]* excited state furnishes the [ReII] complex via reduction of Ru(NH3)63+ to Ru(NH3)62+. This [ReII] species is capable of driving rapid oxidation of trifluorotyrosinate (Figure 2a). Figure 2b shows the TA spectrum of Y· collected 3 μs after excitation, at which point nearly all photochemistry is completed and F3Y· and/or Y· are the only transient species contributing to the absorption feature centered at 412 nm. The overall photochemical yield of Y· is ∼1.9% (calculation included in the Supporting Information). Experiments were performed at protein concentrations such that >97% of [Re]-F3Y356-β2 is complexed to α2 (based on the previously reported KD of 0.7 ± 0.1 μM, measured for [Re]-β2 binding to α2 under the same conditions).30 The control experiment performed with [Re]-Y356F-β2 (black, Figure 2b) shows a minor TA signal, which we have previously observed and ascribed to off-pathway generation of Y·.30 In the absence of an adjacent redox active amino acid, off-pathway radical generation is maximized. Thus, this spectrum (black, Figure 2b) represents a maximum possible contribution to the observed signal and is likely greater than any off-pathway contributions operative when F3Y or Y is present.
Figure 2.
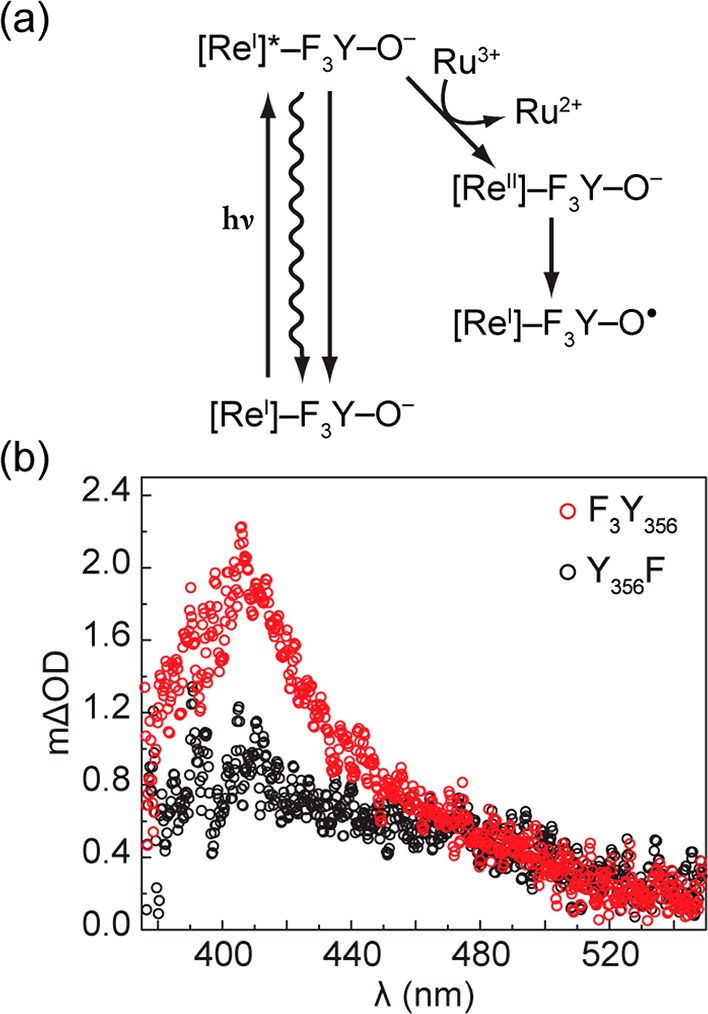
(a) Scheme describing the photochemistry of F3Y· generation; (b) TA spectra collected 3 μs after 355 nm excitation of 50 μM [Re]-F3Y356-β2 (red) or [Re]-Y356F-β2 (black), and 75 μM α2, 1 mM CDP, 3 mM ATP, and 10 mM Ru(NH3)6Cl3, in assay buffer at pH 8.2.
Photochemical Competence for Turnover
To evaluate the relevance of photochemically generated Y· in RNR chemistry, we sought to verify chemical competence for enzymatic turnover via photochemical initiation. Steady-state illumination under single turnover conditions in the presence of radiolabeled substrate ([5-3H]-CDP), effector (ATP), Ru(NH3)6Cl3, and α2 allows quantitation and identification of photogenerated products.30,41,43 Of note, the μ-O-FeIII2/Y122· cofactor of [Re]-F3Y356-β2 has been reduced with inhibitor hydroxyurea to form met-[Re]-F3Y356-β2; thus the normal mechanism for turnover is not viable with this construct. Photochemical production of dCDP is 9 ± 4% that of wt-β2 under identical conditions (Figures 3 and S2), which produces 1.2 dCDP/α2 out of a theoretical maximum of 4. Dark controls and reactions with Y731F-α2 and C439S-α2 variants produce negligible amounts of product.
Figure 3.
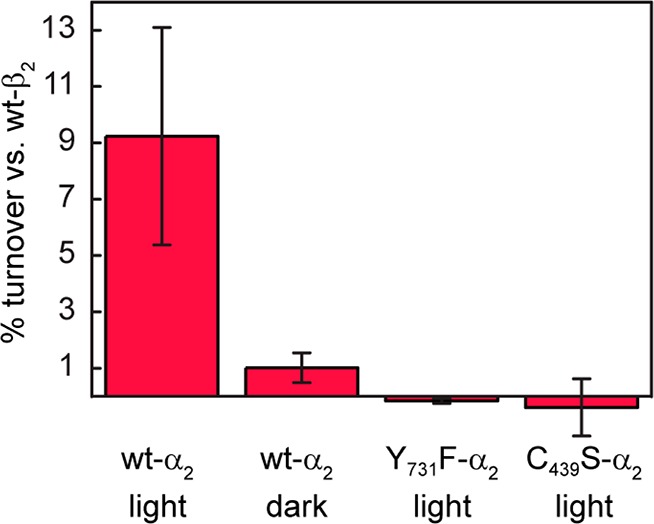
Photochemical turnover of met-[Re]-F3Y356-β2 (10 μM), [5-3H]-CDP (0.2 mM), ATP (3 mM), Ru(NH3)6Cl3 (10 mM), and wt-, Y731F-, or C439S-α2 (10 μM) in assay buffer, pH 7.6 at 25 °C. Numbers are presented as a percentage of product observed with wt-β2. Error bars represent 1 standard deviation for 3–6 independent trials.
Pathway and Isotope Dependence of Y· Lifetime
We set out to explore individual PCET steps by measuring the kinetic behavior of Y· under different conditions. We compared the lifetime of transiently formed Y· (τ) in the presence of CDP, ATP, and wt-α2 with that of τ in the presence of CDP, ATP, and α-variants containing redox-inactive pathway substitutions, as well as in the presence of [3′-2H]-CDP, ATP, and wt-α2. Lifetime data were determined from measurements of three unique samples from a single protein preparation of both wt-α2 and [Re]-F3Y-β2; data sets from two protein preparations were measured. Accordingly, the data in Table 1 are the propagated error for the six measurements across the two trials. Of note, the initiation process in which F3Y356– is oxidized by [ReII] simply generates the radical on the PCET pathway. All fits to kinetics data begin at 3 μs, after the F3Y· has formed. Thus, by removing the proton dependence of the initiation step, we are able to increase the yield of the radical (by relying only on an ET vs PCET for radical initiation), and we do not alter the PCET mechanism at play during the steps of interest that follow. With the exception of τ measured in the presence of Y731F-α2, the lifetime in each case corresponds to a total signal composed of contributions from F3Y356· in β, Y731· and Y730· in α2. Similar signal amplitudes were observed at t = 0 within each trial (Table S1), revealing that Y· generation was similar to the different α2 variants and substrates.
We found that the measured lifetime of τ = 18 ± 1 μs for wt-α2 is extended to 24 ± 1 μs for the Y731F-α2 variant, which cannot produce a radical in α2 (Table 1). This observation suggests that the PCET pathway in α introduces an additional route for Y· decay. The significant difference between these two values provides a means of differentiating on- and off-pathway radical decay. The relative kinetics of the productive, on-pathway contribution to the total decay is calculated according to eq 3, where τo represents the lifetime measured in the presence of Y731F-α2. The resultant rate constant (kobs) is (1.4 ± 0.4) × 104 s–1.
| 3 |
In order to understand which processes in α limit the rate of radical transport, we next blocked the interior end of the PCET pathway by using C439S-α2. A lifetime identical to that measured in the presence of Y731F-α2 was observed (Table 1), revealing that the rate-determining step operative in the presence of wt-α2 occurs either during or after oxidation of residue C439. We note, however, that this observation does not reveal whether or not radical injection into the α subunit occurs in the presence of this variant. It could be envisioned that the downstream perturbation to the PCET pathway imposed by the C439S amino acid substitution precludes radical injection entirely. Thus, either radical injection followed by rapid reverse PCET and quenching occurs or injection is precluded. Both possibilities provide evidence for strict conformational control over PCET events. Throughout our analysis we have assumed that, in the presence of wt-α2 and CDP, new routes for off-pathway Y· decay have not been introduced. In all cases, τo is unrestricted in its definition and simply represents the Y· lifetime via any avenue of nonproductive decay. Thus, in calculating kobs we apply the same τo value for nonproductive decay in the case where the productive pathway (turnover) has also been enabled.
Once C439 is oxidized, the ensuing radical abstracts a hydrogen atom from the 3′-position of substrate (Scheme 1). Subsequent loss of a molecule of water from the 2′-position represents the first irreversible step of turnover. This irreversibility renders any subsequent steps inconsequential to the Y· decay rate. Thus, the rate-determining step with respect to Y· decay in our system (namely, where the native pathway through β is overridden) must be either oxidation of C439 or H· abstraction from C3′ of substrate. To differentiate between these two possibilities we next measured τ in the presence of wt-α2 and [3′-2H]-CDP. Here, a lifetime similar to that observed in the presence of wt-α2 and CDP would suggest that oxidation of C439 by Y730· is rate-determining, because slowing down the subsequent step has no effect on τ. Alternatively, a lifetime intermediate between that observed in the presence of wt-α2 and CDP and that observed in the presence of pathway-blocked variants would indicate that cleavage of the 3′-C–H bond by C439· is rate-limiting. We found that the latter case prevails, and to its extreme extent.
Scheme 1. Mechanistic Model Describing Y· Decay.
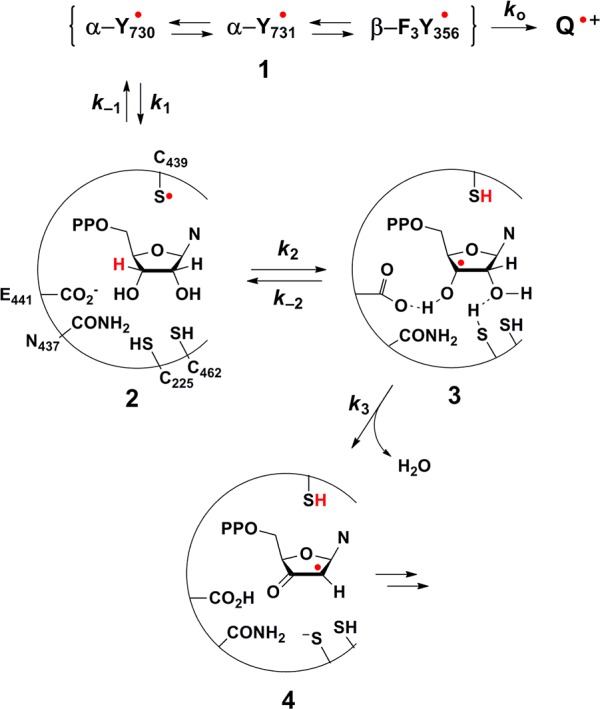
Introduction of deuterium at the 3′-position of substrate completely alters the relative kinetics of the system, resulting in τ statistically identical to τo (Table 1). Further, unlike in the case with C439S-α2, here we are using wt-α2 and so no perturbation to the PCET pathway is incurred. Therefore, in this case we do assume that radical injection into α occurs. Observation of this isotope effect (IE) suggests that we have successfully uncoupled radical translocation from conformational gating, thus allowing for the first direct measure of PCET kinetics within the α2β2 complex.
Discussion
More than 30 years ago [3′-3H]-NDPs were used to investigate the mechanism of RNR.22,23 Small amounts of RNR-mediated 3H2O release and T[V/K] isotope effects provided strong evidence that 3′-C–H bond cleavage occurs during NDP reduction. This study also established that the first irreversible step during a single turnover occurs after hydrogen atom abstraction from substrate and formed the underpinning for the mechanistic model shown in Scheme 1 (intermediates 2–4) and Figure S4.
Stepwise PCET between Tyr-O· → Cys-S· → R3C· akin to the interconversion of 1 → 2 → 3 in Scheme 1 seems contrary to thermodynamic favor (bond dissociation energies of PhO–H, RS–H, and HOCH2–H are ∼86, 91, and 94 kcal/mol, respectively).16,44,45 However, a central tenet of the model proposed in Scheme 1 is that enzymatic coupling of endergonic steps to an irreversible reaction can provide a means of overcoming thermodynamic hurdles. The irreversible and entropically favored release of a molecule of water from the 2′-position of substrate (e.g., 3 → 4 in Scheme 1) is postulated to drive the RNR reaction forward as and when small amounts of intermediate 3 are formed from the reversible steps leading up to it. The loss of a rate enhancement for Y· decay in the presence of [3′-2H]-CDP reveals a primary IE on the cleavage of the substrate 3′-C–H bond, providing direct evidence in support of this model.
The steps in the RNR mechanism that are relevant to the current experiment are outlined in Scheme 1. We note that radical translocation in our system is initiated midstream along the PCET pathway; thus steps within the β subunit are inconsequential. Here, 1 accounts for our experimental observable, a composite signal of unknown relative contributions from F3Y356· in β, Y731· and Y730· in α2. Reoxidation of F3Y356– by Y731· is predicted to be ∼110 mV uphill at pH 8.2 based on the relative potentials of these amino acids in solution.42 Yet, experiments in RNR suggests that Y356 is ∼100 mV easier to oxidize than Y731 within the subunit interface.46 Similarly, Y731· and Y730· have been predicted,47 and experimentally shown,46 to be isoenergetic within the enzyme complex. Thus, all of these radical species have been drawn as reversibly interconverting and reduction of intermediate 1 by an off-pathway quencher, Q, can potentially occur by way of any of these radical intermediates.
On-pathway Y· decay occurs by oxidation of C439 to give intermediate 2. Upon oxidation, C439· reversibly abstracts H· from the 3′-position of substrate to generate 3. General base (E441) and acid (C225) catalysis facilitate dehydration from the 2′-position to yield the 2′-α-ketyl radical intermediate (4). Despite the fact that active site chemical transformations following the formation of 4 ultimately result in the regeneration of the C439· and reverse PCET to reform 1 (Figure S4),48 these processes occur on a much slower time scale (∼100 s–1) than under examination here.49 Accordingly, 1 is not expected to reform during the time course of the experiment. Thus, 4 represents a terminal product with respect to Y· decay and the mechanistic steps relevant to our experimental conditions are limited to the interconversion of species 1–4 in Scheme 1.
Our results comparing τ in the presence of [3′-2H]-CDP versus CDP reveal that the rate enhancement for nonlabeled CDP occurs after oxidation of C439. However, the implications of the reversibility of the oxidation of C439 on the radical lifetimes of 1 must be considered. With little kinetic information regarding the equilibrium between 1 and 2, we turn to small molecule model studies in which the kinetics of bimolecular oxidation of Cys by Tyr· were examined by pulse radiolysis.50 These studies report a rate constant of 2 × 106 M–1 s–1 and that the reverse reaction (oxidation of Tyr by Cys·) is significantly faster.50 These results suggest that a pre-equilibrium may be established between intermediates 1 and 2 in our system and facilitates an estimate for the magnitude of the corresponding equilibrium constant, K±1 (k1/k–1), from thermodynamic values. Electrochemical measurements of tyrosine and glutathione reveal that cysteine and tyrosine have approximately equal midpoint potentials at pH 7.0 (0.94 ± 0.04 V and 0.93 ± 0.02 V, respectively).51 However, calculations based on a trapped form of the active RNR complex suggest that oxidation of C439 by Y730· is endergonic by 3–4 kcal/mol, resulting in K±1 ≈ 10–2.47 Taken together, these studies support that 2 is formed reversibly from 1 and that the resultant equilibrium constant is ≤1. We note that 2 has never been observed or trapped in a class I enzyme, indicating that this intermediate is likely consumed upon its formation. This scenario renders kobs a lower bound for k2 where the extent to which the actual k2 is greater than kobs increases as K±1 decreases from unity.
The reversibility of H atom abstraction may also lead to a case where k2 is greater than kobs. The extent to which this is manifest depends upon the efficiency with which 3 proceeds forward as a fraction of its total decay. This efficiency can be described by a net rate constant equal to k3/(k–2 + k3), which has a maximum value of 1. Reactions similar to both of the processes described by k–2 and k3 have been studied extensively, producing rate constants ranging from 106–108 M–1 s–1,52−56 and 106–108 s–1,57−59 for k–2 and k3, respectively. Though it is difficult to compare bimolecular reactions in solution to those within an enzyme active site, we note that these values are all 102–104-fold faster than the corresponding bimolecular rate constants akin to conversion of 2 → 3 (k2).52−56 Applying the limiting values from these model studies, we find that, at the highest enzymatic efficiency, a factor of 1 is obtained for k3/(k–2 + k3) and thus the reversibility of H· abstraction does not affect kobs. At the lowest enzymatic efficiency, k3/(k–2 + k3) is calculated to be 10–2, rendering k2 102-fold greater than kobs.
In light of the preceding discussion, it is clear that the kobs reported here (1.4 × 104 s–1) is a lower limit for thiyl radical mediated H· abstraction from C3′ of substrate. Chemical precedent for C–H bond cleavage by thiyl radicals has been established by a number of methods including pulse radiolysis,52−56 laser flash photolysis,60 and NMR61 and EPR62 competition experiments. Rate constants for C–H bond cleavage in deoxyribose, tetrahydrofuran, 2-propanol, glucose, and other 2° alcohols and ethers by cysteine, glutathione, and pennicilamine thiyl radicals have been measured by pulse radiolysis to give second-order rate constants in the range (1.2–1.8) × 104 M–1 s–1.52−56 These studies bolster our interpretation that hydrogen atom transfer from C3′ to C439· is rate-limiting within the context of Scheme 1 and provide values that are consistent with the rate constant reported here.
Previous success in unmasking radical translocation kinetics in RNR is limited to only three examples. Replacing β-Y122 with NO2Y, whose high reduction potential and low pKa decouple PT from ET, allowed examination of β-NO2Y122· reduction by stopped-flow absorption and rapid freeze quench EPR spectroscopies.49 This work, though still limited by mixing times and possibly by further conformational gating, allowed the rate constant for ET to NO2Y122· (to form NO2Y–) to be bracketed with a lower limit of 300 s–1.49 The limitations imposed by mixing times are completely eliminated with photoRNRs. Modification of the C-terminal tail of β with an appended [ReI]-2,3,6-F3Y revealed a rate constant of (3 ± 2) × 105 s–1 for radical injection into α2,28 and this observation was further corroborated by our measurement of the rate constant for charge separation (kCS) in a photo-β2 containing the native Y residue at position 356 of β, where a kCS = (4.1 ± 0.1) × 105 s–1 in the presence of wt-α2, CDP, and ATP was observed.30 All of these results are in line with the conclusions presented here: radical injection into the α2 subunit is faster than subsequent PCET and substrate activation steps.
The fact that C–H bond cleavage from substrate by the transiently formed C439· occurs at 104 s–1, along with previous results,28,30,49 reveals that PCET events occur rapidly during radical translocation. Together with recent findings that implicate alignment of the PCET pathway as a target of conformational gating,13,18,47 these results suggest that the reaction profile of the active α2β2 complex remains locked in place as radical translocation and subsequent active site chemical steps transpire. This ability to lock the PCET pathway indicates that RNR capitalizes on the constraints imposed by PT distances in achieving acute control over long-range ET.
A number of studies indicate that the PCET pathway of RNR runs slightly thermodynamically uphill in the forward direction,14,15,31,46,49 and active site chemistry is driven forward by the rapid and irreversible loss of water from the 2′-position of substrate.24,57−59 This reaction landscape presents a mechanism by which RNR avoids the buildup of reactive amino acid radical intermediates over the course of its ∼70 Å round-trip traverse between α and β. Our observation that HAT from C3′ of substrate to C439· is rate-limiting in forward PCET through α provides further evidence that an uphill PCET pathway generates the initial substrate radical.
Conclusions
Jump-starting radical propagation with light has enabled the direct observation of PCET events previously inaccessible, revealing active site chemistry at the heart of RNR catalysis. Despite the fact that RNR turnover is rate-limited by conformational changes occurring at ∼2–10 s–1,8 radical propagation steps are rapid. To unmask PCET events we have constructed a photochemically competent β2 subunit capable of generating observable transient Y· species within the α2β2 complex. With this construct, we have observed an IE on cleavage of the substrate 3′-C–H bond, revealing that this step is rate-limiting with respect to Y· propagation through α and allowing us to report a lower bound for the rate constant associated with this step of (1.4 ± 0.4) × 104 s–1. Unmasking PCET events in the active α2β2 RNR has provided a first measure of active site kinetics in the class Ia enzyme, yielding new evidence for a long-standing model and shedding light on the mechanism by which RNR maintains control and specificity during long-range radical transport.
Acknowledgments
The authors gratefully acknowledge the NIH for funding (GM 47274 D.G.N., GM 29595 J.S.). L.O. acknowledges the NSF for a graduate fellowship.
Supporting Information Available
Experimental methods and instrumentation; calculation of photochemical Y· yield; complete table of data including τ and ΔOD amplitudes for two trials; pKa titration; HPLC analysis of radiolabeled photoproducts; single wavelength kinetics data, fitting, and residuals analysis; scheme depicting the entire model for RNR-mediated substrate turnover. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cukier R. I.; Nocera D. G. Annu. Rev. Phys. Chem. 1998, 49, 337–369. [DOI] [PubMed] [Google Scholar]
- Huynh M.-H. V.; Meyer T. J. Chem. Rev. 2007, 107, 5004–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Acc. Chem. Res. 2009, 42, 1881–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T. R.; Dogutan D. K.; Reece S. Y.; Surendranath Y.; Teets T. S.; Nocera D. G. Chem. Rev. 2010, 110, 6474–6502. [DOI] [PubMed] [Google Scholar]
- Nocera D. G. Inorg. Chem. 2009, 48, 10001–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey J. L.; Winkler J. R.; Gray H. B. Chem. Rev. 2010, 110, 7024–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece S. Y.; Nocera D. G. Annu. Rev. Biochem. 2009, 78, 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J.; Yu G.; Ator M. A.; Stubbe J. Biochemistry 2003, 42, 10071–10083. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. J. Am. Chem. Soc. 2007, 129, 2226–2227. [DOI] [PubMed] [Google Scholar]
- Uhlin U.; Eklund H. Nature 1994, 370, 533–539. [DOI] [PubMed] [Google Scholar]
- Bennati M.; Robblee J. H.; Mugnaini V.; Stubbe J.; Freed J. H.; Borbat P. J. Am. Chem. Soc. 2005, 127, 15014–15015. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Chan C. T. Y.; Mugnaini V.; Stubbe J.; Bennati M. J. Am. Chem. Soc. 2007, 129, 15748–15749. [DOI] [PubMed] [Google Scholar]
- Minnihan E. C.; Ando N.; Brignole E. J.; Olshansky L.; Chittuluru J.; Asturias F. J.; Drennan C. L.; Nocera D. G.; Stubbe J. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 3835–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G.; Yee C. S.; Chang M. C. Y. Chem. Rev. 2003, 103, 2167–2202. [DOI] [PubMed] [Google Scholar]
- Minnihan E. C.; Nocera D. G.; Stubbe J. Acc. Chem. Res. 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J.; van der Donk W. A. Chem. Rev. 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- Brown N. C.; Reichard P. J. Mol. Biol. 1969, 25–38. [DOI] [PubMed] [Google Scholar]
- Wörsdörfer B.; Conner D. A.; Yokoyama K.; Livada J.; Seyedseyamdost M. R.; Jiang W.; Silakov A.; Stubbe J.; Bollinger J. M. Jr.; Krebs C. J. Am. Chem. Soc. 2013, 135, 8585–8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 2522–2523. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 2522–2523. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Xie J.; Cham C. T. Y.; Schultz P. G.; Stubbe J. J. Am. Chem. Soc. 2007, 129, 15060–15071. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Ackles D. J. Biol. Chem. 1980, 255, 8027–8030. [PubMed] [Google Scholar]
- Stubbe J.; Ator M.; Krenitsky T. J. Biol. Chem. 1983, 258, 1625–1630. [PubMed] [Google Scholar]
- Licht S.; Stubbe J. Compr. Nat. Prod. Chem. 1999, 5, 163–203. [Google Scholar]
- Chang M. C. Y.; Yee C. S.; Stubbe J.; Nocera D. G. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 6882–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece S. Y.; Seyedsayamdost M. R.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2007, 129, 8500–8509. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Seyedsayamdost M. R.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2007, 129, 13828–13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder P. G.; Pizano A. A.; Anderson B. L.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2012, 134, 1172–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizano A. A.; Lutterman D. A.; Holder P. G.; Teets T. S.; Stubbe J.; Nocera D. G. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizano A. A.; Olshansky L.; Holder P. G.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2013, 135, 13250–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Young D. D.; Schultz P. G.; Stubbe J. J. Am. Chem. Soc. 2011, 133, 15942–15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando N.; Brignole E. J.; Zimanyi C. M.; Funk M. A.; Yokoyama K.; Asturias F. J.; Stubbe J.; Drennan C. L. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 21046–21051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Högbom M.; Galander M.; Andersson M.; Kolberg M.; Hofbauer W.; Lassmann G.; Nordlund P.; Lendzian F. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3209–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M.; Uhlin U.; Ramaswamy S.; Ekberg M.; Regnström K.; Sjöberg B.-M.; Eklund H. Structure 1997, 5, 1077–1092. [DOI] [PubMed] [Google Scholar]
- Minnihan E. C.; Seyedsayamdost M. R.; Uhlin U.; Stubbe J. J. Am. Chem. Soc. 2011, 133, 9430–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C., Ph.D. Thesis, Massachusetts Institute of Technology, 2012. [Google Scholar]
- Pigiet V. P.; Conley R. R. J. Biol. Chem. 1977, 252, 6367–6372. [PubMed] [Google Scholar]
- Lunn C. A.; Kathju S.; Wallace B. J.; Kushner S. R.; Pigiet V. J. Biol. Chem. 1984, 259, 10469–10474. [PubMed] [Google Scholar]
- Chen H.; Gollnick P.; Phillips R. S. Eur. J. Biochem. 1995, 229, 540–549. [PubMed] [Google Scholar]
- Yokoyama K.; Uhlin U.; Stubbe J. J. Am. Chem. Soc. 2010, 132, 8385–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece S. Y.; Lutterman D. A.; Seyedsayamdost M. R.; Stubbe J.; Nocera D. G. Biochemistry 2009, 48, 5832–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Reece S. Y.; Nocera D. G.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 1569–1579. [DOI] [PubMed] [Google Scholar]
- Steeper J. R.; Steuart C. D. Anal. Biochem. 1970, 34, 123–130. [DOI] [PubMed] [Google Scholar]
- Benson S. W. Chem. Rev. 1978, 78, 23–35. [Google Scholar]
- McMillen D. F.; Golden D. M. Annu. Rev. Phys. Chem. 1982, 33, 493–532. [Google Scholar]
- Yokoyama K.; Smith A. A.; Corzilius B.; Griffin R. G.; Stubbe J. J. Am. Chem. Soc. 2011, 133, 18420–18432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argirević T.; Riplinger C.; Stubbe J.; Neese F.; Bennati M. J. Am. Chem. Soc. 2012, 134, 17661–17670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence C. C.; Bennati M.; Obias H. V.; Bar G.; Griffin R. G.; Stubbe J. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K.; Uhlin U.; Stubbe J. J. Am. Chem. Soc. 2010, 132, 15368–15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkes L. K.; Trujillo M.; Bartesaghi S.; Radi R.; Wardman P. Arch. Biochem. Biophys. 2011, 506, 242–249. [DOI] [PubMed] [Google Scholar]
- Madej E.; Wardman P. Arch. Biochem. Biophys. 2007, 462, 94–102. [DOI] [PubMed] [Google Scholar]
- Sonntag C.; Schöneich C.; Bonifačić M.; Dillinger U.; Asmus K.-D.; Dunster C.; Willson R. L.; Prütz W. A. In Sulfur-Centered Reactive Intermediates in Chemistry and Biology; Catgilialoglu C., Asum K.-D., Eds.; Plenum Press: New York, 1990; Vol. A, series 197, pp 359–399. [Google Scholar]
- Schöneich C.; Bonifacic M.; Asmus K.-D. Free Radic. Res. Commun. 1989, 6, 393–405. [DOI] [PubMed] [Google Scholar]
- Pogocki D.; Schöneich C. Free Radic. Biol. Med. 2001, 31, 98–107. [DOI] [PubMed] [Google Scholar]
- Schöneich C.; Asmus K.-D.; Bonifačić J. Chem. Soc., Faraday Trans. 1995, 91, 1923–1930. [Google Scholar]
- Tamba M.; Quintiliani M. Radiat. Phys. Chem. 1984, 23, 259–263. [Google Scholar]
- Steenken S.; Davies M. J.; Gilbert B. C. J. Chem. Soc., Perkin. Trans. 1986, 2, 1003–1010. [Google Scholar]
- Bansal K. M.; Grätzel M.; Henglein A.; Janata E. J. Phys. Chem. 1973, 77, 16–19. [Google Scholar]
- Lenz R.; Giese B. J. Am. Chem. Soc. 1997, 119, 2784–2794. [Google Scholar]
- Nauser T.; Casi G.; Koppenol W. H.; Schöneich C. J. Phys. Chem. B 2008, 112, 15034–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauser T.; Schöneich C. J. Am. Chem. Soc. 2003, 125, 2042–2043. [DOI] [PubMed] [Google Scholar]
- Mason R. P.; Rao D. N. R.. Sulfur-Centered Reactive Intermediates in Chemistry and Biology; Catgilialoglu C., Asum K.-D., Plenum Press: New York, 1990; Vol. A, series 197, pp 401–408. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.