

Abstract
Anaplastic large cell lymphoma (ALCL) is the most common type of pediatric peripheral T-cell lymphoma. In 70–80% of cases, the chromosomal aberration t(2;5)(p23;q35) results in the juxtaposition of anaplastic lymphoma kinase (ALK) with nucleophosmin (NPM) and the subsequent expression of the NPM-ALK fusion protein. NPM-ALK is a chimeric tyrosine kinase, which induces numerous signaling pathways that drive proliferation and abrogate apoptosis. However, the mechanisms that lead to activation of downstream growth regulatory molecules have not been completely elucidated. Using a mass spectrometry-based phosphoproteomic screen, we identified GSK3β as a signaling mediator of NPM-ALK. Using a selective inhibitor of ALK, we demonstrated that the tyrosine kinase activity of ALK regulates the serine-9 phosphorylation of GSK3β. Expression of NPM-ALK in 293T cells led to an increase of pS9-GSK3β (glycogen synthase kinase 3 beta) compared with kinase-defective K210R mutant NPM-ALK, but did not affect total GSK3β levels. Phosphorylation of pS9-GSK3β by NPM-ALK was mediated by the PI3K/AKT signaling pathway. ALK inhibition resulted in degradation of GSK3β substrates Mcl-1 and CDC25A, which was recovered upon chemical inhibition of the proteasome (MG132). Furthermore, the degradation of Mcl-1 was recoverable with inhibition of GSK3β. ALK inhibition also resulted in decreased cell viability, which was rescued by GSK3β inhibition. Furthermore, stable knockdown of GSK3β conferred resistance to the growth inhibitory effects of ALK inhibition using viability and colony formation assays. pS9-GSK3β and CDC25A were selectively expressed in neoplastic cells of ALK + ALCL tissue biopsies, and showed a significant correlation (P < 0.001). Conversely, ALK-ALCL tissue biopsies did not show significant correlation of pS9-GSK3β and CDC25A expression (P < 0.2). Our results demonstrate that NPM-ALK regulates the phosphorylation of S9-GSK3β by PI3K/AKT. The subsequent inhibition of GSK3β activity results in accumulation of CDC25A and Mcl-1, which confers the advantage of growth and protection from apoptosis. These findings provide support for the role of GSK3β as a mediator of NPM-ALK oncogenesis.
Keywords: NPM-ALK, GSK3, ALCL, oncogenesis
Introduction
Anaplastic large cell lymphoma (ALCL) is an aggressive form of non-Hodgkin lymphoma often characterized by the chromosomal translocation t(2;5)(p23;q35). This translocation results in the expression of the nucleo-phosmin-anaplastic lymphoma kinase (NPM-ALK) fusion protein (Morris et al., 1994). Although the expression of ALK is usually confined to neonatal brain tissue (Iwahara et al., 1997), the translocation places the gene under the regulation of the NPM promoter, which is abundantly and ubiquitously expressed. Furthermore, the dimerization of the fusion protein mediated by the oligomerization domain of NPM (Bischof et al., 1997) leads to its autophosphorylation and constitutive activation (Amin and Lai, 2007; Palmer et al., 2009). NPM-ALK activates diverse growth factor signaling pathways including the JAK/STAT (Zamo et al., 2002), the PI3K/AKT (Slupianek et al., 2001) and the PLCγ pathways (Bai et al., 1998). However, the cellular mechanisms utilized by NPM-ALK to induce oncogenesis have not been completely characterized.
To identify novel signaling proteins regulated by NPM-ALK, we carried out phosphoproteomic studies of t(2;5) + cell lines using the approach outlined previously (Rush et al., 2005). Our studies provide evidence that GSK3β is a novel NPM-ALK-regulated protein. GSK3β is a serine/threonine kinase that is constitutively active in resting cells (Cohen and Frame, 2001; Jope and Johnson, 2004). Upon growth factor signaling, GSK3β is phosphorylated at serine-9, which inhibits its ability to target key substrates for proteasomal degradation. The accumulation of substrates of GSK3β such as CDC25A and Mcl-1 that regulate the cell cycle and apoptosis contributes to the enhanced proliferative capacity of tumor cells (Jinno et al., 1994; Diehl et al., 1997; Zhao et al., 2007; Kang et al., 2008). Here, we show that serine-9 phosphorylation of GSK3β is dependent on NPM-ALK kinase activity and the PI3K/AKT pathway. We provide evidence that NPM-ALK results in stabilization of Mcl-1 and CDC25A through GSK3β inhibition, which in turn enhances cell viability, proliferation and oncogenic potential. Furthermore, tissue biopsies of ALK + ALCLs express pS9-glycogen synthase kinase 3 beta (GSK3β) as well as CDC25A, providing additional support for its functional significance in ALK + ALCLs.
Results and discussion
Given the oncogenic role of constitutively active NPM-ALK tyrosine kinase in ALK + ALCL, we hypothesized that NPM-ALK-expressing cell lines would exhibit a phosphoproteomic signature that would reflect the signaling cascade regulated by the oncogene. To gain insight into these signaling pathways and to identify novel regulators of NPM-ALK-induced oncogenesis, we used a phosphoproteomic approach (Rush et al., 2005) to evaluate the tyrosine phosphorylated proteins in the NPM-ALK-expressing SUPM2 cell line. SUPM2 cell lysates were subjected to phosphopeptide enrichment using phosphotyrosine-specific antibodies, followed by tandem mass spectrometry. NPM-ALK-derived phosphotyrosine peptides in addition to numerous other phosphorylated proteins were identified (data not shown). Among this list was an eleven amino-acid tryptic peptide corresponding to GSK3β protein phosphorylated at Y216 (Figures 1a and b). Of note, this phosphotyrosine peptide has been identified in a similar study contributing to the confidence of this candidate protein (Boccalatte et al., 2009). On the basis of the known role of GSK3β in cellular signaling (Jope and Johnson, 2004), we hypothesized that GSK3β may mediate the oncogenic properties of NPM-ALK. As GSK3β activity is known to be regulated by growth factor signaling through serine9 phosphorylation, we hypothesized that NPM-ALK regulates the serine phosphorylation of GSK3β and inhibits its activity.
Figure 1.
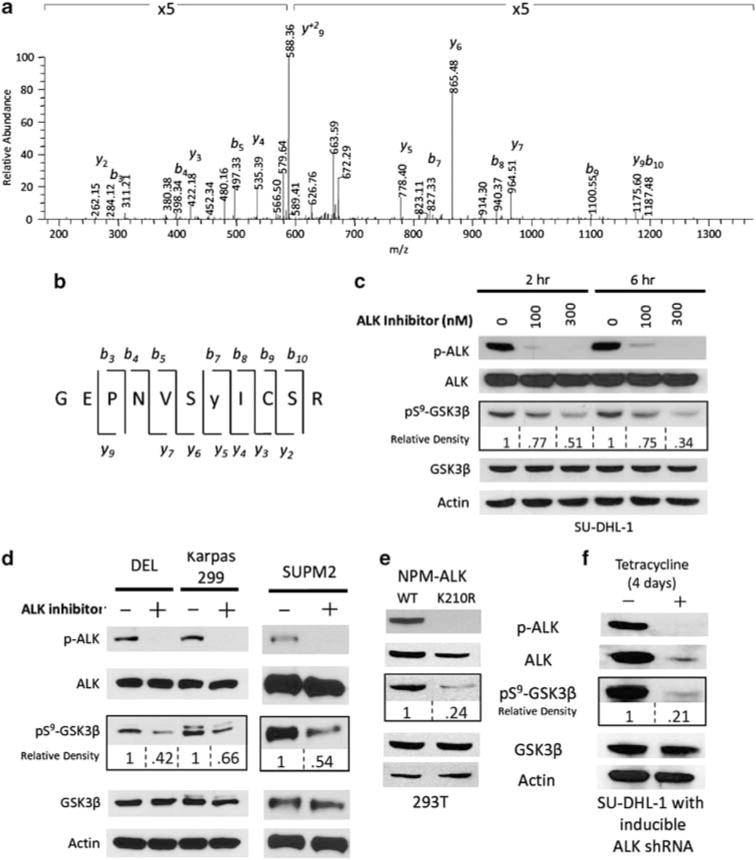
Identification of GSK3β as NPM-ALK-regulated phosphoprotein (a) SU-DHL-1 (ALK + ALCL) cells were subjected to PhosphoScan (Cell Signaling Technologies) enrichment and tandem mass spectrometry as previously described (Rush et al., 2005). Representative mass spectrum of phosphopeptide from GSK3β is shown. Identified b and y ions are indicated. (b) The sequence and cleavage sites for the identified GSK3β phosphopeptide. (c) SU-DHL-1 cells were treated for 2 and 6 h with the ALK inhibitor (Compound 15, Cephalon Inc) at indicated doses in 1% fetal bovine serum media conditions. The synthesis and selectivity of the novel 7-amino-1,3,4,5-tetrahydrobenzoazepinone derivative selective ALK inhibitor (Compound 15) and its properties were described previously (Ott et al., 2010). Cell lysates were resolved on SDS–polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes. Western blot analysis of GSK3β, pS9-GSK3β, p-ALK (Y1604) (rabbit, 1:1000, Cell Signaling) and ALK (mouse, 1:1000, Invitrogen) are shown. Densitometry measurements were conducted with WCIF ImageJ software (Wright Cell Imaging Facility, Toronto Western Research Institute (Toronto, ON, Canada)). (d) DEL, Karpas 299 and SUPM2 cells treated with DMSO or 300 nM ALK inhibitor for 6 h. (e) 293T cells were transiently transfected with NPM-ALK WT or NPM-ALK K210R and harvested 48 h later. Western blot analysis of cell lysates is shown. (f) SU-DHL-1 cells were stably transfected with tetracycline-inducible shRNA against ALK (Ito et al., 2010). After 4 days of tetracycline treatment, cells were harvested and subjected to western blotting. Densitometry of pS9-GSK3β is indicated as relative to control.
In order to determine whether serine phosphorylation of GSK3β is NPM-ALK dependent, we utilized a small molecule (Compound 15) to inhibit ALK kinase (Ott et al., 2010). Treatment of SU-DHL-1 cells with the ALK inhibitor for 2 and 6 h resulted in a marked decrease in the levels of phosphorylated NPM-ALK (Y1604), but not total NPM-ALK (Figure 1c). There was a marked decrease of pS9-GSK3β in both a dose-and time-dependent manner, whereas total GSK3β levels remained unaffected. After 6 h of 300 nM ALK inhibitor treatment, only 34%-of the serine phosphorylated GSK3β remained. Three additional ALCL-derived cell lines (DEL, Karpas 299 and SUPM2) were treated with 300 nM ALK inhibitor for 6 h (Figure 1d), and similar to the effect observed in SU-DHL-1 cells pS9-GSK3β was consistently reduced by ALK inhibition in all of the cell lines. Notably, in contrast to the marked decrease in ALK phosphorylation, the decrease in pS9-GSK3β was not complete. This may be partially explained by the existence of other kinases that phosphorylate GSK3β independent of NPM-ALK. Alternatively, the time points used may not be representative of the maximal loss of pS9-GSK3β. In addition, protein phosphatases that regulate pS9-GSK3β may be weakly active in the context of NPM-ALK. Similarly, 293T cells that were transiently transfected with vectors encoding the kinase-defective mutant (K210R) showed a 76% reduction in GSK3β serine phosphorylation, compared with those that expressed the wild-type NPM-ALK (Figure 1e). Consistent with the previous observation, there was no effect on expression of total GSK3β protein levels. Next, we evaluated the effect of ALK knockdown using stable tetracycline-inducible small hairpin RNA (shRNA) targeting NPM-ALK in SU-DHL-1 cells (Ito et al., 2010). Successful knockdown of NPM-ALK, which was observed after 4 days of tetracycline treatment, led to a marked decrease (79%) in pS9-GSK3β (Figure 1f), whereas the total GSK3β protein levels remained unchanged. These data demonstrate that NPM-ALK kinase activity is required for the serine-9 phosphorylation of GSK3β.
Interrogation of the phosphoproteomic data identified several serine kinases that are known to phosphorylate GSK3β (data not shown). These included the PI3K, PKC and PKA kinases. As the PI3K/AKT pathway is known to be activated by NPM-ALK as well as responsible for GSK3β serine phosphorylation (Cross et al., 1995; Slupianek et al., 2001), we hypothesized that the regulation of pS9-GSK3β by NPM-ALK is mediated by PI3K/AKT. We utilized a selective small molecule inhibitor against PI3K-δ (CAL-101) (Herman et al., 2010) to determine the effect of PI3K/AKT inhibition on pS9-GSK3β in SU-DHL-1 cells. Exposure to CAL-101 for 24 h at increasing concentrations resulted in decreased phosphorylation of pS9-GSK3β and pS473-AKT in a dose-dependent manner (Figure 2a). Importantly, p-ALK was not affected by CAL-101-mediated inhibition of PI3K/AKT. These data show that the PI3K/AKT pathway mediates the serine9 GSK3β phosphorylation by NPM-ALK. However, this doesn’t preclude the possibility that other pathways active in such as WNT (van Noort et al., 2002) or PLCγ (Bai et al., 1998) also contribute to GSK3β phosphorylation.
Figure 2.
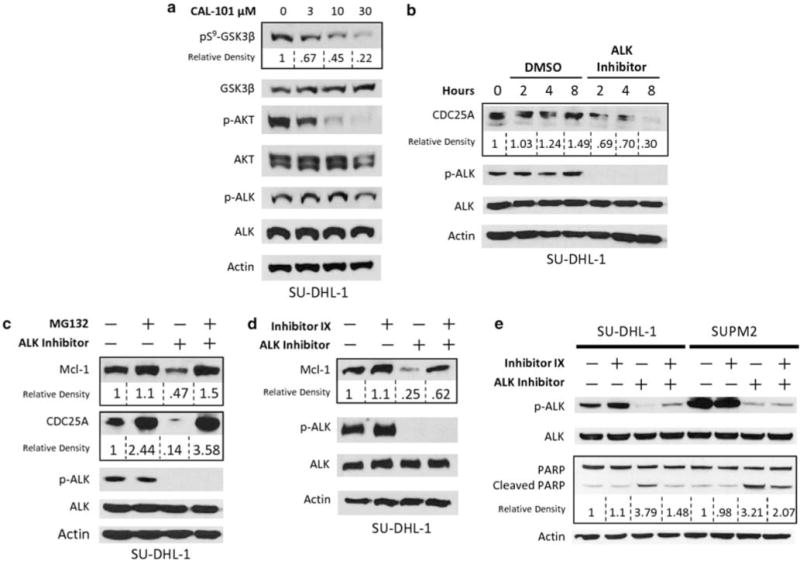
pS9-GSK3β signaling in ALCL (a) SU-DHL-1 cells treated with CAL-101 for 24 h. Characterization of efficacy and selectivity of CAL-101 is previously described (Lannutti et al., 2011). (b) SU-DHL-1 cells were synchronized by double thymidine block. Upon release, cells were treated with DMSO or 300 nM of ALK inhibitor. Cells were lysed at indicated times following release. (c) SUPM2 cells were treated with ALK inhibitor (300 nM) and/or MG132 (10 μM) for 6 h. (d) SU-DHL-1 cells were treated with ALK inhibitor (300 nM) and/or GSK3 inhibitor IX (Calbiochem) (100 nM) for 6 h. (e) SU-DHL-1 cells were treated with ALK inhibitor (100 nM) and/or GSK3 inhibitor IX (100 nM) for 6 h.
The serine9 phosphorylation of GSK3β results in inhibition of its kinase activity (Frame et al., 2001). As active GSK3β is known to target a variety of proteins for proteasomal degradation, we hypothesized that two known substrates of GSK3β, CDC25A and Mcl-1 would be deregulated in ALCL cells. We evaluated the expression of CDC25A in SU-DHL-1 cells after the inhibition of ALK at indicated time points, following release from double thymidine block synchronization (Bostock et al., 1971) (Figure 2b). ALK inhibition resulted in significant reduction of CDC25A levels in a time-dependent manner, with only 30% of the protein remaining after 8 h. In contrast, CDC25A expression remained unchanged through the entire 8 h time period in the dimethyl sulfoxide (DMSO) control condition. These data suggest that NPM-ALK deregulates the normal cyclical expression of CDC25A and leads to stabilization and enhanced expression throughout the cell cycle (Jinno et al., 1994). Similarly, the expression of Mcl-1 and CDC25A were analyzed in SU-DHL-1 cells in the presence of ALK inhibitor and/or proteasome inhibitor (MG132) after release from synchronization using double thymidine block. As shown in Figure 2c, inhibition of ALK resulted in a significant downregulation of Mcl-1 (47% of control) and CDC25A (14% of control), which was recoverable by simultaneous inhibition of the proteasome. These data suggest that NPM-ALK activity enhances the stability of Mcl-1 and CDC25A. Interestingly, MG132 treatment resulted in recovery of Mcl-1 and CDC25A levels that exceeded the expression in the DMSO control (120% and 358%, respectively), suggesting that NPM-ALK does not provide complete proteasomal protection for these proteins.
To determine the role of GSK3β in proteasomal degradation of Mcl-1, SU-DHL-1 cells were treated with the ALK inhibitor and/or a GSK3 inhibitor (inhibitor IX) for 6 h. As shown in Figure 2d, the decrease in Mcl-1 upon ALK inhibition was abrogated by GSK3 inhibition. Thus, NPM-ALK-mediated inhibition of GSK3β results in protection of Mcl-1 from proteasomal degradation. These data provide evidence for the role of GSK3β in enhancing the stability of Mcl-1 in NPM-ALK-expressing cells. As Mcl-1 is a known antiapoptotic protein (Maurer et al., 2006), we asked whether NPM-ALK confers resistance to apoptosis by its inhibition of GSK3β. Western blot analyses were performed on SU-DHL-1 and SUPM2 cell lysates to evaluate poly (ADP-ribose) polymerase (PARP) cleavage as a surrogate marker for apoptosis (Boulares et al., 1999). Cells treated with the GSK3β inhibitor IX alone resulted in no change in PARP cleavage, whereas ALK inhibition greatly reduced ALK phosphorylation and increased the cleavage of PARP (Figure 2e). Interestingly, when both enzymes were inhibited, PARP cleavage was significantly reduced compared with the ALK inhibitor alone. These data indicate that NPM-ALK leads to inhibition of GSK3β, which provide protection against apoptosis.
To determine whether GSK3β is a key mediator of NPM-ALK-induced oncogenesis, we assessed the effect of inhibitor IX on the cell viability of two ALCL-derived cell lines. An effective dose of 100 nM of Inhibitor IX was selected because it represents a concentration well above the IC50 (inhibitory concentration) for GSK3β but below the IC50 for other serine/threonine kinases (Meijer et al., 2003). As expected, the ALK inhibitor (100 nM) resulted in a dramatic reduction of cell viability as determined by WST-1 (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) assay (Figure 3a) to approximately 35% of control in both cell lines. Treatment of ALCL-derived cell lines, SU-DHL-1 and SUPM2 with the GSK3β Inhibitor IX (100 nM) did not affect viability after 24 h. Notably, exposure to both compounds simultaneously (dual) resulted in a viability of 55% and 47% compared with control in SU-DHL-1 and SUPM2 cells, respectively. This represents a significant recovery of viability compared with ALK inhibition (P < 0.01). Importantly, Jurkat cells (ALK-negative T-lymphoblastic leukemia) were not responsive to either the inhibitor IX or the ALK inhibitor, and there was no recovery in cellular viability. To corroborate the results of ALK and GSK3β inhibition on cell proliferation, we carried out flow cytometry-based Annexin V and propidium iodide (PI) staining to quantitatively assess cell viability (Figure 3b). Similar to the results of the WST-1 assay, inhibition of NPM-ALK resulted in a drastic reduction (80%) in the percentage of live (Annexin V and PI negative) cells compared with the DMSO control. Treatment with the GSK3β inhibitor alone did not have an effect on the percentage of live cells. However, inhibition of both GSK3β and NPM-ALK (dual) resulted in a significant increase in the percentage of live cells compared with ALK inhibition alone (34%).
Figure 3.

pS9-GSK3β confers growth advantage for ALCL cells (a) SU-DHL-1 (ALK + ALCL), SUPM2 (ALK + ALCL) and Jurkat (ALK- ALL) cells were treated with ALK inhibitor and/or inhibitor IX (GSK3 inhibitor) for 24 h followed by WST-1 assay. (b) SU-DHL-1 cells were treated with indicated inhibitors (100 nM) for 24 h. After 15 min incubation with PI and Annexin V, cells were subjected to flow cytometric quantification of live cells (PI negative and Annexin V negative). (c) Karpas 299 cells were pretreated with indicated inhibitors (100 nM) for 6 h, then seeded in to methylcellulose media (MethoCult, Stemcell Technologies). After 14 days, colonies were stained with iodonitrotetrazolium chloride overnight and counted. (d) After 6 h drug treatment but before seeding into methylcellulose media, Karpas 299 cell viability was assessed by trypan blue staining. (e) Validation of GSK3β stable knockdown in Karpas 299 and SU-DHL-1 cells (shRNA sequence: 5′-CCGGCCCAAATGTCAAACTACCAAACTCGAGTTTGGTAGTTTGAC ATTTGGGTTTTT-3′). (f) WST-1 assay after 48 h treatment with 300 nM ALK inhibitor in SU-DHL-1 and Karpas 299 cells with or without GSK3β knockdown Each ALK inhibitor condition normalized to its DMSO control. (g) Karpas 299 cells with or without GSK3β knockdown was subjected to methylcellulose colony formation assay after treatment with ALK inhibitor for 6 h. Colony numbers normalized to DMSO for each cell condition. (*P < 0.05, **P < 0.01).
To address the role of GSK3β in oncogenic potential of ALCL cells, Karpas 299 cells were treated with the ALK inhibitor and inhibitor IX for 6 h before seeding into methylcellulose media. After 14 days, colonies were stained and enumerated (Figure 3c). Whereas inhibition of ALK led to a significant reduction of the number of colonies compared with control (51%), dual inhibition of both GSK3β and ALK caused a recovery in the number of colonies (79% compared with DMSO). We verified that 6 h pre-treatment did not affect viability before seeding into the methylcellulose media by trypan blue staining. This showed that the same number of viable cells was used for all conditions that were evaluated (Figure 3d).
To further evaluate the role of GSK3β in NPM-ALK-mediated oncogenesis, stable GSK3β knockdown cell lines were created by lentiviral transfection of shRNA targeted against GSK3β (Figure 3e) in two ALCL-derived cell lines. SU-DHL-1 and Karpas 299 cells stably expressing scramble or shGSK3β vectors were subjected to WST-1 assay following treatment with the ALK inhibitor (Figure 3f). Both cell lines expressing scramble shRNA showed a decrease in viability upon ALK inhibition. In contrast, the cells with knockdown of GSK3β (shGSK3β) were resistant to the growth inhibitory effect of ALK inhibition. Similarly, cells expressing scramble shRNA displayed a signification reduction in number of colonies following ALK inhibition, whereas those with knockdown of GSK3β (shGSK3β) were resistant to ALK inhibition and showed more colonies (Figure 3g). These data support the hypothesis that NPM-ALK-mediated phosphorylation of GSK3β promotes proliferation and oncogenic transformation. It is of interest to note that the rescue mediated by inhibition of GSK3β was not complete This may be because of the presence of several GSK3β-independent signaling pathways that are regulated by NPM-ALK such as JAK/STAT or ERK (Ruchatz et al., 2003; Marzec et al., 2006; Lim et al., 2009). This idea is also evident in the observation that pS9-GSK3β was not completely abolished by ALK inhibition (Figure 1c), yet a dramatic decrease in cell viability was observed.
Immunohistochemistry (IHC) was performed on 16 ALK-positive ALCL (ALK + ALCL) tissue biopsies and 16 ALK-negative ALCL (ALK-ALCL) tissue biopsies. ALK + ALCL samples showed strong staining for ALK in both the cytoplasm and nucleus of neoplastic cells (Figure 4a), whereas ALK-ALCL samples showed no expression of ALK (Figure 4e). A total of 14 of the 16 ALK + ALCL samples showed strong expression of pS9-GSK3β in the neoplastic cells with weak expression in adjacent endothelial cells (Figures 4b and d). Additionally, 13 out of 16 ALK + ALCL cases also showed expression of CDC25A, highlighting the nuclear membranes of the neoplastic cells (Figure 4c). Reactive lymphocytes did not express significant levels of pS9-GSK3β (Figure 4b) or CDC25A (Figure 4c). Using χ-squared-squared distribution to test whether the observed distribution is statistically different from the expected randomized distribution, we found that the ALK + ALCL cases displayed a significant correlation of pS9-GSK3β and CDC25A expression (P < 0.001) (Figure 4d). In contrast, only 5 out of 16 of the ALK-ALCL cases showed expression of pS9-GSK3β in the neoplastic cells whereas the infiltrating neutrophils showed strong expression (Figures 4f and h). CDC25A expression in ALK-ALCL was present in 11 out of 16 cases. Figure 4g shows a representative case of ALK-ALCL lacking CDC25A expression. In contrast to the ALK + ALCL cases, the ALK-ALCL cases displayed a lack of significant correlation of pS9-GSK3β and CDC25A expression distribution (P < 0.2) (Figure 4h). Reactive tonsillar tissues demonstrated weak expression of pS9-GSK3β in epithelial cells and scattered interfollicular activated T-lymphocytes (Figure 4i). CDC25A expression was strongly expressed in the epithelial cells and in scattered interfollicular activated T-lymphocytes (Figure 4j). These findings confirm the presence of the inhibited form of GSK3β and the accumulation of one of its downstream substrates in human ALCL tumor samples. Overexpression of CDC25A transcript has been observed in various lymphomas including ALK + ALCLs (Fernandez-Vidal et al., 2009), however its expression in ALCL tumor tissue samples using IHC has not been reported previously.
Figure 4.
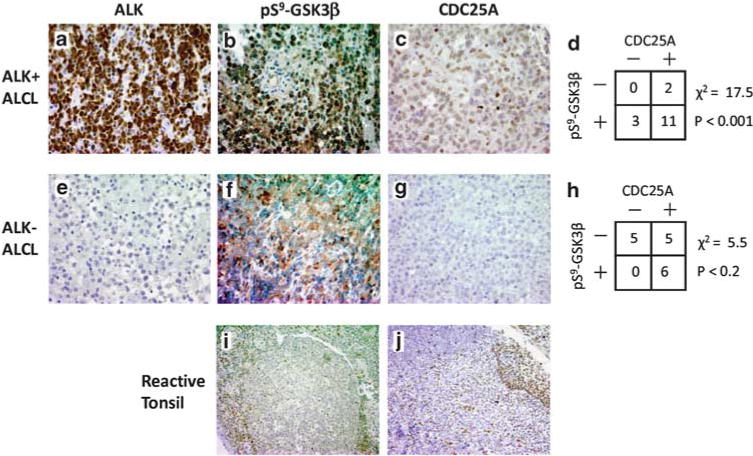
pS9-GSK3β and CDC25A are expressed in human tumors. ALK + ALCL tissue samples were obtained from the archives of the Hematopathology Section, Department of Pathology, University of Michigan, with institutional approval (HUM00023256). (a–c) Representative case of ALK + ALCL. (e–g) Representative case of ALK-ALCL. (i, j) Reactive tonsil. (a–e) IHC staining for ALK (mouse, 1:100, Dako). (b, f and i) IHC staining for pS9-GSK3β (rabbit, 1:50, Cell Signaling Technology). (c, g and j) IHC staining for CDC25A (mouse, 1:500, Abcam). (d) Tabulated results for 16 ALK + ALCL tumor samples analyzed for immunohistochemical staining for CDC25A and pS9-GSK3β. IHC results were evaluated using parameters of staining intensity (0–3) and percentage of immunoreactive neoplastic cells. Positivity was defined by ≥30% of neoplastic cells showing reactivity. (h) Tabulated results for 16 ALK-ALCL tumor samples analyzed for immunohistochemical staining for CDC25A and pS9-GSK3β.
In conclusion, our data show that NPM-ALK regulates the inhibitory serine phosphorylation of GSK3β in ALCL cells, providing a growth advantage and the maintenance of a proliferative phenotype. Our studies uncover a novel mechanism of NPM-ALK-mediated oncogenesis. Detailed elucidation of such deregulated signaling events may be exploited in the design of targeted and combinatorial therapeutic strategies for the treatment of ALK-driven neoplasia.
Materials and methods
Cell lines and inhibitors
SU-DHL-1, DEL, Karpas299 and SUPM2 cells were cultured in Roswell Park Memorial Institute medium 1640 + 10% fetal bovine serum. 293T cells were cultured in DMEM + 10% fetal bovine serum. All cells were maintained in humidified incubators containing 5% CO2. SU-DHL-1 cells stably expressing tetracycline-inducible shRNA targeting ALK were generated as previously described (Ito et al., 2010). Transient transfections were done with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Stable GSK3β knockdown SU-DHL-1 and Karpas299 cells were generated by lentiviral transfection with MISSION shRNA sequence: 5′-CCGGCCCAAATGTC AAACTACCAAACTCGAGTTTGGTAGTTTGACATTTG GGTTTTT-3′ (Sigma-Aldrich, St Louis, MO, USA). The 7-amino-1,3,4,5-tetrahydrobenzoazepinone derivative selective ALK inhibitor (Compound 15, Cephalon, Inc, Frazer, PA, USA) was previously described (Ott et al., 2010). CAL-101 was acquired from Chemietek (Indianapolis, IN, USA). MG132 was acquired from Sigma-Aldrich. GSK3 Inhibitor IX was acquired from Calbiochem (San Diego, CA, USA). All compounds were dissolved in DMSO.
Phosphoproteomics
SU-DHL-1 cells were lysed and proteins were trypsinized to produce peptides of manageable length for mass spectrometry analysis. Phosphotyrosine containing peptides were purified using the PhosphoScan kit (Cell Signaling Technologies, Danvers, MA, USA) and subjected to tandem mass spectrometry for protein identification (Rush et al., 2005).
Western blotting and antibodies
Cell lysates were resolved on 10% SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose. Antibodies used were: GSK3β, pS9-GSK3β, p-ALK (Y1604), Mcl-1, AKT, p-AKT (S473), PARP (rabbit, 1:1000, Cell Signaling), ALK (mouse, 1:1000, Invitrogen) and CDC25A (mouse, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
WST-1, colony formation and flow cytometry
Cell proliferation and viability was assessed by WST-1 assay as per manufacturer’s protocol (Roche Applied Science, Indianapolis, IN, USA). Colony formation assay was performed with MethoCult methylcellulose-based media as per manufacturer’s protocol (Stemcell Technologies, Vancouver, British Columbia, Canada). After 14 days, colonies were stained with iodonitrotetrazolium chloride overnight and counted. Apoptosis and viability were assessed by flow cytometry following cell staining with Annexin V and PI (Vermes et al., 1995).
Immunohistochemistry
Tissue samples were obtained from the archives of the Hematopathology Section, Department of Pathology, University of Michigan, with institutional approval (HUM00023256). IHC staining was conducted with the following antibodies: ALK (mouse, 1:100, Dako, Carpinteria, CA, USA), pS9-GSK3β (rabbit, 1:50, Cell Signaling Technology) and CDC25A (mouse, 1:500, Abcam, Cambridge, MA, USA).
Acknowledgments
This study was supported in part by funding from the NIH (5R01CA140806-02) awarded to MS Lim, MD/PhD and (R01DE119249 and R01CA136905) KSJ Elenitoba-Johnson, MD. Mangeng Cheng, PhD of Cephalon, Inc provided the small molecule ALK inhibitor.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Author Contributions: SRP McDonnell prepared manuscript, designed and conducted experiments. SR Hwang conducted experiments. V Basrur, PhD and KP Conlon performed mass spectrometry analysis. D Fermin, PhD carried out informatics analysis of mass spectrometry data. E Way, MD and MS Lim, MD/PhD evaluated the results of IHC studies. C Murga-Zamalloa, MD generated SU-DHL-1 and Karpas 299 GSK3β knockdown cells. Z Zeng, PhD and Y Zu, MD generated the SU-DHL-1 cells with tetracycline-inducible shRNA. KSJ Elenitoba-Johnson, MD and MS Lim, MD/PhD designed experiments and prepared manuscript.
References
- Amin HM, Lai R. Pathobiology of ALK + anaplastic large-cell lymphoma. Blood. 2007;110:2259–2267. doi: 10.1182/blood-2007-04-060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol. 1998;18:6951–6961. doi: 10.1128/mcb.18.12.6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof D, Pulford K, Mason DY, Morris SW. Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol. 1997;17:2312–2325. doi: 10.1128/mcb.17.4.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccalatte FE, Voena C, Riganti C, Bosia A, D’Amico L, Riera L, et al. The enzymatic activity of 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase is enhanced by NPM-ALK: new insights in ALK-mediated pathogenesis and the treatment of ALCL. Blood. 2009;113:2776–2790. doi: 10.1182/blood-2008-06-161018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock CJ, Prescott DM, Kirkpatrick JB. An evaluation of the double thymidine block for synchronizing mammalian cells at the G1-S border. Exp Cell Res. 1971;68:163–168. doi: 10.1016/0014-4827(71)90599-4. [DOI] [PubMed] [Google Scholar]
- Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, et al. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vidal A, Mazars A, Gautier EF, Prevost G, Payrastre B, Manenti S. Upregulation of the CDC25A phosphatase down-stream of the NPM/ALK oncogene participates to anaplastic large cell lymphoma enhanced proliferation. Cell Cycle. 2009;8:1373–1379. doi: 10.4161/cc.8.9.8302. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate-binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–2088. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Zhao N, Zeng Z, Chang CC, Zu Y. Synergistic growth inhibition of anaplastic large cell lymphoma cells by combining cellular ALK gene silencing and a low dose of the kinase inhibitor U0126. Cancer Gene Ther. 2010;17:633–644. doi: 10.1038/cgt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, et al. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, et al. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MS, Carlson ML, Crockett DK, Fillmore GC, Abbott DR, Elenitoba-Johnson OF, et al. The proteomic signature of NPM/ALK reveals deregulation of multiple cellular pathways. Blood. 2009;114:1585–1595. doi: 10.1182/blood-2009-02-204735. [DOI] [PubMed] [Google Scholar]
- Marzec M, Kasprzycka M, Liu X, Raghunath PN, Wlodarski P, Wasik MA. Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene. 2006;26:813–821. doi: 10.1038/sj.onc.1209843. [DOI] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- Ott GR, Tripathy R, Cheng M, McHugh R, Anzalone AV. Discovery of a potent inhibitor of anaplastic lymphoma kinase with in vivo antitumor activity. ACS Med Chem Lett. 2010;1:493–498. doi: 10.1021/ml100158s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420:345–361. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchatz H, Coluccia AM, Stano P, Marchesi E, Gambacorti-Passerini C. Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp Hematol. 2003;31:309–315. doi: 10.1016/s0301-472x(03)00007-9. [DOI] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 2001;61:2194–2199. [PubMed] [Google Scholar]
- van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21:1038–1047. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, et al. Glycogen synthase kinase 3alpha and 3βeta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]