

Abstract
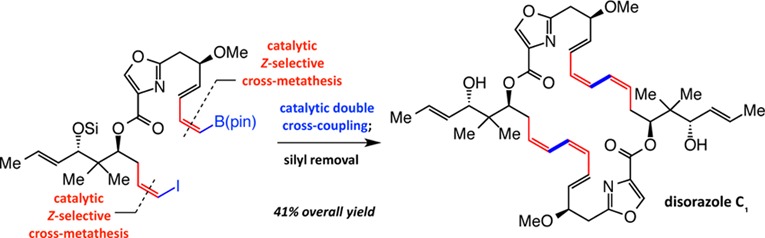
A convergent diastereo- and enantioselective total synthesis of anticancer and antifungal macrocyclic natural product disorazole C1 is reported. The central feature of the successful route is the application of catalytic Z-selective cross-metathesis (CM). Specifically, we illustrate that catalyst-controlled stereoselective CM can be performed to afford structurally complex Z-alkenyl–B(pin) as well as Z-alkenyl iodide compounds reliably, efficiently, and with high selectivity (pin = pinacolato). The resulting intermediates are then joined in a single-step operation through catalytic inter- and intramolecular cross-coupling to furnish the desired 30-membered ring macrocycle containing the critical (Z,Z,E)-triene moieties.
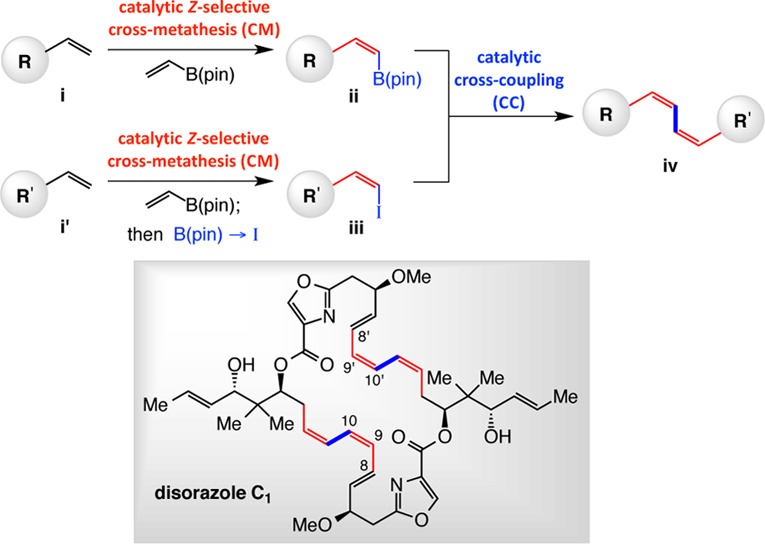
We recently introduced a direct method for preparation of alkyl- and aryl-substituted Z-(pinacolato)alkenylboron [alkenyl–B(pin)] compounds through catalytic stereoselective cross-metathesis (CM; e.g., i → ii, Scheme 1);1 these transformations proceed in the presence of terminal alkenes, relatively robust and commercially available vinyl–B(pin), and an in situ-generated Mo- or W-based monoaryloxide pyrrolide (MAP) complex.2 A benefit of the CM protocol is that it may be combined with catalytic cross-coupling (CC)1,3 to furnish reliable access to a range of otherwise difficult-to-obtain Z alkenes. Another critical advantage of the stereoselective CM is that the resulting Z alkenylboron products can be efficiently and stereoselectively converted to the corresponding alkenyl iodides (e.g., i′ → iii). In this way, both substrates in a catalytic CC reaction to generate a (Z,Z)-1,3-diene (cf. iv) may originate from terminal alkenes (e.g., i and i′). The proposed strategy has significant implications vis-à-vis the design of convergent routes for synthesis of biologically active complex molecules that contain one or more (Z,Z,E)-1,3,5-triene fragments.4 Herein, we disclose the application of the above plan to the diastereo- and enantioselective preparation of anticancer and antifungal natural product disorazole C1.5
Scheme 1. Principal Strategy and the Targeted Natural Product.
pin = pinacolato.
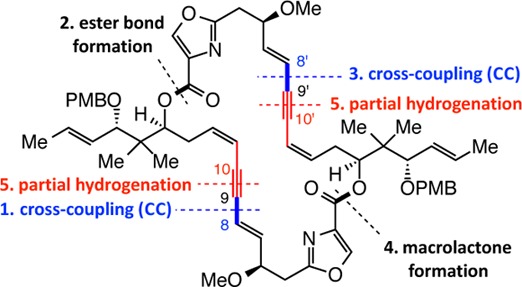
The only extant total synthesis of disorazole C1, which also established its relative and absolute stereochemistry, is due to the seminal work of Wipf and Graham (Scheme 2).6 The route involved two Sonogashira-type CC reactions (C8–C9), one performed prior to generation of one of the ester linkages and the other carried out subsequently (C8′–C9′). A Yamaguchi-type lactone formation furnished the macrocyclic ring; this was followed by the unveiling of the C9–C10 and C9′–C10′ Z alkenes through partial hydrogenation of the alkyne units. The 2004 study illustrated that any approach toward disorazole C1 and related compounds must be mindful of the sensitivity of the polyene segments. By masking the latter moieties as ene-yne-ene units until the last event of the total synthesis, complications were largely sidestepped that would have otherwise negatively impacted the final stages of the 20-step sequence.
Scheme 2. Previous Synthesis of Disorazole C16.
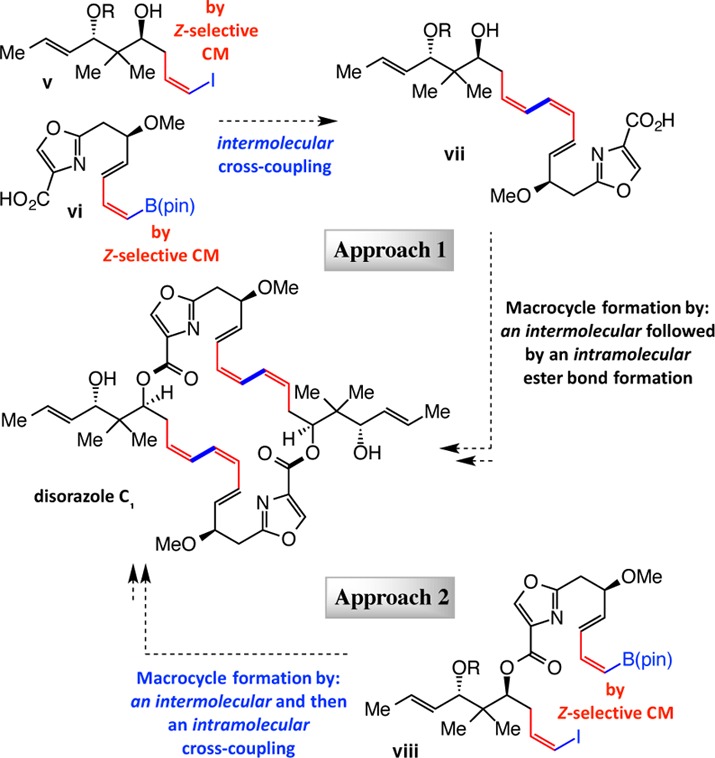
We envisioned two approaches for the assembly of disorazole C1 (Scheme 3). We hoped to identify convergent pathways by utilizing the aforementioned CM and CC processes and by exploiting the symmetric structure of the natural product. In one route (Approach 1), a Z-alkenyl iodide (v) and a Z-alkenyl–B(pin) (vi) would be subjected to catalytic CC conditions and the resulting (Z,Z,E)-triene (vii) would be converted to disorazole C1 through a double ester bond formation. In addition to the Wipf precedence,6 synthesis of related macrocycles with diene-yne or ene-yne-ene moieties by sequential formation of ester bonds has been reported by Meyers7 and Hoffmann,8 respectively. In the earlier instance,7 partial hydrogenation of the alkyne units was unsuccessful and removal of silyl protecting groups could not be performed in high yield; in the other case, data regarding reduction of alkynes or unmasking of [2-(trimethylsilyl)ethoxy]methyl (SEM) groups were not provided.8 Additionally, single-step double esterification has been shown to be reasonably effective in accessing a derivative of disorazole Z (26% yield), although this member of the natural product family contains two Z,E,E triene groups within a 26-membered macrocycle (vs Z,Z,E units in a 30-membered ring in disorazole C1).9 In this latter initiative, a pair of t-Bu(Me)2Si (TBS) ethers within the macrocyclic product could be unmasked but only in 22% yield. We hoped that with a short and convergent route for accessing the requisite substrates, an effective double ester bond synthesis and an appropriately mild deprotection would be identified. The other strategy to be probed involved double CC of a precursor such as viii (Approach 2, Scheme 3). To the best of our knowledge, this type of a transformation has not been previously applied to the preparation of this or related types of polyene-containing natural products.10 One positive attribute of the above pathways is that closely related building blocks would be required so that exploration of one would serve to facilitate examination of the other.
Scheme 3. Synthesis Strategies to be Explored in This Study.
We began by developing a concise route for synthesis of alkenyl iodide 7 (Scheme 4). Although a related derivative of this fragment (TES vs TMS) has previously been prepared, we opted to identify a more efficient pathway.11 The first key operation required stereoselective addition of an allyl unit to enantiomerically enriched aldehyde 1.9 After screening a variety of conditions involving assorted catalysts and reagents, we found that use of allylsilane 2 in the presence of 5.0 mol % Sc(OTf)3, as outlined by Leighton,12 delivered the most desirable outcome [>98% yield, 10:1 diastereomeric ratio (dr)]. Use of the original and closely related reagent derived from norephedrine13 generated a nearly equal mixture of diastereomers.14 Protection of the secondary alcohol as a TMS ether (92% yield) was followed by a Z-selective CM with vinyl–B(pin) (performed on g-scale; 72% yield); this sequence, with complex 4a serving as the MAP complex, furnished 5 with complete Z selectivity. As long as vinyl–B(pin) was available, none of the competitive RCM, leading to the formation of cyclohexene 6, could be detected; this was despite the conformational rigidity provided by the gem-dimethyl unit and suggests that B(pin)-substituted Mo alkylidene, generated preferentially,15 serves as the propagating species (vs that derived from the terminal alkene of TMS-protected 3). Z-Alkenyl iodide 7 was then prepared in 79% yield (>98% Z).16
Scheme 4. Preparation of Z-Alkenyl Iodide Fragment 7.
DMAP = 4-dimethylaminopyridine.
Next, we set out to identify an efficient sequence that would furnish (Z,E)-dienyl–B(pin) fragment 13 (Scheme 5). This task emerged as significantly more challenging, largely because introducing the sensitive dienyl moiety of 12 through a catalytic enantioselective or auxiliary-based diastereoselective aldol addition with pentadienyl aldehyde proved to be particularly difficult (extensive product decomposition and/or inferior stereoselectivity).17 A relatively efficient and diastereoselective variant involving an N-acetylthiazolidinethione suffered substantial intramolecular acylation during attempts to obtain the derived methyl ether.17 We consequently designed an alternative scheme that would expose the dienyl moiety through an appropriately mild procedure at a later stage. Conversion of enantiomerically enriched allylic alcohol 8(18) to serine-derived amide 9 was accomplished in 3 steps and 61% overall yield (Scheme 5). Cyclization with DAST, followed by oxidation initiated by BrCCl3 and DBU19 generated oxazole 10. Installation of the 1,3-diene moiety was accomplished by the following sequence: CM between 10 and commercially available 4-bromo-1-butene in the presence of 5.0 mol % Ru carbene 11(20) and subsequent treatment with DBU to facilitate elimination.21E-Diene 12 was accordingly obtained in 74% overall yield and 93% E selectivity. Catalytic CM involving 12 and vinyl–B(pin), performed with 10 mol % in situ-generated 4a, afforded (Z,E)-1,3-diene 13 in 76% yield and 92% Z selectivity.
Scheme 5. Preparation of (Z,E)-Dienyl–B(pin) Fragment 13.
TFFH = fluoro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate. DAST = (diethylamino)sulfur trifluoride. DBU = 1,8-diazabicycloundec-7-ene.
Z-Alkenyl iodide 14 was obtained as a single diastereomer after removal of the TMS group in 7 (85% yield).17 Coupling of 14 with (Z,E)-1,3-dienyl–B(pin) 13 by the use of 10 mol % Pd(PPh3)4 and 1.1 equiv of Ag2O22 generated the desired (Z,Z,E)-triene, which was then converted to carboxylic acid 15 (Scheme 6). The latter transformation was complete in 1.5 h, but 15 proved to be relatively unstable, likely due to incompatibility of the sensitive triene moiety with the acidic functionality; attempts at purification through silica gel chromatography resulted in complete material loss. We therefore used unpurified 15 to investigate a number of conditions, such as those originally introduced by Yamaguchi23 and Shiina,24 to promote formation of macrocyclic 16 or at least the corresponding dimeric ester intermediate. Extensive efforts, including conditions effective for similar intermolecular ester bond forming reactions,25 led only to unidentifiable mixtures.
Scheme 6. Examination of Catalytic CC/Double Ester Formation Strategy.
At this point, we turned to evaluating Approach 2 (cf. Scheme 3). We were heartened by the relative facility with which tetraene 15 could be obtained by an intermolecular CC process. However, after extensive screening studies, we were able to prepare 17 in no more than 30% overall yield (from methyl ester 13 and alcohol 14; Scheme 7a). In considering ways to improve efficiency, we surmised that the second-stage coupling, and not the initial ester hydrolysis, might be the problematic process. We envisioned that, as shown in complex ix (Scheme 7a), the nucleophilicity of the purported carboxylate intermediate might be hampered by the Lewis acidic B(pin) unit of another molecule of the carboxylic acid derived from 13. We therefore chose to generate the carboxylic ester bond first without the possibility of interference by the organoboron group and then perform a Z-selective CM with vinyl–B(pin). Ester bond formation with the carboxylic acid derived from 1,3-diene 12 and the sizable alcohol 14 was indeed significantly more efficient, delivering ester 18 in 83% overall yield [vs 30% with acid of alkenyl–B(pin) 13]. The stage was thus set for catalytic CM with the highly functionalized polyene 18 (Scheme 7b), a transformation that proceeded readily with 10 mol % Mo MAP complex 4b (cf. Scheme 4; performed on ∼0.2 g of 18) and ∼7 equiv of vinyl–B(pin) to afford 17 with 95:5 Z/E selectivity. Silica gel chromatography furnished (Z,E)-dienyl–B(pin) 17 in 91% yield as a single stereoisomer.
Scheme 7. Examination of Ester Bond Formation/Double CC Strategy.
DCC = N,N′-dicyclohexylcarbodiimide.
We searched for conditions that would allow the single-step inter- and intramolecular catalytic CC to provide the 30-membered ring structure of disorazole C1. Major side products included oligomeric compounds as well as 15-membered lactone 19 (Scheme 8), derived from intramolecular CC of 17. As might be expected, choice of Pd complex (especially to preserve to the stereochemical integrity of the Z,Z,E trienes),26 base, and solvent proved to be critical.17 Our investigations eventually led us to establish that, in the presence of 5.0 mol % Pd[(o-tol)3P]2 and 1.0 equiv of Cs2CO3 in MeOH at ambient temperature for 12 h, the catalytic double CC is most efficient, affording 16 in 60% yield (>98% conv). Importantly, with MeOH as solvent, although some oligomeric products were formed, <2% of 19 was detected.17 The present system is more challenging than a previous study involving the “stitching” of di(alkenyl iodide) and a distannyl alkene, where neither reactant can undergo unimolecular cyclization (cf. 19).27 Moreover, in a formerly disclosed double-CC approach to a 16-membered ring triene, intramolecular ring closure would mean the formation of an energetically prohibitive E,E-cyclooctadienoate.10b
Scheme 8. Synthesis of the Macrocycle and Completion of the Synthesis by Catalytic Double CC.
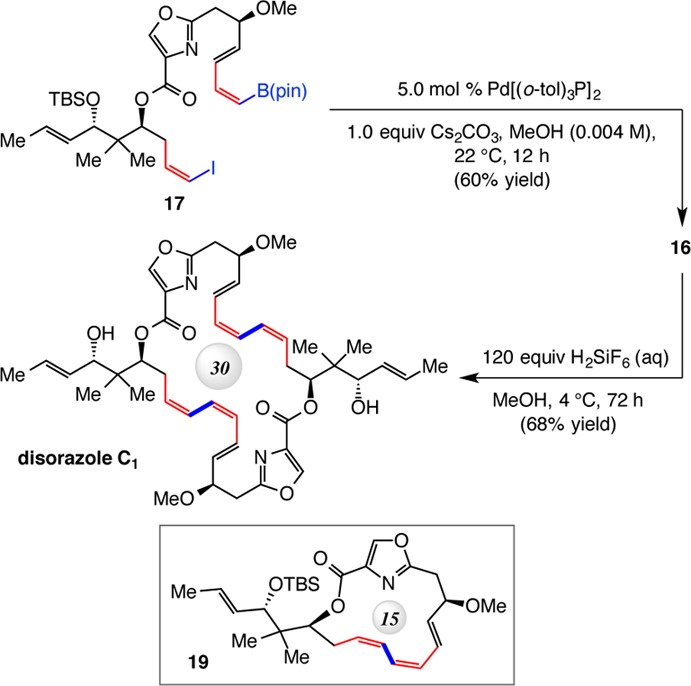
Considering the documented difficulties vis-à-vis deprotecting procedures with related compounds,7−9 we were mindful of the remaining transformation. Significant screening was required here as well, leading us to determine that subjection of 16 to aqueous H2SiF6 and MeOH28 for 72 h at 4 °C could deliver disorazole C1 in 68% yield. The present approach, which relies on the combination of catalytic CM/CC is significantly more efficient than previous approaches utilized to access Z,E,E-triene-containing complex molecules and that entail the use of Wittig-type processes.29
This study illustrates that CM reactions with high oxidation-state MAP complexes can involve highly functionalized substrates and be performed at relatively late stages of a multistep sequence. We show that, in combination with catalytic CC, CM reactions furnish efficient and stereoselective access to delicate conjugated trienes that contain two Z alkenes and reside in acyclic or macrocyclic natural products.4 The total synthesis is convergent, contains a longest linear sequence of 12 steps (8 → 12 → disorazole C1; 8.0% overall yield), and lends itself to analogue preparation.
Acknowledgments
Funding was provided by the NIH (GM-59426).
Supporting Information Available
Experimental procedures and spectral and analytical data for all products. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Kiesewetter E. T.; O’Brien R. V.; Yu E. C.; Meek S. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2013, 135, 6026. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent application to complex molecule synthesis, see:; b Yu M.; Schrock R. R.; Hoveyda A. H. Angew. Chem., Int. Ed. 2014, 10.1002/anie.201409120. [DOI] [Google Scholar]
- For the original report on Z-selective CM with Mo-based MAP complexes, see:; a Meek S. J.; O’Brien R. V.; Llaveria J.; Schrock R. R.; Hoveyda A. H. Nature 2011, 471, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a more recent study, see:; b Mann T. J.; Speed A. W. H.; Schrock R. R.; Hoveyda A. H. Angew. Chem., Int. Ed. 2013, 53, 8395. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent review on catalyst-controlled stereoselective olefin metathesis, see:; c Hoveyda A. H. J. Org. Chem. 2014, 79, 4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on the utility of catalytic CC in natural product synthesis, see:Nicolaou K. C.; Bulger P. G.; Sarlah D. Angew. Chem., Int. Ed. 2005, 44, 4442. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Congreve M. S.; Holmes A. B.; Hughes A. B.; Looney M. G. J. Am. Chem. Soc. 1993, 115, 5815. [Google Scholar]; b Batsanov A. S.; Knowles J. P.; Whiting A. J. Org. Chem. 2007, 72, 2525. [DOI] [PubMed] [Google Scholar]; c Hopkins C. D.; Wipf P. Nat. Prod. Rep. 2009, 26, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Smith A. B.; Dong S.; Brenneman J. B.; Fox R. J. J. Am. Chem. Soc. 2009, 131, 12109. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Burke C. P.; Swingle M. R.; Honkanen R. E.; Boger D. L. J. Org. Chem. 2010, 75, 7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For isolation, biological activity and selected approaches to preparation of disorazole C1, see:; a Jansen R.; Irschik H.; Reichenbach H.; Wray V.; Höfle G. Leibigs Ann. Chem. 1994, 759. [Google Scholar]; b Ref (4c)
- Wipf P.; Graham T. H. J. Am. Chem. Soc. 2004, 126, 15346. [DOI] [PubMed] [Google Scholar]
- Hillier M. C.; Price A. T.; Meyers A. I. J. Org. Chem. 2001, 66, 6037. [DOI] [PubMed] [Google Scholar]
- Niess B.; Hartung I. V.; Haustedt L. O.; Hoffmann H. M. R. Eur. J. Org. Chem. 2006, 1132. [Google Scholar]
- Schäckel R.; Hinkelmann B.; Sasse F.; Kalesse M. Angew. Chem., Int. Ed. 2010, 49, 1619. [DOI] [PubMed] [Google Scholar]
- For a total synthesis involving a CC to generate a (Z,Z,E)-triene within a 12-membered ring lactone, see:; Molander G. A.; Dehmel F. J. Am. Chem. Soc. 2004, 126, 10313. [DOI] [PubMed] [Google Scholar]; For a related double-CC of a (E,E)-dienoate, see:; b Paterson I.; Lombart H.-G.; Allerton C. Org. Lett. 1999, 1, 19. [Google Scholar]
- Hopkins C. D.; Schmitz J. C.; Chu E.; Wipf P. Org. Lett. 2011, 13, 4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kubota K.; Leighton J. L. Angew. Chem., Int. Ed. 2003, 42, 946. [DOI] [PubMed] [Google Scholar]; b Kim H.; Ho S.; Leighton J. L. J. Am. Chem. Soc. 2011, 133, 6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnaird J. W. A.; Ng P. Y.; Kubota K.; Wang X.; Leighton J. L. J. Am. Chem. Soc. 2002, 124, 7920. [DOI] [PubMed] [Google Scholar]
- This is unlike allyl addition to a structurally similar aldehyde, but one that lacks the more distal silyl ether moiety. See:Feng Y.; Jiang X.; De Brabander J. K. J. Am. Chem. Soc. 2012, 134, 17083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend E. M.; Kilyanek S. M.; Schrock R. R.; Müller P.; Smith S. J.; Hoveyda A. H. Organometallics 2013, 32, 4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrill C.; Grubbs R. H. J. Org. Chem. 2003, 68, 6031. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for details.
- Seiser T.; Kamena F.; Cramer N. Angew. Chem., Int. Ed. 2008, 47, 6483. [DOI] [PubMed] [Google Scholar]
- Phillips A. J.; Uto Y.; Wipf P.; Reno M. J.; Williams D. R. Org. Lett. 2000, 2, 1165. [DOI] [PubMed] [Google Scholar]
- Garber S. B.; Kingsbury J. S.; Gray B. L.; Hoveyda A. H. J. Am. Chem. Soc. 2000, 122, 8168. [Google Scholar]
- Lipshutz B. H.; Ghorai S.; Bošković Ž. V. Tetrahedron 2008, 64, 6949. [Google Scholar]
- a Reddy Y. K.; Falck J. R. Org. Lett. 2002, 4, 969. [DOI] [PubMed] [Google Scholar]; b Gao D.; O’Doherty G. A. Org. Lett. 2010, 12, 3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanaga J.; Hirata K.; Saeki H.; Katsuki T.; Yamaguchi M. Bull. Chem. Soc. Jpn. 1979, 52, 1989. [Google Scholar]
- Shiina I.; Kubota M.; Oshiumi H.; Hashizume M. J. Org. Chem. 2004, 69, 1822. [DOI] [PubMed] [Google Scholar]
- Boden E. P.; Keck G. E.. J. Org. Chem. 1985, 50, 2394.Addition of DMAP·hydrochloride minimizes conversion to the corresponding acylurea. [Google Scholar]
- Lu G.-P.; Voigtritter K. R.; Cai C.; Lipshutz B. H. J. Org. Chem. 2012, 77, 3700. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Chakraborty T. K.; Piscopio A. D.; Minowa N.; Bertinato P. J. Am. Chem. Soc. 1993, 115, 4419. [Google Scholar]
- Pilcher A. S.; DeShong P. J. Org. Chem. 1993, 58, 5130. [Google Scholar]
- Brodmann T.; Janssen D.; Kalesse M. J. Am. Chem. Soc. 2010, 132, 13610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.