

Abstract
Background
Traumatic brain injury (TBI) initiates a neuroinflammatory response that increases the risk of TBI-related mortality. Acute alcohol intoxication at the time of TBI is associated with improved survival. Ethanol is recognized as a systemic immunomodulator that may also impart neuroprotection. The effects of alcohol on TBI-induced neuroinflammation, however, are unknown. We hypothesized that ethanol treatment prior to TBI may provide neuroprotection by diminishing the neuroinflammatory response to injury.
Materials and methods
Mice underwent gavage with ethanol (EtOH) or water (H2O) prior to TBI. Animals were subjected to blunt TBI or sham injury (Sham). Posttraumatic rapid righting reflex (RRR) and apnea times were assessed. Cerebral and serum samples were analyzed by ELISA for inflammatory cytokine levels. Serum neuron-specific enolase (NSE), a biomarker of injury severity, was also measured.
Results
Neurologic recovery from TBI was more rapid in H2O-treated mice compared with EtOH-treated mice. However, EtOH/TBI mice had a 4-fold increase in RRR time compared with EtOH/Sham, whereas H2O/TBI mice had a 15-fold increase in RRR time compared with H2O/Sham. Ethanol intoxication at the time of TBI significantly increased posttraumatic apnea time. Preinjury EtOH treatment was associated with reduced levels of proinflammatory cytokines IL-6, KC, MCP-1, and MIP-1α post TBI. NSE was significantly increased post injury in the H2O/TBI group compared with H2O/Sham but was not significantly reduced by EtOH pretreatment.
Conclusions
Alcohol treatment prior to TBI reduces the local neuroinflammatory response to injury. The decreased neurologic and inflammatory impact of TBI in acutely intoxicated patients may be responsible for improved clinical outcomes.
Keywords: Traumatic brain injury, Alcohol, Inflammation, Neuroinflammation
1. Introduction
Traumatic brain injury (TBI) remains the most common cause of death and disability in children and young adults. One and a half million Americans annually suffer a head injury, resulting in 50,000 deaths and pervasive physical or cognitive disability in nearly 100,000 individuals, with an annual total lifetime cost of $60 billion [1-3]. Because of the paucity of recent significant therapeutic innovations to improve the clinical outcome of head-injured patients, there has been ongoing research into understanding and modulating the pathophysiology of TBI. Posttraumatic cerebral and systemic inflammatory responses are increasingly recognized as playing an integral role in the progression of cerebral cellular damage following head injury, thereby contributing to the risk of mortality following TBI. Experimental TBI models have demonstrated that attenuation of the posttraumatic inflammatory cascade may diminish the severity and mortality of cerebral injury [4,5].
Alcohol consumption increases the risk of all types of trauma. Up to 50% of all patients suffering TBI are intoxicated at the time of injury [6]. In addition, high serum ethanol levels can be associated with increased occurrence of posttraumatic hypotension and apnea [6-8]. Recent clinical studies have shown that ethanol intoxication at the time of TBI, in contrast to conventional wisdom, reduces the risk of mortality and may be associated with improved neurologic functional recovery [1,6,9,10]. Experimental animal studies suggest that ethanol may provide neuroprotection following TBI [11-13]. The mechanism of this neuroprotective effect, however, is not completely understood.
Acute alcohol exposure suppresses the systemic proinflammatory cytokine release following inflammatory challenges such as traumatic injury [14]. The effect of preinjury ethanol intake on the neuroinflammatory response to blunt TBI has not been investigated. In the current studies, we tested the hypothesis that ethanol treatment prior to TBI may provide neuroprotection by diminishing the local neuroinflammatory response to injury.
2. Materials and methods
2.1. Animals
Male C57/BL6 mice weighing 22–26 g were purchased from Charles River Laboratories (Wilmington, MA), fed standard laboratory diet and water ad libitum, and acclimated for 2–3 wk in a climate-controlled room with a 12-h light-dark cycle. Experiments were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
2.2. Acute ethanol exposure
Mice were randomized to receive either ethanol (EtOH) or water (H2O) as control prior to undergoing TBI or sham injury (n = 5 each sham group, n = 8 each TBI group). Ethanol was administered as a single 5 g/kg dose of a 32% (vol/vol) ethanol solution in water by intragastric lavage following light sedation in 2% isoflurane (EtOH group) [15]. At the chosen intragastric ethanol dose of 5 g/kg for this study, blood alcohol concentration rises sharply to a maximum level of 0.25%–0.3% 1 h after administration, with a steady subsequent decline to zero >8 h after the initial dose. This dose of ethanol provides moderately intoxicating but not incapacitating behavioral effects, including stupor and impaired sensation, memory, and motor function [15,16]. Weight-based volumes of 0.45–0.6 mL were delivered with a 20-gauge gavage needle. Vehicle controls were dosed with water in weight-matched volumes (H2O group).
2.3. Head injury model
Blunt traumatic brain injury was induced 1 h after intragastric EtOH or H2O administration utilizing a weight-drop device. Mice were anesthetized for 2 min with 2% isoflurane in 100% oxygen at 1 L/min. Animals were then placed in a prone position on a platform below the head injury device so that the central region between coronal and lambdoid sutures was centered beneath the guide tube [17]. Moderately severe closed head injury was induced by striking the head from a 2-cm height with a 415-g cylindrical weight with a 1-cm-wide contact surface placed within a plastic guide tube. Rebound impact was prevented. This injury was not associated with a skull fracture or extra-axial hemorrhage. Sham-injury mice were anesthetized and positioned below the guide tube but not subjected to head injury.
Following traumatic weight impact, head-injured and sham mice underwent acute neurologic evaluation using the righting reflex response (RRR). The RRR was defined as the animal’s ability to right itself to a prone position three times consecutively after being placed supine immediately after injury [18]. Time to achieve righting was recorded for each animal. Postinjury apnea duration was also recorded. No occurrences of postinjury convulsions or visible limb paralysis were noted.
2.4. Serum and tissue analysis
Animals were sacrificed at intervals over the first 24 h following TBI or sham. Blood was obtained by cardiac puncture and the brain was excised. Serum and left cortical brain samples were analyzed at each time point for levels of inflammatory cytokines using customized multiplex enzyme-linked immunosorbent assay (ELISA) (Quansys, Logan, UT). Fifteen cytokines and chemokines were evaluated in the ELISA analysis, including GM-CSF, IFNγ, IL-1β, IL-4, IL-6, IL-10, IL-12p70, IL-17, IP-10, KC, MCP-1, MDC, MIP-1α, RANTES, and TNFα. Cortical samples were homogenized in 1 mL phosphate-buffered saline containing a complete protease inhibitor cocktail (Roche, Indianapolis, IN). Supernatants were centrifuged three times at 12,000g for 15 min each. Serum was extracted from blood samples by centrifugation at 8000 rpm for 10 min in serum separator tubes and stored at −80°C until analysis. The multiplex ELISA was performed per manufacturer instructions. Cerebral cytokine levels were normalized to cortical protein content using a BCA Protein Assay Kit (Thermo Scientific, Rockford, IL).
Neuron-specific enolase (NSE) was measured as a serum biomarker of head injury severity using a commercially available human ELISA kit (Immuno-Biological Laboratories, Inc, Minneapolis, MN).
Statistical analysis was performed by two-tailed t-test or one-way analysis of variance with Tukey post hoc analysis for multiple comparisons. Spearman rank order was used to test correlations between quantitative variables for significance using SigmaStat 3.5 (Systat, Chicago, IL). Data are reported as mean ± SEM. A P value of <0.05 was considered significant.
3. Results
3.1. Effects of ethanol on the neurologic response to TBI
Mice pretreated with H2O prior to sham injury demonstrated minimal loss of the RRR, whereas mice pretreated with EtOH prior to sham injury had a significant delay in the RRR, consistent with acute alcohol intoxication (Fig. 1A). After injury, mice sustaining TBI demonstrated a significantly longer loss of the RRR compared with their respective sham groups. Interestingly, although the RRR after EtOH pretreatment and subsequent TBI was significantly longer than after H2O ingestion, postintoxication TBI had a less profound additional impact on neurologic recovery than head injury after H2O alone (4-fold increase RRR time versus 15-fold increase RRR time, EtOH/TBI : EtOH/Sham versus H2O/TBI : H2O/Sham; Fig. 1A).
Fig. 1.
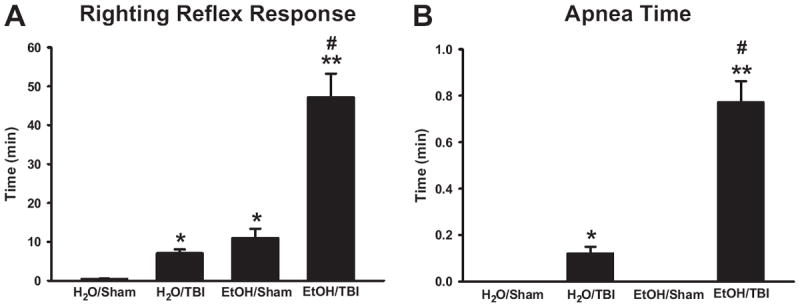
Righting reflex response recovery time (A) and posttraumatic apnea time (B) following H2O or EtOH administration prior to blunt head injury. *P < 0.001 versus H2O/Sham, **P < 0.001 versus EtOH/Sham, #P < 0.001 versus H2O/TBI.
TBI significantly increased the duration of postinjury apnea time as compared with sham-treated mice in both EtOH and H2O pretreatment groups (Fig. 1B). Furthermore, preinjury administration of EtOH significantly increased postinjury apnea time compared with H2O pretreatment (Fig. 1B).
No mortality occurred beyond the time of initial head injury. Any deaths were noted immediately after weight-drop impact without postinjury neurologic recovery and were not considered for further statistical analysis. EtOH administration alone did not result in any mortality.
3.2. Ethanol decreases the systemic inflammatory response after TBI
Serum levels of the proinflammatory mediators IL-6 and KC were increased after induction of TBI, peaking at 3 h (Fig. 2A and B). Pretreatment of mice with EtOH decreased serum IL-6 after TBI as compared with control mice (Fig. 2A and B). Treatment of mice with EtOH alone had no effect on serum cytokine levels (Fig. 2A and B). Analysis of serum levels of the 13 additional cytokines and chemokines revealed no further differences among the four experimental groups.
Fig. 2.
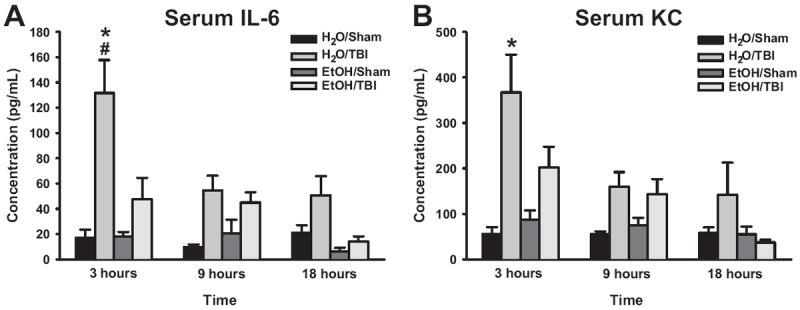
Serum proinflammatory cytokine levels of IL-6 (A) and KC (B) during the first 24 h following blunt head injury. *P < 0.05 versus H2O/Sham, #P < 0.05 versus H2O/TBI.
3.3. Ethanol attenuates the neuroinflammatory response after TBI
Evaluation of brain cytokine levels demonstrated significant increases in IL-6, MCP-1, MIP-1α, and KC after TBI (Fig. 3). IL-6 and KC were significantly increased in mice within 9 h of TBI (Fig. 3A and B). Pretreatment with ethanol completely abrogated the increased expression of IL-6 and KC in brain tissue (Fig. 3A and B). Cerebral levels of MCP-1 and MIP-1α were significantly increased 9 and 18 h after TBI (Fig. 3C and D, respectively). Similar to the effect on IL-6, pretreatment with ethanol completely inhibited these increases (Fig. 3C and D).
Fig. 3.

Cerebral tissue levels of IL-6 (A), KC (B), MCP-1 (C), and MIP-1α (D) during the first 24 h after TBI. *P < 0.05 versus H2O/Sham, #P < 0.05 versus EtOH/TBI.
3.4. Serum NSE release post TBI
To determine if the attenuated neuroinflammatory response observed in the EtOH/TBI groups resulted in reduced markers of brain injury, serum NSE levels were measured during the first 24 h after TBI. Mice undergoing TBI after H2O pretreatment demonstrated significantly increased levels of NSE as compared with sham-treated mice (Fig. 4). Reductions in serum NSE in the EtOH/TBI groups were not significantly different from H2O/TBI groups. EtOH administration alone without subsequent TBI had no effect on serum NSE, indicating that direct cerebral injury is necessary to cause NSE release from damaged neurons.
Fig. 4.
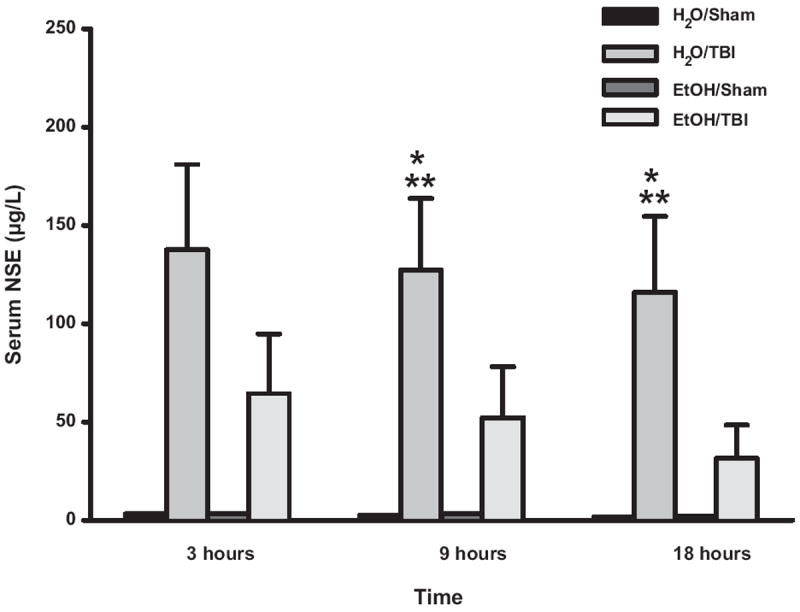
Serum NSE levels after TBI or sham injury. *P < 0.05 versus H2O/Sham, **P < 0.05 versus EtOH/Sham.
In order to increase our understanding of the relationship between NSE release, cerebral neuroinflammation, and initial injury severity, we examined the relationship between serum NSE levels and RRR time. Serum NSE levels significantly correlated to the initial RRR time for both the H2O/TBI and EtOH/TBI groups. In addition, serum NSE levels also significantly correlated to the cerebral levels of proinflammatory cytokines IL-6, KC, MCP-1, and MIP-1α following TBI for both H2O and EtOH pretreatment groups (Table).
Table.
NSE correlations.
| Spearman rank-order coefficient (r) | P value | |
|---|---|---|
| NSE versus RRR | ||
| H2O/TBI | 0.65 | <0.001 |
| EtOH/TBI | 0.60 | <0.001 |
| NSE versus cerebral IL-6 | ||
| H2O/TBI | 0.60 | <0.001 |
| EtOH/TBI | 0.49 | <0.001 |
| NSE versus cerebral KC | ||
| H2O/TBI | 0.36 | <0.01 |
| EtOH/TBI | 0.34 | 0.01 |
| NSE versus cerebral MCP-1 | ||
| H2O/TBI | 0.62 | <0.001 |
| EtOH/TBI | 0.55 | <0.001 |
| NSE versus cerebral MIP-1α | ||
| H2O/TBI | 0.59 | <0.001 |
| EtOH/TBI | 0.31 | 0.03 |
4. Discussion
In the present study, we examined the effects of ethanol treatment prior to traumatic brain injury. We demonstrated that a murine model of concussive blunt head injury induces a systemic as well as a cerebral inflammatory response and characterized the time course of the inflammatory profile over the first 24 h post injury. Using this model of TBI, we investigated the effects of preinjury ethanol treatment on the systemic and cerebral inflammatory status, neurologic recovery, and neuronal damage following blunt head injury. Our data suggest that the presence of acute alcohol intoxication prior to TBI may provide neuroprotection via modulation of the systemic and neuroinflammatory responses to head injury.
Evaluation of posttraumatic neurologic recovery using RRR time demonstrated that TBI and ethanol administration alone each result in similar neurodepressive insults. Our results provide evidence that, in our model, TBI itself impacts neurologic recovery more profoundly when occurring without preceding ethanol use. Without further invasive interventions such as intubation, however, the neurologic impact of post-intoxication TBI in this model was limited by the risk of death with increased severity of brain injury. Our findings support previous work in preinjury intoxication models that have demonstrated diminished posttraumatic cognitive impairments and more rapid return to baseline task performance in animals undergoing ethanol pretreatment prior to TBI by direct cerebral contusion [11,12]. Furthermore, our data demonstrate that posttraumatic neurologic status following head injury may be significantly altered by preinjury ethanol intake. These results support the clinical observation that preinjury alcohol use may confound initial Glasgow Coma Score (GCS) assessment with an artificially low score due to the effects of alcohol rather than accurately characterizing the underlying brain injury [9].
Posttraumatic apnea resulting in early systemic hypoxia and reduced cerebral oxygenation contributes to the development of secondary brain injury. In contrast to primary brain injury, which encompasses the cerebral damage induced by the original insult, secondary brain injury results from progressive vasogenic and cellular edema that causes ongoing neuronal ischemia and is responsible for the majority of inpatient mortality following TBI [19]. Our results provide strong evidence that preinjury ethanol intoxication is a significant risk factor for post-TBI apnea. In addition, the posttraumatic apnea in this head injury model may also delay clearance of the inhaled anesthetic used for sedation during induction of TBI, thereby further contributing to prolonged post-TBI neurologic recovery. Others have shown increased duration of postinjury apnea caused by lower ventilation and hypercapnic response sensitivity in a porcine model of TBI [7,8]. These data suggest that preinjury ethanol use may exacerbate secondary brain injury, leading to worsened outcome from TBI.
Assessment of the posttraumatic systemic inflammatory response demonstrated significant reduction of serum IL-6 by ethanol pretreatment. Following H2O pretreatment, TBI induced a rapid increase of serum IL-6 and KC levels to peak values at 3 h post injury with sustained elevation of serum levels to 24 h. By contrast, IL-6 levels were significantly reduced in head-injured animals undergoing EtOH pretreatment. In addition, posttraumatic systemic IL-6 and KC profiles in intoxicated TBI mice demonstrated a delayed peak elevation to 6 h post TBI with a normalization to sham serum levels by 18 h. Previous studies have similarly demonstrated a diminished serum IL-6 and KC release in animals subjected to ethanol treatment prior to inflammatory challenge [14,20]. Our data extend previous findings by demonstrating a reduced systemic proinflammatory response to TBI following acute ethanol intoxication. Previous studies concerning alcohol administration suggest that this diminished systemic inflammatory response may be related, at least in part, to increased corticosteroid release following ethanol administration [21].
Analysis of the cerebral expression of proinflammatory cytokines and chemokines following TBI demonstrated a significant attenuation of the acute neuroinflammatory response in acutely intoxicated animals. Post-TBI levels of the proinflammatory mediators IL-6, KC, MCP-1, and MIP-1α in the cortex were significantly reduced by ethanol pretreatment. These findings are important and have potential clinical impact. Following TBI, sustained elevations of IL-6 and IL-8 (the human analogue of murine KC) in serum and cerebrospinal fluid have been clinically associated with increased mortality and worsened neurologic outcomes [22-24]. Additionally, MCP-1 expression has been shown to be elevated at the site of cerebral contusion in experimental head injury models as well as human clinical reports [25,26]. Posttraumatic cerebral MIP-1α has not been studied to date in TBI models or human clinical studies. Taken together, our data demonstrate significant reductions of critical neuroinflammatory cytokines by preinjury ethanol intoxication. Future studies should explore the optimal timing and dose of periinjury ethanol for maximal reduction of the postinjury neuroinflammatory response, potentially investigating the administration of ethanol as a therapeutic immunomodulator in the postinjury period.
Neuron-specific enolase is a recognized serum biomarker of TBI released by direct neuronal injury. NSE correlates clinically with the degree of head injury as determined by Injury Severity Score and radiographic findings, as well as with ultimate neurologic outcome [27-29]. Although preinjury administration of EtOH did not significantly reduce post-TBI serum NSE release, levels of NSE in the EtOH/TBI groups were not statistically different from those in the EtOH/Sham groups at any time point. Following TBI, serum NSE levels were significantly increased in the H2O-pretreated animals at 9 and 18 h post injury compared with sham. These findings, however, may support previous clinical work that demonstrated no change in plasma NSE levels with increasing alcohol intoxication [30]. The serum profile of cerebral NSE release correlated with that of cerebral proinflammatory cytokine expression. These results suggest that an ongoing neuroinflammatory response exacerbates neuronal injury induced by a closed head injury. Although this is the first study to demonstrate a correlation between cerebral proinflammatory cytokine release and serum NSE levels, a single clinical study has identified correlations between cerebrospinal fluid NSE and IL-6 as well as serum NSE and IL-6 levels [31]. Our data further show a correlation between serum NSE levels and postinjury neurologic status, as measured by RRR in the murine TBI model. Although the clinical correlation between GCS and serum NSE remains controversial in the literature, some studies have observed that patients with lower GCS have a higher initial NSE level [27].
5. Conclusions
TBI following acute alcohol intoxication affects the initial neurologic assessment of the head-injured patient and may lead to overestimation of brain injury severity based on GCS alone. Preinjury ethanol intoxication increases the risk for posttraumatic apnea and, therefore, secondary brain damage following closed head injury. Conversely, alcohol intoxication prior to TBI attenuates both the systemic and local neuroinflammatory response to TBI. Serumbiomarkers, such as NSE, may be utilized as surrogate markers for neurologic status and ongoing cell damage secondary to neuroinflammation after the initial injury. The decreased neurologic and inflammatory impact of TBI in acutely intoxicated patients may be responsible for improved clinical outcomes. Despite the recently demonstrated improvement in clinical outcomes of intoxicated head-injured patients, the multitude of effects of preinjury ethanol ingestion on secondary brain injury remain to be comprehensively studied both at the benchtop and at the bedside.
Acknowledgments
This work was supported in part by awards from NIH training grant T32 GM08478.
References
- 1.Salim A, Ley EJ, Cryer HG, Margulies DR, Ramicone E, Tillou A. Positive serum ethanol level and mortality in moderate to severe traumatic brain injury. Arch Surg. 2009;144:865. doi: 10.1001/archsurg.2009.158. [DOI] [PubMed] [Google Scholar]
- 2.Finkelstein E, Corso P, Miller T. The incidence and economic burden of injuries in the United States. New York: Oxford University Press; 2006. [Google Scholar]
- 3.Traumatic brain injury in the United States a report to Congress. Vol. 2004. Atlanta: Georgia Centers for Disease Control; 2001. [Google Scholar]
- 4.Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response to head injury. Injury. 2007;38:1392. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury-an inflammatory disease? Brain Res Rev. 2005;48:388. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 6.Tien HC, Tremblay LN, Rizoli SB, et al. Association between alcohol and mortality in patients with severe traumatic head injury. Arch Surg. 2006;141:1185. doi: 10.1001/archsurg.141.12.1185. discussion 1192. [DOI] [PubMed] [Google Scholar]
- 7.Zink BJ, Feustel PJ. Effects of ethanol on respiratory function in traumatic brain injury. J Neurosurg. 1995;82:822. doi: 10.3171/jns.1995.82.5.0822. [DOI] [PubMed] [Google Scholar]
- 8.Zink BJ, Walsh RF, Feustel PJ. Effects of ethanol in traumatic brain injury. J Neurotrauma. 1993;10:275. doi: 10.1089/neu.1993.10.275. [DOI] [PubMed] [Google Scholar]
- 9.O’Phelan K, McArthur DL, Chang CW, Green D, Hovda DA. The impact of substance abuse on mortality in patients with severe traumatic brain injury. J Trauma. 2008;65:674. doi: 10.1097/TA.0b013e31817db0a5. [DOI] [PubMed] [Google Scholar]
- 10.Vickery CD, Sherer M, Nick TG, et al. Relationships among premorbid alcohol use, acute intoxication, and early functional status after traumatic brain injury. Arch Phys Med Rehabil. 2008;89:48. doi: 10.1016/j.apmr.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 11.Janis LS, Hoane MR, Conde D, Fulop Z, Stein DG. Acute ethanol administration reduces the cognitive deficits associated with traumatic brain injury in rats. J Neurotrauma. 1998;15:105. doi: 10.1089/neu.1998.15.105. [DOI] [PubMed] [Google Scholar]
- 12.Kelly DF, Lee SM, Pinanong PA, Hovda DA. Paradoxical effects of acute ethanolism in experimental brain injury. J Neurosurg. 1997;86:876. doi: 10.3171/jns.1997.86.5.0876. [DOI] [PubMed] [Google Scholar]
- 13.Tureci E, Dashti R, Tanriverdi T, Sanus GZ, Oz B, Uzan M. Acute ethanol intoxication in a model of traumatic brain injury the protective role of moderate doses demonstrated by immunoreactivity of synaptophysin in hippocampal neurons. Neurol Res. 2004;26:108. doi: 10.1179/016164104773026633. [DOI] [PubMed] [Google Scholar]
- 14.Greiffenstein P, Mathis KW, Stouwe CV, Molina PE. Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcohol Clin Exp Res. 2007;31:704. doi: 10.1111/j.1530-0277.2007.00355.x. [DOI] [PubMed] [Google Scholar]
- 15.Carson EJ, Pruett SB. Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol Clin Exp Res. 1996;20:132. doi: 10.1111/j.1530-0277.1996.tb01055.x. [DOI] [PubMed] [Google Scholar]
- 16.Livy DJ, Parnell SE, West JR. Blood ethanol concentration profiles a comparison between rats and mice. Alcohol. 2003;29:165. doi: 10.1016/s0741-8329(03)00025-9. [DOI] [PubMed] [Google Scholar]
- 17.Tang Y, Yukihiro N, Hasegawa T, Nabeshima T. A concussive-like brain injury model in mice (I) impairment in learning and memory. J Neurotrauma. 1997;14:851. doi: 10.1089/neu.1997.14.851. [DOI] [PubMed] [Google Scholar]
- 18.Dixon CE, Lyeth G, Povlishock JT, et al. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 19.Ghajar J. Traumatic brain injury. Lancet. 2000;356:923. [Google Scholar]
- 20.Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33:220. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottesfeld Z, Moore AN, Dash PK. Acute ethanol intake attenuates inflammatory cytokines after brain injury in rats: a possible role for corticosterone. J Neurotrauma. 2002;19:317. doi: 10.1089/089771502753594882. [DOI] [PubMed] [Google Scholar]
- 22.Chiaretti A, Genovese O, Aloe L, et al. Interleukin 1beta and interleukin 6 relationship with paediatric head trauma severity and outcome. Childs Nerv Syst. 2005;21:185. doi: 10.1007/s00381-004-1032-1. [DOI] [PubMed] [Google Scholar]
- 23.Kushi H, Saito T, Makino K, Hayashi N. IL-8 is a key mediator of neuroinflammation in severe traumatic brain injuries. Acta Neurochir Suppl. 2003;86:347. doi: 10.1007/978-3-7091-0651-8_74. [DOI] [PubMed] [Google Scholar]
- 24.Whalen MJ, Carlos TM, Kochanek PM, et al. Interleukin-8 is increased in cerebrospinal fluid of children with severe head injury. Crit Care Med. 2000;28:929. doi: 10.1097/00003246-200004000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Rhodes JK, Sharkey J, Andrews PJ. The temporal expression, cellular localization, and inhibition of the chemokines MIP-2 and MCP-1 after traumatic brain injury in the rat. J Neurotrauma. 2009;26:507. doi: 10.1089/neu.2008.0686. [DOI] [PubMed] [Google Scholar]
- 26.Stefini R, Catenacci E, Piva S, et al. Chemokine detection in the cerebral tissue of patients with posttraumatic brain contusions. J Neurosurg. 2008;108:958. doi: 10.3171/JNS/2008/108/5/0958. [DOI] [PubMed] [Google Scholar]
- 27.Guzel A, Er U, Tatli M, et al. Serum neuron-specific enolase as a predictor of short-term outcome and its correlation with Glasgow Coma Scale in traumatic brain injury. Neurosurg Rev. 2008;31:439. doi: 10.1007/s10143-008-0148-2. discussion 444. [DOI] [PubMed] [Google Scholar]
- 28.Pelinka LE, Hertz H, Mauritz W, et al. Nonspecific increase of systemic neuron-specific enolase after trauma clinical and experimental findings. Shock. 2005;24:119. doi: 10.1097/01.shk.0000168876.68154.43. [DOI] [PubMed] [Google Scholar]
- 29.Vos PE, Lamers KJ, Hendriks JC, et al. Glial and neuronal proteins in serum predict outcome after severe traumatic brain injury. Neurology. 2004;62:1303. doi: 10.1212/01.wnl.0000120550.00643.dc. [DOI] [PubMed] [Google Scholar]
- 30.Mussack T, Biberthaler P, Kanz KG, et al. Immediate S-100B and neuron-specific enolase plasma measurements for rapid evaluation of primary brain damage in alcohol-intoxicated, minor head-injured patients. Shock. 2002;18:395. doi: 10.1097/00024382-200211000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Pleines UE, Morganti-Kossmann MC, Rancan M, Joller H, Trentz O, Kossmann T. S-100 beta reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J Neurotrauma. 2001;18:491. doi: 10.1089/089771501300227297. [DOI] [PubMed] [Google Scholar]