

Abstract
Mice carrying mutations in both the dystrophin and utrophin genes die prematurely as a consequence of severe muscular dystrophy. Here, we demonstrate that intravascular administration of recombinant adeno-associated viral (rAAV) vectors carrying a microdystrophin gene restores dystrophin expression in the striated musculature of these animals, considerably reducing skeletal muscle pathology and extending lifespan. These findings suggest rAAV vector-mediated systemic gene transfer may be useful for treatment of serious neuromuscular disorders such as Duchenne muscular dystrophy (DMD).
Miniaturized dystrophin expression cassettes that restore sarcolemmal organization of the dystrophin-glycoprotein complex can be highly functional in transgenic mice1. However, delivering potentially therapeutic constructs throughout the musculature of animals with pre-existing muscular dystrophy using traditional methods has proven inefficient. Recently, we established that intravascular administration of recombinant adeno-associated viral vectors pseudotyped with the serotype-6 capsid (rAAV6) can transduce the striated musculature of adult mice2. This advance enables assessment of systemic microdystrophin delivery in animal models of disease. Historically the dystrophin-deficient mdx mouse3 has been employed as the primary model of DMD, although this animal does not experience the severe, body-wide dystrophy that shortens lifespan by 75% in patients4-6. The robustness of mdx mice is attributed to compensatory over-expression of the dystrophin-related protein utrophin, as knockout of both dystrophin and utrophin in mice causes progressive muscle wasting, impaired mobility and premature death5, 6. In this study, we tested the hypothesis that systemic administration of rAAV6-microdystrophin can ameliorate the pathology associated with severe muscular dystrophy in dystrophin/utrophin double-knockout (dko) mice.
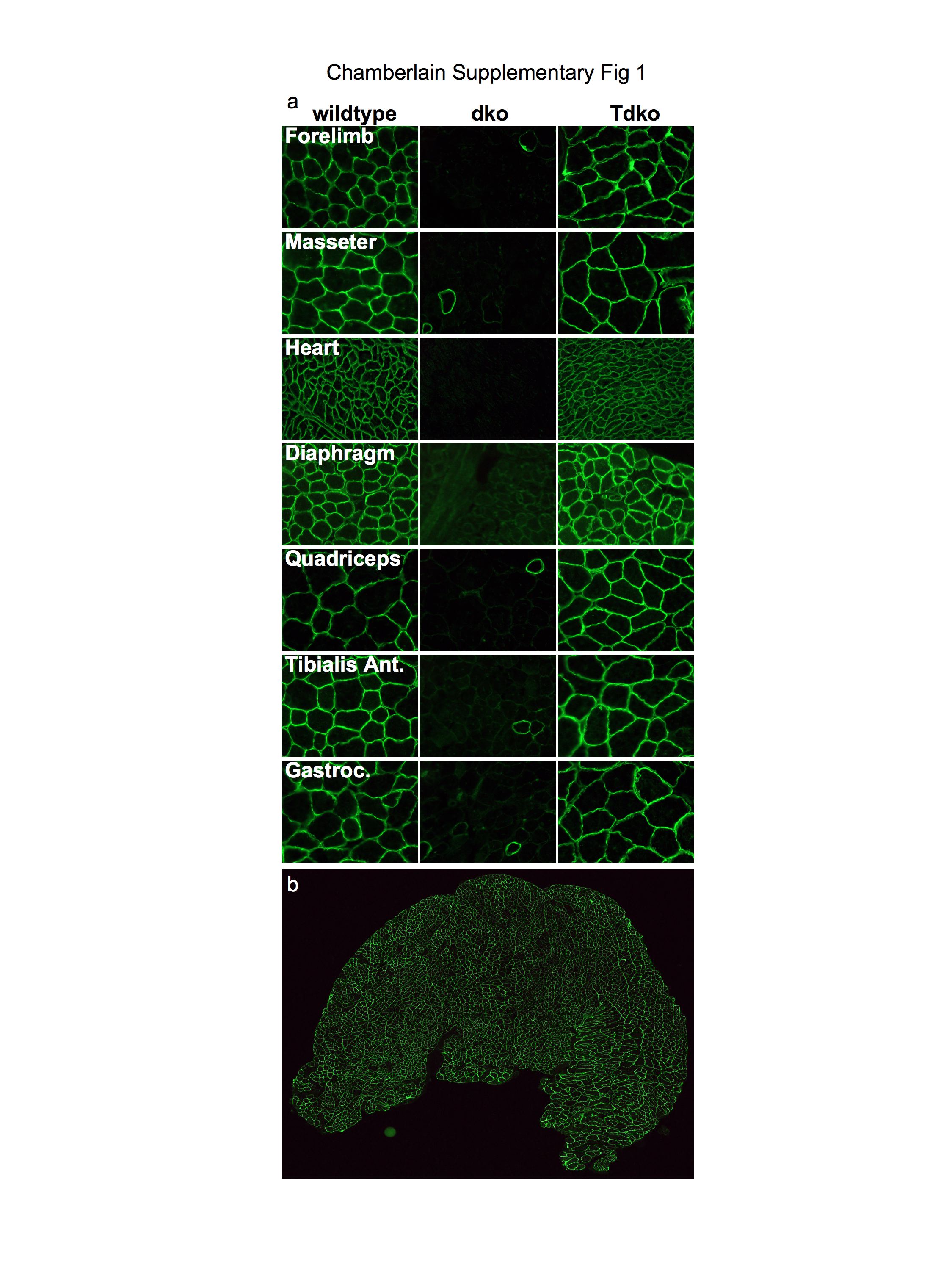
A single intravenous administration of ~3×1012 vector genomes of rAAV6-microdystrophin was administered to one month-old dko mice as described previously2, resulting in uniform, body-wide expression of dystrophin for at least one year (Supplementary Fig. 1 online). Because muscle deterioration leading to respiratory failure is the primary cause of death in patients with DMD7, we examined the effects of treatment upon mouse diaphragm (DIA) muscles4. Dystrophin expression in DIA muscles examined 18 weeks after treatment was widespread (Fig. 1a,b) and was associated with a significant reduction in the prevalence of smaller regenerating muscle cells compared with untreated muscles (Fig. 1c). Treatment also reduced the frequency of centrally-nucleated myofibers – a feature of muscle regeneration – by ~85% compared with the DIA muscles of untreated mice (Fig. 1d). Importantly, the DIA muscles of treated animals demonstrated more than two-fold increased normalized force-producing capacity compared with the muscles of untreated mice (Fig. 1e). Using a protocol we developed for subjecting muscles to progressively increased strain under contraction (Supplementary Methods online), we also determined that the DIA muscles of treated animals exhibited improved resistance to contraction-induced injury (the principal mechanism of muscle pathology attributed to dystrophin deficiency1, 8), that was essentially equal to that demonstrated by the muscles of wildtype animals (Fig. 1f).
Figure 1.

Systemic administration of rAAV6-microdystrophin enhances the structural and functional properties of respiratory muscles in dystrophic mice. (a) Restoration of dystrophin expression (green, left column) throughout the diaphragm muscle of a treated mouse (Tdko) contributes to improved muscle fiber size and organization (laminin and nuclei - red and blue respectively in middle panel; eosin and hematoxylin – pink and purple respectively in right column), as compared with the muscles of an untreated dystrophic mouse (dko), and a wildtype mouse (wt). Scale bar represents 100 μm. (b) Treatment restored dystrophin expression in the vast majority of diaphragm myofibers of treated mice (red), in contrast with no expression in the muscles of untreated mice (grey) and comprehensive expression in the muscles of wildtype mice (black). (c) Dystrophin-positive myofibers of treated diaphragm muscles (yellow) were less frequently small in size (mean myofiber diameter; wildtype, 22.1 ± 0.7, dystrophic, 14.9 ± 0.7, treated - dystrophin-positive fibers, 23.2 ± 1 μm), and (d) centrally-nucleated compared with the myofibers in the muscles of untreated mice. (e) Treatment also improved the peak isometric force-producing capacity of diaphragm muscles as normalized for cross-sectional area. (f) Contractile performance after consecutive eccentric contractions of progressively increasing strain (identified as % beyond optimal muscle length, and expressed in terms of force relative to respective initial output), demonstrates that treatment essentially corrects the susceptibility to contraction-induced injury otherwise observed in dystrophic diaphragm muscles.
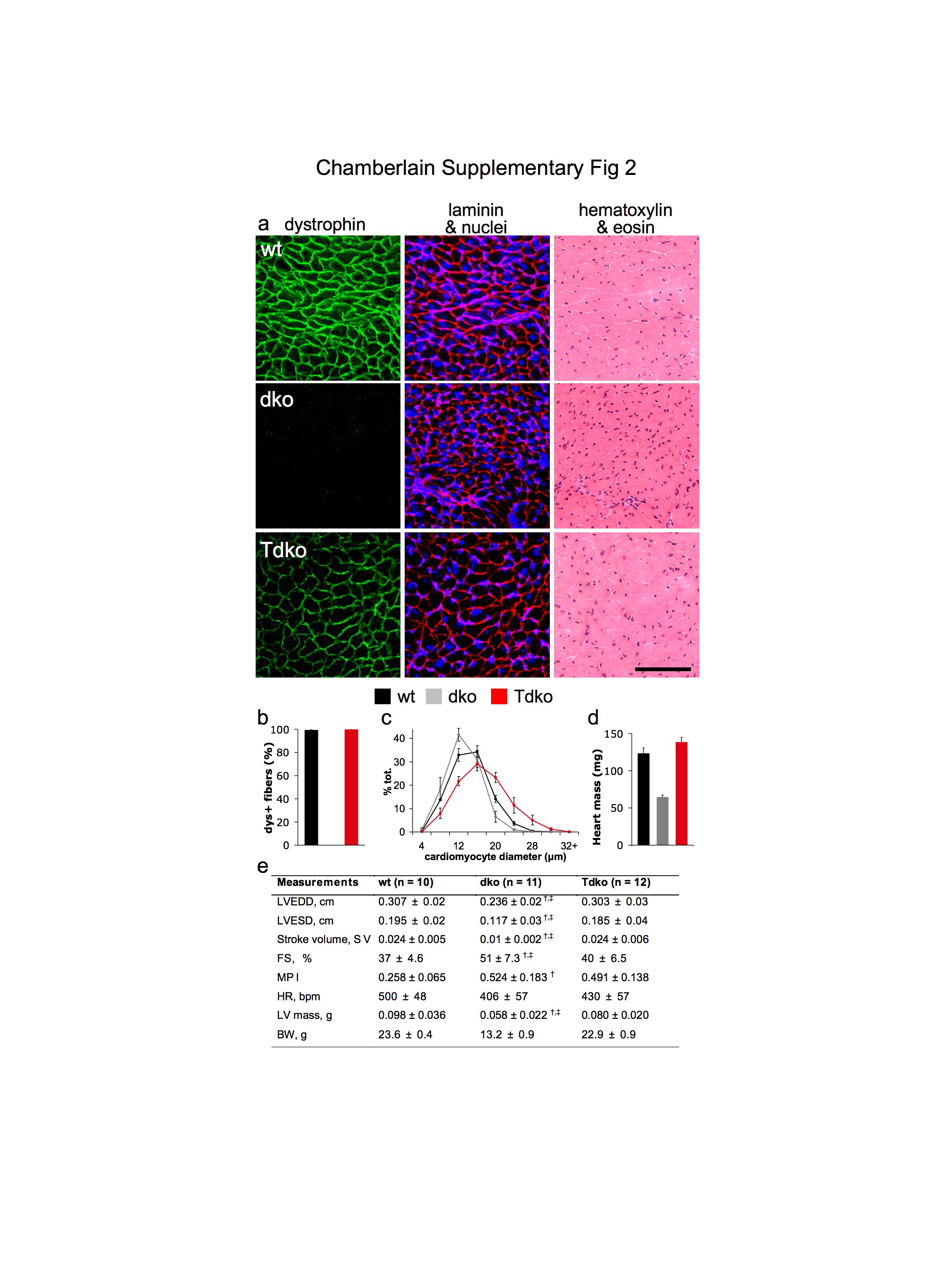
Among patients with DMD that utilize mechanical respiratory support, cardiac dysfunction attributed to dystrophin deficiency is an increasingly prevalent cause of death. Though prior reports suggest that dko mice succumb to skeletal muscle dysfunction before developing life-threatening cardiac disease9, we evaluated transduction of cardiac tissue in treated dko mice (Supplementary Fig. 2 online). Heart and body masses in untreated dko mice were approximately half those of wildtype animals. Though the hearts of untreated dko mice lacked dystrophin and comprised somewhat smaller cardiomyocytes than non-dystrophic hearts, the only indication of reduced cardiac function that we identified via echocardiography was an increased myocardial performance index - an inverse correlate of contractility10. In treated mice, dystrophin expression was restored throughout the myocardium (Supplementary Fig. 2a,b online). Treated animals exhibited moderately increased cardiomyocyte size, but considerably increased heart dimensions and mass, consistent with the approximately doubled body mass. (Supplementary Fig. 2c-e online). Treatment did not significantly alter myocardial performance index. These data are an encouraging demonstration that systemic rAAV6-microdystrophin administration can transduce the entire heart of an adult dystrophic mammal. As traditional methods cannot transduce the myocardium to any practical degree without invasive access11, 12 - a risk factor for frail patients - adaptation of this methodology may prove useful for the treatment of conditions associated with heart disease.
DMD patients experience incapacitating limb muscle degeneration, so we assessed the properties of the tibialis anterior (TA) hindlimb muscles of treated mice. Restoration of dystrophin expression (Fig. 2a) increased mean myofiber size by ~85% (Fig. 2b) and reduced the frequency of centrally-nucleated muscle fibers by ~60% (Fig. 2c) compared with the TA muscles of untreated mice. As a consequence of these effects, TA muscle mass was increased by more than 90% in treated dystrophic mice (Fig. 2d). rAAV6-microdystrophin administration corrected 65% of the deficit in force-producing capacity otherwise demonstrated by the TA muscles of untreated mice (Fig. 2e). Furthermore, treated TA muscles also exhibited improved resistance to contraction-induced injury that was essentially equal to that demonstrated by wildtype muscles within the physiological range13 (up to 15% strain, Fig. 2f).
Figure 2.

Systemic rAAV6-microdystrophin administration improves limb muscle function and extends the lifespan of dystrophic mice. (a) Dystrophin expression was restored in the majority of the myofibers comprising the Tibialis anterior muscles of mice examined 18 weeks after treatment (red), compared with the muscles of untreated dystrophic (grey) and wildtype (black) mice. (b) Dystrophin-positive myofibers in the muscles of treated mice (yellow) tended to be larger in diameter, (c) and less frequently centrally-nucleated than cells in the muscles of untreated mice. (d) Treatment was associated with an increased TA muscle mass, (e) and peak isometric force-producing capacity compared with untreated muscles (though unchanged specific force; wt, 240 ± 11, dko, 149 ± 5, Tdko, 145 ± 6 kN/m2). (f) Contractile performance expressed in terms of absolute force (upper) and performance relative to initial output (lower) demonstrates that the muscles of treated mice exhibit improved force output and resistance to contraction-induced injury when subjected to consecutive eccentric contractions of progressively increasing strain (identified as % beyond optimal muscle length). (g) Creatine kinase activity as measured in serum is markedly reduced in treated mice compared with untreated mice, yet remains moderately elevated compared with non-dystrophic animals. (h) Body mass (upper panel) in treated mice is increased compared with untreated animals, and comparable with the mass of wildtype mice. In cohorts monitored for lifespan (lower panel), treated mice (n = 8) live significantly longer than untreated mice (n = 70). (i) Untreated mice typically exhibit significant muscle wasting and kyphotic posture by 16 weeks of age, while treated mice retain considerably greater muscularity.
Having studied specific striated muscles that influence physical condition, we considered whole-body indices of disease state. Treatment corrected more than 75% of the discrepancy in serum creatine kinase levels (a measure of myocellular degeneration) normally observed between untreated and non-dystrophic mice (Fig. 2g). Where untreated dko mice exhibited reduced body mass and died prematurely (80% mortality at 15 weeks of age) compared with wildtype mice, treated littermates demonstrated increased body mass beyond 2 weeks post-treatment, and considerably extended life span (at the time of writing, cohort mean age greater than 40 weeks, Fig. 2h). The progressive muscle wasting observed in untreated dystrophic mice impaired posture and ambulation (Fig. 2i and Supplementary Video 1 online), while treated mice retained sufficient muscularity and mobility to readily use a voluntary running wheel where untreated mice could not (Fig. 2i and Supplementary Video 2 online).
This is the first study to demonstrate that an intervention can restore dystrophin expression in the respiratory, cardiac and limb musculature of dystrophin/utrophin double-knockout mice, resulting in improved muscle function and an extended lifespan. The widespread and persistent transduction achieved without serious pathogenic effects following administration of rAAV6 vectors in this manner suggests that systemic gene replacement strategies may be beneficial for treatment of serious neuromuscular disorders such as DMD.
Supplementary Material
{kind=link}
{kind=link}
ACKNOWLEGEMENTS
We thank Joshua R. Sanes for the utrophin-null mice, and Yan Tesch (University of Washington, Seattle) for assistance with initial set-up of the dystrophic mouse breeding colony. Supported by a Development Grant from the Muscular Dystrophy Association (to PG), by grants from the Muscular Dystrophy Association (to JSC) and the National Institutes of Health (to JSC, SCF and MEA, and CEM).
REFERENCES
- 1.Harper SQ, et al. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 2.Gregorevic P, et al. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulfield G, et al. Proc. Natl. Acad. Sci. U. S. A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stedman HH, et al. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 5.Grady RM, et al. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 6.Deconinck AE, et al. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 7.Gozal D. Pediatr. Pulmonol. 2000;29:141–150. doi: 10.1002/(sici)1099-0496(200002)29:2<141::aid-ppul9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 8.Petrof BJ, et al. Proc. Natl. Acad. Sci. U. S. A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rafael JA, et al. Nat. Genet. 1998;19:79–92. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 10.Collins KA, et al. Physiol. Genomics. 2003;13:227–239. doi: 10.1152/physiolgenomics.00005.2003. [DOI] [PubMed] [Google Scholar]
- 11.Greelish JP, et al. Nat. Med. 1999;5:439–443. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- 12.Yue Y, et al. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawkins D, Bey M. J. Biomech. 1997;30:63–70. doi: 10.1016/s0021-9290(96)00094-2. [DOI] [PubMed] [Google Scholar]
- 14.Allen JM, et al. Mol. Ther. 2000;1:88–95. doi: 10.1006/mthe.1999.0010. [DOI] [PubMed] [Google Scholar]
- 15.Gregorevic P, et al. Am. J. Pathol. 2002;161:2263–2272. doi: 10.1016/S0002-9440(10)64502-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.