

Abstract
TDP-43 proteinopathies are clinically and genetically heterogeneous diseases that had been considered distinct from classical amyloid diseases. Here, we provide evidence for the structural similarity between TDP-43 peptides and other amyloid proteins. Atomic force microscopy and electron microscopy examination of peptides spanning a previously defined amyloidogenic fragment revealed a minimal core region that forms amyloid fibrils similar to the TDP-43 fibrils detected in FTLD-TDP brain tissues. An ALS-mutant A315E amyloidogenic TDP-43 peptide is capable of cross-seeding other TDP-43 peptides and an amyloid-β peptide. Sequential Nuclear Overhauser Effects and double-quantum-filtered correlation spectroscopy in nuclear magnetic resonance (NMR) analyses of the A315E-mutant TDP-43 peptide indicate that it adopts an anti-parallel β conformation. When added to cell cultures, the amyloidogenic TDP-43 peptides induce TDP-43 redistribution from the nucleus to the cytoplasm. Neuronal cultures in compartmentalized microfluidic-chambers demonstrate that the TDP-43 peptides can be taken up by axons and induce axonotoxicity and neuronal death, thus recapitulating key neuropathological features of TDP-43 proteinopathies. Importantly, a single amino acid change in the amyloidogenic TDP-43 peptide that disrupts fibril formation also eliminates neurotoxicity, supporting that amyloidogenesis is critical for TDP-43 neurotoxicity.
INTRODUCTION
Intense efforts have been made to understand the molecular pathogenesis of neurodegenerative diseases, including dementia [e.g. Alzheimer's disease (AD) and frontotemporal lobar degeneration (FTLD)], motor neuron diseases [e.g. Amyotrophic lateral sclerosis (ALS)], Parkinson's disease (PD) and spongiform encephalopathies (e.g. prion diseases). One common characteristic of these neurodegenerative diseases is the aggregation and misfolding of disease-associated proteins in the affected neurons (1,2). Such proteins include amyloid-β in AD, α-synuclein in PD, prion proteins in spongiform encephalopathy and more recently, TDP-43 in TDP-43 proteinopathies. A shared biochemical feature of these proteins is the propensity to form amyloid fibrils [for recent reviews see (1,3) and references within; (4)]. However, the structural basis for TDP-43 amyloidogenesis and its role in neurotoxicity remain to be elucidated.
TDP-43 is a characteristic constituent of protein inclusion bodies that define TDP-43 proteinopathies, a group of devastating neurodegenerative disorders, including FTLD-TDP and ALS-TDP (5). As a multifunctional DNA/RNA binding protein, TDP-43 plays critical roles in RNA processing, including post-transcriptional regulation, alternative splicing and microRNA biogenesis (reviewed in 2,3). Previous data have demonstrated that ALS-associated mutations cluster in the carboxyl terminal glycine-rich domain of TDP-43 (reviewed in 3). Although the RNA recognition motifs in the RNA-binding domain of TDP-43 have been well characterized (6,7), the structure of its mutation-rich disease-associated carboxyl terminal domain was unclear. In this study, we utilized nuclear magnetic resonance (NMR) to solve the atomic structure of an amyloidogenic core TDP-43 peptide containing an ALS-associated mutation, A315E. Our data show that TDP-43 shares structural characteristics with other classical amyloid proteins and that the amyloidogenicity of TDP-43 correlates with its neurotoxicity.
RESULTS
Identification and characterization of a minimal amyloidogenic core at the carboxyl domain of TDP-43
Recent studies have shown that synthetic peptides corresponding to the Q286-Q331 segment in the carboxyl-terminal domain of TDP-43 (Fig. 1A) form amyloid fibrils and cause neurotoxicity (4,8). To gain structural insights into the amyloid fibril formation of TDP-43 and examine the molecular basis for its neurotoxicity, we used shorter TDP-43 peptides spanning this amyloidogenic region to define the minimal core region sufficient for fibril formation. An ALS-associated mutant, A315T, is associated with more severe neurotoxicity and neurodegeneration than the wild-type (Wt) TDP-43 in animal models [(4); for recent reviews, see (9,10)]. TDP-43 protein detected in inclusion bodies is often hyperphosphorylated [(11) and references within], and A315T/A315E mutations have been identified in ALS patients (12–14). For these reasons, we also synthesized the TDP-43 peptides containing A315T or A315E mutations (Fig. 1A).
Figure 1.

Amyloid fibril formation of TDP-43 peptides as detected by AFM and EM. All peptides were dissolved at 62.5 μm in PBS for 8 h before detection using AFM. (A) A diagram illustrating positions and sequences of different TDP-43 peptides within the C-terminal glycine-rich domain of TDP-43, showing the sequence and ALS-associated mutations in our previously defined amyloidogenic region of TDP-43 (aa286–331) (4). The amino acid residues affected by the disease-associated mutations are highlighted in yellow. In red or green are residues that are identical or similar, respectively, in the prion protein with its residues 60–120 aligned to TDP-43(aa286–331). It should be noted that the phosphorylated A315T peptide, T6, was synthesized but not pursued further because it behaved less consistently from batch to batch, possibly due to incomplete/heterogeneous phosphorylation. The T6 peptide was therefore not included in the subsequent experiments. (B) Time-lapse AFM images of corresponding TDP-43 peptides at different time points. Only a few fibrils were detected at 0.5-h time point in the A315T-group (shown in the red circle), whereas many granule-like aggregates were detected in the A315E-peptide at this time point. By 1 h, numerous short fibrils were found in the A315E-peptide sample. By 3–8 h, the A315E-peptide formed abundant fibrils, whereas the Wt- or A315T-peptide formed fewer, although thicker and longer fibrils. (C) EM images of different TDP-43 variant peptides at different magnifications. The Wt- and A315T- peptides formed mostly straight fibrils, whereas the fibrils formed from the A315E-peptide showed the twisted-helical features with the average distance between twists ∼287.3 nm (ranging 88.3–642.3 nm; the arrows mark two twists). The average diameter (nm) for the fibrils in different groups are: Wt, 9.3 (range: 4.1–21.1); A315T, 10.1 (range: 3.5–21.6) and A315E, 10.9 (range: 4.9–17.3). Introduction of the G314V mutation into either the Wt- or A314E-mutant peptides eliminated the fibril formation of these peptides (see also Supplementary Material, Fig. S2). The scale bars: 200 nm.
First, we employed atomic force microscopy (AFM) to examine the fibril formation of these peptides (Fig. 1). Following extended incubation of peptides for up to 2 weeks, the TDP-43 derived peptides, T1 (aa320–331), T2 (aa286–299) and T3 (aa300–306), all failed to form any fibril-like aggregates, similar to the control T0 peptide containing the reversed sequence of amino acid 286–298. In contrast, T4 (Wt, aa307–319), T5 (A315T-mutant, aa307–319) and T7 (A315E-mutant, aa307–319) peptides formed well-defined fibrils following 8-h incubation. We next used time-resolved AFM to characterize the fibril formation. As early as 0.5-h post-incubation, evenly distributed small aggregates were observed in the A315E-mutant, whereas little or no aggregates were detected in other samples including Wt-, A315T-mutant and truncated T8 (aa310–319) peptides. At this time point, only thin fibrils were detected in the A315T sample (as highlighted by the red circle; Fig. 1B). Aggregation of Wt- or A315E-mutant TDP-43 peptides proceeded from initially few small aggregates, to short protofibrils, and finally fibrils. The A315T-peptide showed a faster initial phase of fibril formation, with very few thin fibrils by 0.5 h and longer fibrils by 1 h (Fig. 1B). At the 1-h point, the Wt- and A315E-peptides formed short but more numerous fibrils. By 3 h, the Wt- and A315T-mutant peptides formed thick and long fibrils, whereas the A315E-peptide displayed homogeneous thick fibrils with higher density. By 8 h, the aggregates of A315E-peptide presented as dense fibrils covering almost the entire surface. In contrast, the T8-peptide formed granular and flaky structures after 8-h incubation (Fig. 1B).
Electron microscopy (EM) was carried out to examine fibrils formed by these TDP-43 peptides. The Wt-peptide formed fibril bundles that were heterogeneous in thickness and length. The A315T-fibrils often aggregated into stacks of straight fibrils, whereas the A315E-fibrils were more evenly distributed in thickness and length. At higher magnifications, the A315E-fibrils showed a twisted-helical feature with the average distance between twists ∼287.3 nm (ranging 88.3–642.3 nm). Molecular dynamic simulation predicted that the A315E-peptide would have high probability of forming β-sheet structure and that the glycine residue at the position 314 (G314) may be important for the β-structure (Supplementary Material, Fig. S1). Consistent with the prediction, changing the G314 residue to valine abolished the fibril formation of the A315E-TDP-43 peptide, as shown by EM (Fig. 1C) or AFM (Supplementary Material, Fig. S2).
Detection of TDP-43 immunoreactive fibrils in FTLD-TDP brain samples
To investigate whether TDP-43 forms fibril-like aggregates in vivo, we used immuno-electron microscopy (IEM) to examine brain tissues obtained from patients pathologically diagnosed with FTLD-TDP. IEM using a specific anti-TDP-43 antibody together with the secondary antibody conjugated with gold particles revealed TDP-43 immunoreactive fibrils in all three cases of FTLD-TDP brains examined (Fig. 2), whereas none of the control brains showed such fibril structures. It is interesting that the TDP-43 immunoreactive fibrils detected in the FTLD-TDP brain tissues and the amyloid fibrils formed by the TDP-43 peptide in vitro show similar filamentous structures (Fig. 1C). While this paper was in preparation, an elegant EM study was published, showing the filamentous TDP-43 positive structures detected in the spinal cords of a subset of ALS-TDP patients, although such structures were not detected in FTLD-TDP brain samples in their study (15). The filamentous structures detected by Robinson et al. (15) are similar in morphology to the TDP-43 immunoreactive fibrils detected in our FTLD-TDP brain samples.
Figure 2.

ImmunoEM reveals TDP-43-positive fibrils in brain tissues of patients affected by FTLD-TDP. The de-identified postmortem brain samples from FTLD-TDP patients and control subjects were described previously (4), and subjected to immunostaining using the specific anti-TDP-43 followed by the secondary anti-Rabbit-immunogold (10 nm). Three FTLD-TDP cases examined all showed TDP-43 immunoreactive fibril-like structures, whereas such fibrils were not detected in six control brain samples. The fibril-like structures detected in these FTLD-TDP cases resemble the fibrils formed by the TDP-43 peptides shown in Figure 1C.
TDP-43 amyloidogenic peptides can self-seed and cross-seed to induce the formation of amyloid fibrils
Thioflavin-T (ThT) is a fluorescent dye that binds amyloid fibrils. Using a time-resolved ThT binding assay, we characterized kinetic properties of the TDP-43 fibril formation. Consistent with AFM and EM results, neither T1, T2 nor T3- TDP-43 peptides exhibited detectable ThT binding, whereas Wt-, A315T- and A315E-peptides, but not T8, showed enhanced ThT binding, increasing with the incubation time (Fig. 3A). Importantly, the Wt TDP-43 peptide only started to form ThT-positive fibrils after a lag-phase of ∼2.5 h at the concentration range tested, whereas A315T- or A315E-mutant peptides formed ThT-reactive fibrils almost immediately after the initiation of the ThT-binding reaction. At lower peptide concentrations (62.5 and 125 μm, Fig. 3A, upper and middle panels), the ThT fluorescence intensity for the Wt-peptide was lower than that of A315T- or A315E-peptides, whereas increasing the peptide concentration to 250 μm (Fig. 3A, lower panel) dramatically enhanced ThT binding of the Wt-peptide (higher than those of A315T- or A315E-peptides). For A315T- and A315E-peptides, increasing the peptide concentration also enhanced the ThT fluorescence intensity.
Figure 3.

The time course of fibril formation for TDP-43 peptides by themselves and in self- or cross-seeding reactions, as monitored by ThT binding. (A) Synthetic peptides of TDP-43 were incubated at different concentrations, 62.5, 125 and 250 μm, and amyloid fibril formation was detected by fluorescence enhancement upon ThT binding. (B) Different TDP-43 peptides were incubated with a series of pre-formed seeds in PBS buffer (pH 7.4) at a concentration of 62.5 μm with seeds pre-formed by Wt, A315T and A315E. ‘x’ represents the ThT fluorescence intensity of the pre-formed seeds only. It should be noted that only the A315E-peptide seeds were capable of inducing the fibril formation of T8-peptide and shortening the lag phase of the fibril formation of the Wt-peptide. (C and D) Two amyloid beta protein-derived peptides, Aβ 9–14 and Aβ 1–40 were examined at different concentrations (62.5 and 125 μm) for ThT binding. The addition of the A315E-mutant TDP-43 peptide seeds promoted the amyloid fibril formation of the Aβ 1–40 peptide.
To examine whether different TDP-43 peptides (Wt-, A315T- or A315E-mutant) could induce the fibril formation as seeds, we tested whether pre-formed fibrillar seeds of one peptide were capable of inducing fibril formation of the other TDP-43 peptides. Because the fibril formation of A315T- or A315E-peptides has no lag phase, it was difficult to see the differences between un-seeded and seeded kinetics of ThT binding. For the Wt-TDP-43 peptide, both self-seeding and cross-seeding shortened the lag-phase of fibril formation (Fig. 3B) in comparison with reactions without any seeds (Fig. 3A). The lag phase of fibril formation for the Wt-peptide was shortened in the presence of the A315E-mutant peptide seed (Fig. 3B, lower panel). Interestingly, only the A315E-peptide (Fig. 3B, lower panel), but not the Wt- or A315T-peptides (Fib. 3B, upper and middle panels), was capable of cross-seeding to promote amyloid fibrils formation of the truncated T8-peptide. These results indicate that the A315E-mutant TDP-43 peptide is capable of cross-seeding, facilitating amyloid fibril formation of TDP-43 peptides that otherwise do not form fibrils by themselves. This cross-seeding activity was high when the neutral alanine at the position 315 was changed to glutamate in the A315E mutant, suggesting that phosphorylation at the residue 315T or substitution by an acidic residue may increase the ability of TDP-43 to promote recruitment of TDP-43 derivatives into the TDP-43 aggregates.
Recent studies have revealed significant clinical and pathological overlap between AD and TDP-43 proteinopathy (16–18). To test the effects of TDP-43 peptides on the fibril formation of amyloid-β peptides, we used Aβ9–14, a peptide derived from amyloid-β protein that was not capable of forming fibrils (unpublished observation) and a 40-mer Aβ1–40 peptide in the ThT cross-seeding assay. The Aβ1–40 peptide formed amyloid fibrils at relatively high concentrations (19). At the concentration of 62.5 μm, no amyloid fibrils were detectable after 40-h incubation of Aβ9–14 (Fig. 3C) and amyloid fibrils formation was very slow for the Aβ1–40 peptide in comparison with the TDP-43 peptides (Fig. 3D). Interestingly, addition of the A315E-mutant TDP-43 peptide seeds accelerated fibril formation and enhanced ThT binding of Aβ1–40 peptide at both 62.5 and 125 μm concentrations, demonstrating that the A315E-mutant TDP-43 peptide was capable of cross-seeding the Aβ1–40 peptide (Fig. 3D). This observation suggests that TDP-43 amyloid fibril formation might play a role in amyloid-β pathology in AD patients.
Circular dichroism and Fourier transformation infrared analyses support that the amyloidogenic TDP-43 peptides adopt β-conformation
Circular dichroism (CD) spectrum analysis of these TDP-43 peptides was carried out (Fig. 4A). The A315E-peptide showed a clear positive peak at 193 nm, suggesting the presence of β-structure; whereas the Wt-TDP peptide exhibited a broader peak in this region, suggesting a possible mixture of α-helix and β-sheet (20). It is notable that the far-UV CD profile of the A315E-peptide is similar to that of chymotrypsin which is mostly composed of β-sheet structure (21,22) (Fig. 4A). Consistent with this, the Wt- and A315E-peptides were ranked higher than other TDP-43 peptides in β-sheet content predicted using the Young's algorithm, whereas the A315T peptide ranked highest in predicted β-turn content (23) (see Supplementary Material, Table S1). We next performed Fourier transformation infrared (FTIR) spectroscopy to further examine these TDP-43 peptides (24). For the Wt- or A315T-peptides, a major band at 1677 cm−1 together with a minor 1626 cm−1 peak was observed, whereas the major band shifted to 1626 cm−1 for A315E- or T8 peptides (Fig. 4B). It has been established that the 1677 cm−1 peak is attributed to the amide bond I vibrations, whereas 1626 cm−1 peak suggests the presence of β-sheet structure (25). Interestingly, one major peak at 1626 cm−1 together with three minor ones at 1696, 1672 and 1525 cm−1 were detected in the profile of the A315E-peptide, suggesting the presence of an anti-parallel β-sheet structure (25–27). Both CD and FTIR data support the presence of β-conformation in the TDP-43 peptides.
Figure 4.
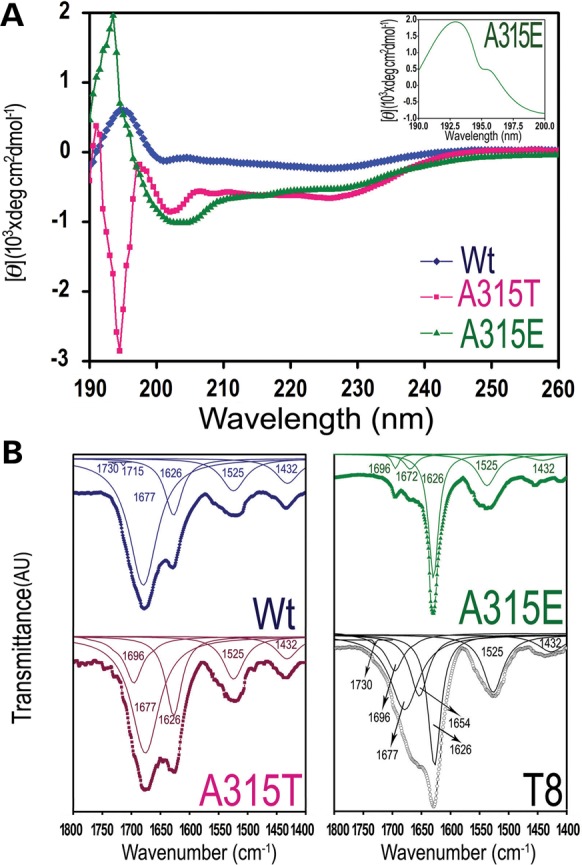
CD and FTIR analyses of the amyloidogenic TDP-43 peptides. (A) CD spectra of the Wt-, A315T-, A315E- and T8- TDP-43 peptides with the zoom-in spectrum of the A315E-peptide shown in the inset. The clear positive peak at 193 nm in the A315E-peptide suggests the presence of the β-sheet structure. (B) FTIR spectra of the TDP-43 peptides. The thick dotted lines are original curves, and the thin solid lines are corresponding peak-differentiation curves. The peak positions in the differentiated peaks are indicated.
NMR structure of the A315E-mutant TDP-43 peptide
Next, we used NMR spectroscopy to examine these TDP-43-(M307-N319) core peptides in solution. In spite of extensive efforts, NMR analyses of either the Wt- or A315T-mutant TDP-43 peptides did not yield information sufficient for resolving their structures. On the other hand, analyses of the A315E-mutant TDP-43 core peptide in solution provided detailed structural information, except for the N-terminal MGGG region. In addition to sequential Nuclear Overhauser Effects (NOEs), a set of the long-range NOEs between M311 and I318 was detected (Fig. 5A), indicating that the peptide may either adopt an intra-molecular folded structure or form inter-molecular interactions in an extended conformation. Several sets of NOEs between M311 and F313 and between F316 and I318, respectively, were also observed (Fig. 5A). Thus, the residues M311, F313, F316 and I318 are likely to pack with each other to form a hydrophobic cluster. Double-quantum-filtered correlation spectroscopy (DQF-COSY) was then used to measure coupling constants and to distinguish between intra-molecular and inter-molecular interactions. If the peptide adopted an intra-molecular fold-back structure in which residues F313, G314, E315 and F316 formed a β-turn, then one of the G314 coupling constants would be smaller than 6 Hz. However, the coupling constants between the amide and alpha protons of G314 were measured as 12.6 and 11.4 Hz, respectively (Fig. 5B), indicating an extended structure rather than an intra-molecular fold-back conformation. Therefore, the long-range NOE obtained from the inter-molecular contacts was calculated accordingly (Fig. 5C). In this model, two molecules of TDP-43-(M307-N319) peptides pack in an anti-parallel manner (i.e. head-to-tail packing) with the four hydrophobic residues (M311, F313, F316, I318) packed with each other between chains in an inter-molecular manner (Fig. 5D). This NMR structure of the ALS-associated mutant (A315E) TDP-43 peptide provides a structural basis for the similarity between TDP-43 and previously characterized classical amyloid peptides.
Figure 5.

The solution structure of the A315E-mutant TDP-43 core peptide as revealed by NMR. (A) The A315E-TDP-43 (M307-N319) core peptide sequence is shown with corresponding amino acid residues marked on the top according to the full-length TDP43 (ref Seq ID: NM_007375; NP_031401). For convenience of labeling, the amino acid residue numbers in (C) and (D) are corresponding positions in the peptide from the amino-terminus. Selected regions of the 2-D 1H-NOESY spectra showing the NOE cross peaks formed between the hydrophobic residues of the peptide. Notably, several sets of the NOEs between M311 and I318, between M311 and F313, and between F316 and I318 were observed, indicating that the residues M311, F313, F316 and I318 are likely to form a hydrophobic cluster. (B) The regions of the DQF-COSY spectra showing the coupling constants of G314. Groups of COSY peaks for the G314 residue were enclosed in red circles with the coupling constants shown above or below the corresponding circles. (C) The stereo-view of the backbones of 10 superimposed NMR-derived structures of the A315E-mutant TDP-43 core peptide in the inter-molecular interaction model. The two peptide chains, chain A (in blue) and chain B (in red) are packed in a head-to-tail anti-parallel manner with amino acid residue numbers and the N- and C-termini of the peptides labeled. (D) A ribbon-diagram of a representative NMR structure of the A315E- TDP-43 amyloidogenic core peptide in the inter-molecular interaction model. The side-chains of the residues involved in forming hydrophobic clusters are drawn in an explicit atomic model in green with backbones of the chain A in blue and the chain B in red.
The amyloidogenic TDP-43 peptide induces TDP-43 protein redistribution to the cytoplasm
One prominent pathological feature of TDP-43 proteinopathy is the clearance of the nuclear TDP-43 in the affected tissues (28). Because the amyloidogenic TDP-43 peptides were capable of acting as seeds for fibril formation and could be taken up by neurons to induce neurotoxicity (see Figs 3 and 8), we tested whether these TDP-43 peptides might affect the subcellular distribution of the endogenous TDP-43 protein. Immunostaining of HEK293 cells treated with different TDP-43 peptides showed that the Wt-TDP-43 peptide led to a moderate decrease in the nuclear TDP-43, whereas the A315T-mutant, especially the A315E-mutant TDP-43 peptide induced more prominent redistribution of nuclear TDP-43 to the cytoplasm (Fig. 6A). TDP-43 redistribution to the cytoplasm was obvious in cells treated with the A315T- or A315E-mutant TDP-43 peptide by ∼24 h following peptide treatment (Fig. 6A–D; Supplementary Material, Fig. S3). Consistently with immunostaining, Western blotting of protein lysates following cytoplasm-nucleus fractionation showed that treatment by Wt- or the A315T- or A315E-mutant TDP-43 peptide led to increased cytoplasmic and decreased nuclear TDP-43 (Fig. 6B–D). Interestingly, the G314V variant of A315E-mutant TDP-43 peptide failed to induce TDP-43 redistribution (Fig. 6A–D), suggesting that fibril formation of the TDP-43 peptide may correlate with its ability to induce TDP-43 redistribution. We further examined whether these TDP-43 peptides had similar effects in cultured neurons (Fig. 6E–G). TDP-43 redistribution to the cytoplasm was detected in ∼40% of neurons 24 h after addition of the A315E-mutant TDP-43 peptide to the culture media, whereas the Wt- or A315T-mutant TDP-43 peptides only show slight effects when compared with the control group. By 48 h, all of the amyloidogenic TDP-43 peptides, including Wt-, A315T- and A315E-mutant, significantly increased the percentage of neurons showing reduced nuclear TDP-43 staining and increased cytoplasmic TDP-43 signals, with the A315E-mutant exhibiting more prominent effect (Fig. 6).
Figure 8.

The amyloidogenic TDP-43 peptides induce neurotoxicity when added to the cultured neurons. (A and B) TUNEL staining of the cortical neurons shows neurotoxicity of the Wt- or the A315T- or A315E-mutant TDP-43 peptides. The percentage of TUNEL-positive neurons showing apoptotic nuclei is quantified and shown in (B). The neurons were cultured on coverslips, stained and quantified as described previously (4). Healthy neurons in the control group are shown in subpanels A1–A4 in (A). The arrows mark the TUNEL-positive apoptotic neurons with condensed nuclei, whereas the arrowheads mark those with fragmented nuclei. (C–H) Neuronal cultures in microfluidic chambers demonstrate that the TDP-43 peptides applied to the axonal compartment are sufficient to cause axonal toxicity and neuronal death. The data represent three independent experiments with >200 neurons quantified in each group (***P < 0.0001; **P < 0.001, one-way ANOVA with Bonferroni multiple post test). (C) A diagrammatic illustration of the microfluidic chambers with different compartments and application of the peptides in the axonal compartments. On the right side are the fluorescent microscopic images showing the axons in microgrooves following immunostaining using the anti-Tuj1 antibody with nuclei in the cell body compartment detected by Hoechst dye staining. The cell body compartment (Cell), axonal compartment (Axon) are illustrated together with the proximal (Prox.) and distal (Dist.) axonal microgrooves. (D and E) 72 h following application of fluorescently labeled peptides (FITC-control peptide, Cy2-Wt- or Cy3-A315E-TDP-43 peptides) in the axonal compartment, samples were fixed and stained using mouse monoclonal ant-Tuj1 antibody with anti-mouse Cy5-conjugated secondary antibody. Axonal continuity index was calculated using the algorithm that we developed (33), indicating that TDP-43 peptides caused significant axonal damage with more severe effects detected in the A315E-mutant group. (F–H) Following application of the non-labeled peptides to the axonal compartment [the control peptide (Ctr), Wt-, or A315E- or A315E/G314V-mutant TDP-43 peptides], samples were immunostained with anti-Tuj1 and counter-stained with Hoechst dye to reveal nuclear morphology. Arrows mark the condensed apoptotic nuclei, whereas arrowheads label nuclei with normal morphology. The axonal continuity index and apoptotic nuclei were quantified, showing that the A315E-mutant peptide induced more severe neurotoxicity than the Wt-peptide and that G314V mutation eliminated both axonal toxicity and nuclear damage caused by the A315E-TDP-43 peptide. The data represent three independent experiments with axons in >60 microgrooves quantified (***P < 0.0001; **P < 0.001, one-way ANOVA with Bonferroni post test).
Figure 6.

The amyloidogenic TDP-43 peptides induce TDP-43 redistribution. (A) HEK293 cells were treated with the control (Ctr) or Wt-, A315T-, A315E- or A315E/G314V (A315E/gv)-mutant TDP-43 peptide (50 μm) for 24 h and then fixed for immunostaining using anti-TDP-43 to detect the endogenous protein. Nuclei (Nu) were revealed by Hoechst dye staining. The A315E-mutant TDP-43 peptide induced prominent redistribution of the TDP-43 immunostaining signals from the nuclei, whereas the A315E/G314V mutant peptide failed to induce TDP-43 redistribution. Z-stack projections of confocal images are shown. Arrowheads mark the cells with reduced nuclear and increased cytoplasmic TDP-43 staining signals, whereas arrows label the cells with almost no detectable nuclear TDP-43 signals. (B–D) Biochemical fractionation experiment to detect cytoplasmic and nuclear TDP-43 levels, cytoplasmic fractions (lanes 1–5) and nuclear fractions (lanes 6–10) 24 h following treatment of HEK293 cells with different TDP-43 peptides [Ctr: lanes 1, 6; Wt: lanes 2, 7; A315T: lanes 3, 8; A315E: lanes 4, 9 or A315E/G314V (A315E/gv): lanes 5, 10]. RhoA and PCNA were used to monitor cytoplasmic and nuclear proteins, respectively, in the fractions. (C and D) Quantification of western blotting signals in corresponding lanes of (B) (***P < 0.001; **P < 0.01; *P < 0.05; one-way ANOVA with Bonferroni test). Signals were normalized to that in the control lane. (E) Immunofluorescent staining images of cortical neurons (div3) 48 h following treatment with the control or different TDP-43 peptides as indicated. (F and G) Quantification of percentage of neurons that showed TDP-43 negative nuclei 24 and 48 h following peptide treatment. By 24 h following the peptide treatment, only the A315E-mutant peptide reduced TDP-43 nuclear staining, whereas other TDP-43 peptides did not show significant effects when compared with the control. By 48 h following peptide treatment, the amyloidogenic peptides (Wt, A315T and A315E) induced redistribution of TDP-43 in the cortical neurons. The arrows mark the neurons showing TDP-43 redistribution. 100 neurons were scored in each group. The data represent mean ± SEM in three independent experiments (***P < 0.001; **P < 0.01; *P < 0.05; One-way ANOVA with Bonferroni post hoc test).
Amyloidogenic TDP-43 peptides induce axonal damage and neuronal death with A315T- or A315E-mutant showing enhanced neurotoxicity
To model TDP-43 proteinopathies, we had made transgenic flies expressing either Wt- or A315T-mutant TDP-43 protein (4,29). When targeted to photoreceptors, expression of the Wt-TDP-43 led to progressive retinal degeneration, and that of A315T-mutant TDP-43 resulted in faster progression and more severe retinal damage (Fig. 7A–C). When expressed in the mushroom body (MB) neurons, the A315T-mutant TDP-43 induced more severe axonal damage than the Wt-TDP-43, although the TDP-43 protein expression levels in the Wt- and A315T mutant flies were equivalent (see Supplementary Information in reference 4) (Fig. 7D). Remarkably, axonal impairment was detectable in any of three MB lobes; however, the damage was not symmetrical or homogeneous, often affecting one lobe much more aggressively than others. This suggests that axon damage may occur in ‘grouped’ fashion, similar to that detected in motor neurons of ALS patients (30,31). It also suggests that neurotoxic TDP-43 derivatives may act in a non-cell autonomous fashion to spread among the neighboring cells, consistent with what was proposed in previous studies (4,8,29,32). To test this notion, we added the TDP-43 peptides to cortical neurons cultured in vitro and asked if the extracellularly delivered TDP-43 derivatives could induce neurotoxicity (Fig. 8). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) was used to evaluate neuronal death in cultures treated with either the control, or Wt-, A315T-, or A315E-mutant TDP-43 peptides. The 13-mer core amyloidogenic TDP-43 peptides added to the culture media were capable of inducing neuronal death, with the A315T- and A315E-mutant TDP-43 peptides showing greater neurotoxicity than the Wt-TDP-43 peptide (Fig. 8A and B).
Figure 7.
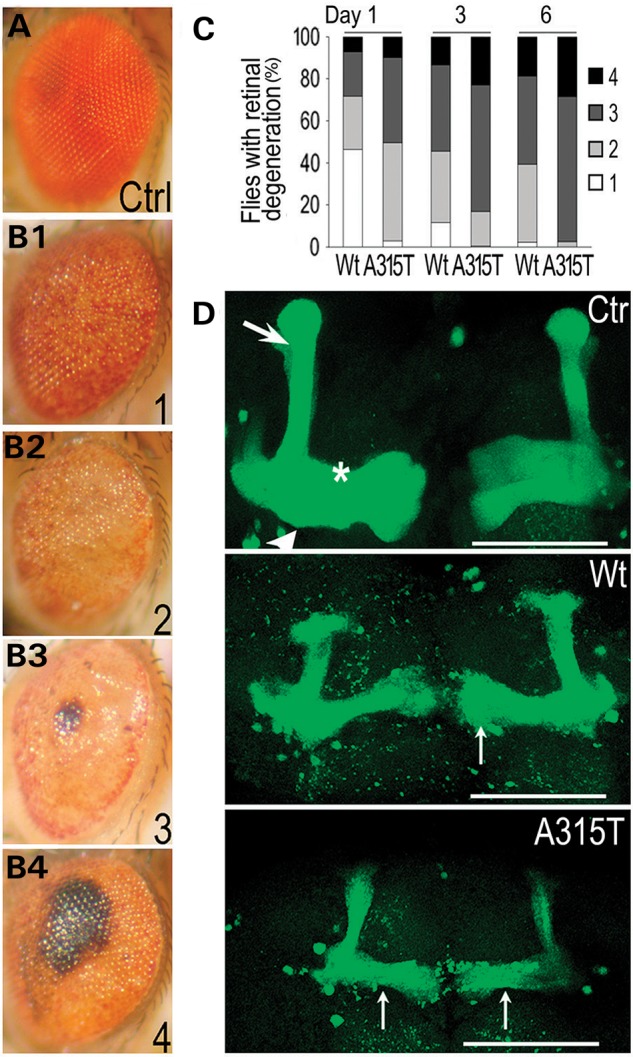
The A315T-mutant TDP-43 expressing transgenic flies show more severe neurodegeneration than those expressing the wild-type TDP-43. (A–C) Expression of the Wt- or A315T-mutant human TDP-43 in photoreceptors led to progressive retinal degeneration in transgenic flies. A GMR-gal4 driver was used to express either control vector or Wt- or A315T-mutant TDP-43; the Wt- and A315T-mutant TDP-43 proteins were expressed at an equivalent level in photoreceptor cells (see Supplementary Information in reference 4). It should be noted that technical difficulties have prevented us from obtaining transgenic flies expressing the A315E mutant TDP-43 protein. Fly eyes were examined with retinal degeneration phenotypes scored at Day 1, 3 and 6 after eclosion as in our previous study (29). Briefly, the retinal degeneration was classified as levels 1–4 as shown in (B1)–(B4) [when compared with the normal eye from the control fly shown in (A)]. The percentage of flies showing different levels of retinal damage was quantified and shown in (C). Expression of the A315T-mutant TDP-43 resulted in more severe retinal degeneration than the Wt-TDP-43. (D) Axonal degeneration induced by the expression of the Wt- or A315T-mutant TDP-43 in fly mushroom bodies (MBs). Confocal microscopic images are shown of MBs from day-5 transgenic flies. The control flies (OK107-Gal4/UAS-mGFP/UAS-RFP) exhibited the normal MB lobe structure. Arrow: α/α′ lobes; arrowhead: β/β′ lobes; star: γ lobe. OK107-Gal4/UAS-mGFP/UAS-Wt-hTDP43-RFP flies began to lose MB lobes at Day 1. By Day 5, significant axonal damage and reduction in MB size were detected. Such axonal damage and MB lobe loss was more pronounced in flies expressing the A315T-mutant (OK107-Gal4/UAS-mGFP/UAS-A315T-hTDP43-RFP). The thin arrows mark the abnormal axonal varicosities with uneven distribution of mGFP signals. Scale bars: 50 μm.
To test whether the TDP-43 peptides were taken up by axons to cause neurotoxicity, we employed our modified axonal microfluidic chambers for neuronal culture (33) (Fig. 8C). In these microfluidic chambers, the fluidic pressure was maintained in such a way that no diffusion of the fluorescent peptide into the cell body compartment was detected even 72 h after addition of the fluorescently labeled peptides to the axonal compartment and the central flow channel (Supplementary Material, Fig. S4). By this time, significant axonal damage was detected in Wt- or A315E-mutant peptide treated groups when compared with the control, with the A315E-mutant peptide eliciting more severe axonal toxicity (Fig. 8D and E). Such axonal damage was not caused by the fluorescent moiety because non-fluorescent TDP-43 peptides induced similar detrimental effects on axons (Fig. 8F and G). Furthermore, we found that TDP-43 peptides applied to the axonal compartment led to nuclear death, indicating that extracellular TDP-43 peptides can cause not only axonal damage but also neuronal death. Importantly, the non-amyloidogenic G314V-mutant lost both axonotoxicity and its ability to induce neuronal apoptosis (see Fig. 1C; Supplementary Material, Fig. S2; Fig. 8G and H). Such microfluidic-chamber neuronal cultures allowed spatially contained and temporally resolved experimental design and enabled us to demonstrate that the extracellular application of the amyloidogenic core TDP-43 peptides was sufficient to elicit axonal damage and neuronal death.
DISCUSSION
Beta-conformation is important for biological activities of numerous proteins and critical for many human diseases (1,34,35). The formation of insoluble or aggregated proteins often involves conversion of soluble and properly folded peptides into misfolded ones containing β-structures. Although crystal structures have not yet been determined for the full-length proteins for many of those disease-associated ‘misfolded-proteins’, significant structural insights have come from careful studies of derivatives or fragments of these proteins (35). For example, fragments of several amyloid proteins have been studied by NMR or X-ray crystallography: VQIVYK of the human tau protein (36,37), GNNQQNY of the yeast prion Sup35 (38), MVGGVV/GGVVIA of the beta-amyloid protein (36), SNQNNF of the human prion protein (36). One major common structural feature of these amyloidogenic peptides is the β-sheet structure that allows both backbone stacking and side-chain hydrogen bonding. Side chains protruding from the two β-sheets form a tightly self-complementing steric zipper (35–38). GNNQQNY, SNQNNF and GGVVIA form parallel structures whereas MVGGVV forms an anti-parallel structure (36). NMR and high resolution AFM have also been extensively used to analyze amyloid structures (39–41). The β-amyloid fibrils consist of dimeric units stacked in sync as crossed β-sheets (1,40,42–45). When compared with the known amyloid proteins, the TDP-43 amyloidogenic region appears most similar to prion and amyloid-β peptides (1) but has its distinct properties in both primary sequence and 3-dimensional structure.
In this study, we examined the molecular and structural features of TDP-43 core amyloidogenic peptides by AFM, EM, CD, FTIR and NMR. Interestingly, the wild type TDP-43 peptide can form amyloid fibrils. Furthermore, the ALS-mutants, A315T and A315E, form fibrils at accelerated rates. For many other amyloid peptides, amyloid fibril formation displays a sigmoidal kinetic pattern with a lag phase. In our case, only the Wt-TDP-43 peptide showed a typical time course of fibril formation, whereas both the A315T- and A315E-mutant peptides formed ThT-positive fibrils soon after initiation of the reaction. Such enhanced amyloid fibril formation of the A315T- or A315E- mutant is associated with their increased neurotoxicity when compared with the Wt-TDP-43 peptide (Fig. 8). Many amyloid-like proteins can self-seed in fibril formation, such as the recombinant human α-synuclein (46), mouse PrP (89–230) (47) or synthetic amyloid-β peptides 1–40 and 1–42 (48,49). Some amyloid peptides can cross-seed the fibril formation of heterologous peptides. For example, α-synuclein can cross-seed fibril formation of synthetic amyloid-β peptides 1–40 and 1–42 (49). Our experiments show that the A315E-peptide was capable of cross-seeding amyloid fibrils formation of other TDP-43 peptides, including Wt and the shortened Wt-peptide (T8). This raised the possibility that in the ALS patients with the A315T/E mutation, increased phosphorylation of the mutant TDP-43 protein or the A-to-E mutation might accelerate amyloid fibril formation of Wt-TDP-43 protein, leading to increased protein aggregation and neurotoxicity. Our data may provide one mechanism to explain the relatively early onset, increased severity and accelerated disease progression in those ALS patients. In addition, our observation that A315E-mutant TDP-43 peptide was able to cross-seed amyloid-β 1–40 in fibril formation suggests an explanation for the neuropathological and clinical overlap between TDP-43 proteinopathy and AD.
Using NMR, we demonstrated that the A315E-mutant TDP-43 peptide forms the extended β-conformation in an anti-parallel manner. Although the NMR analysis of the wild type TDP-43 did not allow resolution of its structure, it is possible that the Wt-TDP-43 peptide favors a folded-back conformation, as supported by molecular dynamic simulation and scanning tunneling microscopy (STM) studies (50) (Supplementary Material, Fig. S1). The anti-parallel β-structure of the A315E-mutant TDP-43 peptide is similar to the classes 5–8 among the eight classes of steric zippers in which the β-strands within the sheets are anti-parallel (35,36). Although detailed structural features of the TDP-43 amyloid fibrils need further investigation, our work offers an interesting glimpse of the molecular structure at the core amyloidogenic region of TDP-43. Our data suggest that at least some ALS-associated mutations may alter the conformation of TDP-43, rendering the mutant more prone to self-propagate into fibrils, leading to enhanced neurotoxicity. Our work presented here helps us understand the structural basis for TDP-43 neurotoxicity and for the amyloid fibril formation of TDP-43 at the atomic level. An α-helical structure was reported based on the NMR analysis of an adjacent region in TDP-43; and α-helix to β-sheet transformation was proposed based entirely on the aggregation and CD spectral change of bacterially expressed TDP-43 fragments (51). However, no definitive evidence was presented for the β-conformation, and no data were provided to support the functional significance of their proposed ‘α-helix to β-sheet transformation’.
Interestingly, a recent study uncovered mutations among multi-system proteinopathy and ALS patients in genes encoding another two RNA binding proteins, hnRNPA1 and A2 (52). For hnRNPA1 and hnRNPA2, the mutant hexapeptides (SYNVFG and NYNVFG) are highly amyloidogenic. The 13-mer mutant peptide (MGGGMNFGEFSIN) of TDP-43 that we identified shares sequence similarity with the hnRNPA1/A2 hexamers, especially in the region of NFGEFS. It is possible that two F residues in the TDP-43 peptides may share certain critical structural feature for amyloidogenesis with those two aromatic resides (Y and F) in the hnRNPA1/A2 hexamer sequences. This notion needs to be tested in future investigation.
Fibrils formed by hnRNPA1 (D262V or D262N) and hnRNPA2 (D290V) mutant hexapeptides not only seeded their own assembly, but also cross-seeded fibrillization of their respective wild-type counterparts. However, hnRNPA1 fibrils did not cross-seed assembly of hnRNPA2, or vice versa; nor did hnRNPA1/A2 cross-seed TDP-43 (52). This demonstrated relative specificity of cross-seeding by the hnRNPA1/A2 peptides. Similar to the mutant hexapeptides of hnRNPA1 and hnRNPA2, the A315E-mutant peptide of TDP-43 can cross-seed shorter wild-type peptide of TDP-43 (T8) (see Fig. 3B, lower panel). Interestingly, the A315E-mutant TDP-43 peptide was also capable of enhancing amyloid fibril formation of amyloid-β 1–40 peptide (see Fig. 3D).
Previous studies revealed interesting structural features of RRM domain of TDP-43 (6,7,53). It was reported that TDP-43 formed dimers through amino acid 1–183 region (53) and the RRM2 domain (6). The dimeric interface of RRM2 was observed in the crystal structure of the RRM2-DNA complex (6), suggesting that dimer formation may be required for nucleotide binding. This self-association property of TDP-43 may contribute to its aggregation and inclusion formation. Notably, the inclusion formation of full-length TDP-43 with the RRM1-C/S mutation (an ALS mutation) requires the C-terminal domain (54), highlighting the critical importance of the glycine-rich carboxyl domain in the TDP-43 aggregation in the pathogenesis of TDP-43 proteinopathies.
Prior to our previous study (4) demonstrating the biochemical and sequence similarity of TDP-43 to prion proteins, TDP-43 proteinopathy had been believed to be a group of thioflavin-S-negative diseases, distinct from classical amyloid diseases. Our data prompted re-evaluation of thioflavin-S staining in TDP-43 proteinopathy cases, leading to the finding that TDP-43 positive inclusions in most cases of FTLD-TDP surveyed, but not FTLD-FUS or ALS-SOD1 cases, are thioflavin-S positive (55). Our work here, using combined approaches, has now revealed the structural basis for the similarity between TDP-43 and classical amyloid proteins. Importantly, our data show that the amyloid fibril formation correlates with the neurotoxicity of TDP-43, because the single G314V change eliminates both amyloid fibril formation and neurotoxicity in cultured neurons.
The subcellular localization of TDP-43 is crucial for its biological function and pathogenic roles. Various genetic mutations and cellular stresses can affect the balance of nuclear to cytoplasmic ratio of TDP-43, disrupting the predominantly nuclear localization of TDP-43 [reviewed in (3)]. Indeed, one of prominent neuropathological features of TDP-43 proteinopathies, the nuclear clearance of TDP-43, has been suggested as an early event in the pathogenesis of TDP-43 proteinopathies (28). Aberrant cleavage or truncation of TDP-43 promotes cytoplasmic aggregation and toxicity of TDP-43 (56,57). Our previous work indicates that cytoplasmic localization of TDP-43 correlates with its neurotoxicity (58). A recent study showed that muscle-specific overexpression of the fly TDP-43 homolog (TBPH) led to nuclear clearance of TBPH and protein aggregates (59). Our data suggest that the human TDP-43 derivatives present in the extracellular compartment could be taken up by neurons and induce redistribution of the endogenous nuclear TDP-43 to the cytoplasm. This has a number of implications. First, our data support a non-cell autonomous mechanism by which the TDP-43 protein products released by damaged or dead neurons or glia cells could be taken up by neighboring neurons or axons to cause horizontal spread of the TDP-43 proteinopathy (32). This is consistent with the result that neural/axonal damage in our transgenic flies appeared in a ‘grouped’ fashion (see Fig. 7D), similar to motor neuron damage detected in ALS patients (30,31). Second, our observation that amyloidogenic TDP-43 peptides are capable of inducing redistribution of nuclear TDP-43 to the cytoplasm provides a link for the two hypotheses that have been proposed for the pathogenesis of TDP-43 proteinopathy: the loss-of-function of nuclear TDP-43 versus the gain-of-toxicity of cytoplasmic TDP-43 aggregates/inclusions. Third, the possible horizontal transmission of TDP-43 peptides and their neurotoxicity should motivate us to develop sensitive methods for detecting such TDP-43 gene products in the cerebral spinal fluid or plasma of patients affected by TDP-43 proteinopathy. This may help in developing biomarkers for TDP-43 proteinopathy. Finally, searching for chemical compounds or modifier genes that could suppress the formation of the amyloidogenic TDP-43 species, or inhibit amyloid fibril formation/seeding or block the uptake of neurotoxic TDP-43 peptides or alternatively promote the clearance of neurotoxic TDP-43 gene products may provide therapeutic benefits for patients affected by devastating TDP-43 proteinopathies.
MATERIALS AND METHODS
Ethics statement
De-identified postmortem brain samples from patients affected by TDP-43-proteinopathy and control subjects were obtained from the Cognitive Neurology & Alzheimer's Disease Center (CNADC) at Northwestern University following institutional and NIH guidelines. All experiments involving animal tissue samples were carried out following institutional and NIH guidelines.
Peptide synthesis and sample preparation
The TDP-43 synthetic peptides [T1 (P320-Q331): PAMMAAAQAALQ; T2 (Q286-L299): QGGFGNSRGGGAGL; T3 (G200-N306): GNNQGSN; T4 (M307-N319-Wt): MGGGMNFGAFSIN; T5 (M307-N319-A315T): MGGGMNFGTFSIN; T7 (M307-N319-A315E): MGGGMNFGEFSIN; T8 (M310-N319):GMNFGAFSIN], Wt/G314V (M307-N319: MGGGMNFVAFSIN), A315E/G314V (M307-N319: MGGGMNFVEFSIN), or the control 13mer peptide T0: LGAGGGRSNGFGG (containing reversed sequence of amino acid 286–298)] and amyloid-β peptides 9–14 and 1–40 were purchased from Shanghai Science Peptide Biological Technology Co., Ltd or ProteinTech Group (Chicago, USA). These sequences are derived from the corresponding amino acid residues 286–331 of human TDP-43 protein (4). The peptides were purified twice using HPLC to achieve the purity of >99%. The peptides were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Sigma-Aldrich) initially for extended time, and the solution was transferred into a sterile microcentrifuge tube. All the peptides are solved in HFIP at a concentration of 20 mg/ml as stock solutions. All the samples for FTIR spectrum, circular dichroism spectrum and AFM experiments are diluted to specified concentrations from the stock solutions.
Far-UV CD spectrum measurement
The stock solutions of TDP-43 peptides (20 mg/ml in HFIP) were diluted to 0.2 mg/ml with 20 mm PBS buffer (pH 7.4). Circular dichroism experiment was performed on a J-810 CD spectrometer (Jasco, Japan) at room temperature using a quartz cell with 0.1 cm path.
FTIR spectrum measurement
Infrared spectra were recorded on a PerkinElmer FTIR spectrometer at a resolution of 4 cm−1. FTIR spectroscopic measurements were performed in the transmission mode with BaF2 windows. The solution was dropped onto the BaF2 surface followed by air dry prior to FTIR measurements.
AFM measurement
The aqueous solutions of Wt, A315T, A315E, Wt/G314V, A315E/G314V and T8 with concentration of 62.5 μm were prepared with PBS (pH 7.4). Thirty microliters of the peptide solutions incubated with shaking at 37°C for 0, 0.5, 1, 3, 5 and 8 h were drop-casted on freshly cleaved mica surface. After 10 min adsorption, excessive solutions are withdrawn from the mica surface followed by AFM observations. Tapping-mode AFM studies were performed on a Dimension 3100 AFM (Bruker, USA) under ambient conditions. Commercial silicon tips with a nominal spring constant of 2.0 N/m and resonant frequency of 442.5 kHz were used in all the AFM imaging.
Thioflavin T binding, self-seeding and cross-seeding Assays
Synthetic peptides of TDP-43 were dissolved in DMSO at a concentration of 12.5 mm, then diluted to PBS buffer (pH 7.4) at different concentrations (62.5, 125 and 250 μm, respectively) and spun at 13.3 krpm for 10 min to remove any insoluble material. Amyloid fibril formation was detected by fluorescence enhancement of thioflavin T (ThT, Sigma-Aldrich) upon binding. Fibril formation reactions were set up in triplicate in 96-well black plates with round bottom (Corning Costar). Each well had one 1/8-inch glass sphere (Fisher Scientific) and 150 μl of peptide in PBS buffer supplemented with 200 μm ThT. Plates were sealed with a Greiner EASYseal (Greiner Bio-one) and continuously shaken at 37°C except for 1 min each 10 min as required for data acquisition. ThT fluorescence changes were monitored at 440 nm (excitation)/480 nm (emission) on a Varioskan Flash plate reader (Thermo Scientific).
Self-seeding and cross-seeding assays: different peptides were incubated at a concentration of 250 μm at 37°C with a 1/8-inch glass sphere and continuously shaking in PBS buffer (pH 7.4) for 24 h, by which time fibril formation was complete as determined by ThT assay and EM. Mature fibrils were then sonicated using a sonicator for 1 min (5-s on and 5-s off) at 10% of the maximal amplitude to produce the seeds used in the seeding assay. Corresponding TDP-43 synthetic peptide (at 62.5 μm) was incubated with a small amount [5% (w/w)] of pre-prepared seeds in PBS buffer (pH 7.4) on ice. Amyloid fibril formation was initiated by shaking at 37°C with its kinetics monitored by ThT binding assay as described above.
NMR sample preparation, data collection and structure determination
The sequences of all peptides used were confirmed by mass spectroscopy. The wild type or A315E-mutant of TDP-43 synthetic peptide (M307-N319) (∼2 mg) was first dissolved in 5 μl DMSO-d6, and then diluted to the final concentration of 2.5 mm in PBS (pH 6.5–6.8) and spun at 13.3 krpm for 10 min to remove any insoluble substance. All the NMR spectra were acquired at 25°C on an Agilent 600 MHz DD2 spectrometer equipped with a triple resonance cold probe. The total correlation spectroscopy (TOCSY) and the Nuclear Overhauser Effect Spectroscopy (NOESY) were carried out to collect spectra for the sequential backbone and the side chain resonance assignments (60), whereas the DQF-COSY was used to measure coupling constants. Approximate inter-proton distance restraints were derived from the 2D 1H-NOESY spectra (with 300 ms mixing time). The spectra were processed with the program NMRPipe (61) and analyzed by Sparky (http://www.cgl.ucsf.edu/home/sparky/). Structures were calculated using the program CNS (62,63). A total of 200 structures were calculated and the final 20 structures with the lowest total energy and least experimental violations were selected to represent the peptide structure. The structure ensemble was displayed and analyzed with the software MolMol (64) and PyMol (http://www.pymol.org/).
Primary neuronal culture, microfluidic chambers, peptide treatment and immunofluorescent staining
Cortical neuronal cultures on coverslips were performed as described (4). Briefly, synthetic TDP-43 peptides were diluted to 200 μm in Neurobasal medium and incubated at room temperature for 8 h before treatment. Peptides are then added to E18 rat cortical neurons that have been cultured in vitro for 6 days (Div6) to a final concentration of 20 μm. At 48 h, half of medium was changed with freshly prepared medium containing 20 μm peptides. Cells were fixed 96 h post-peptide treatment and stained with anti-Tuj1, TUNEL and the Hoechst dye.
Axonal microfluidic chambers were prepared as described (33). Briefly, cortical neurons were isolated from E18 rat embryos in HBSS buffer (Gibco) following disassociation in the presence of papain (Sigma) at 37°C for 15 min. Approximately 5 × 104 cells were seeded into the cell compartment of the microfluidic chambers assembled on the poly-d-lysine (PDL, Sigma)-coated coverslips. Neuronal cultures were incubated at 37°C, 5% CO2 in Neurobasal medium supplemented with 2% B-27 (Gibco). Neurons were cultured 7 days in vitro (DIV) before the peptide treatment. Stock solutions of the control or corresponding TDP-43 peptides at 5 mm were prepared in dimethyl sulfoxide (DMSO) and diluted to 20 μm with the culture medium before adding to neuronal cultures. After rinsing the central flow chamber and axonal chamber once with the peptide solution at the working concentration, 50 μl of corresponding peptide solution was added to the top well in the central flow channel and 100 μl to the axonal well. Sufficient culture media was added in the cell body compartment to maintain a positive hydraulic pressure against peptide diffusion during the entire treatment process. Neuronal axons were incubated with TDP-43 peptides for 72 h before fixation for microscopy. Axonal continuity was quantified using the AxonQuant algorithm that we developed (33).
To examine TDP-43 distribution, HEK293 or primary rat cortical neurons were fixed at different time points following peptide treatment and immunostained with the specific anti-TDP-43 antibody (ProteinTech Group) with secondary antibody conjugated to Cy3. The cortical neurons were immunostained using anti-Tuj1 antibody to reveal axonal morphology in conjunction with anti-TDP-43 to examine TDP-43 distribution. Tuj1-positive neurons were scored for TDP-43 nuclear and cytoplasmic distribution by immunostaining.
ImmunoEM
De-identified postmortem brain samples from patients affected by TDP-43-proteinopathy and control subjects were obtained from the CNADC at Northwestern University, as described previously (4). The brain tissues were fixed in 2% paraformaldehyde with 0.2% glutaraldehyde and embedded in 12% gelatin. The gelatin-embedded blocks were prepared and stored in 2.3 m sucrose at 4°C. Ultrathin sections (70-nm) were cut at −120°C using dry diamond knives. Following blocking, the sections were immunostained with polyclonal anti-TDP-43 antibody (ProteinTech Group; 1:100) and anti-rabbit IgG antibody conjugated to 10 nm-colloidal gold particles. All EM images were obtained using a Tecnai Spirit (120 kV) electron microscope.
SUPPLEMENTARY MATERIAL
AUTHOR CONTRIBUTIONS
J.Y.W. and C.W. conceived and designed the experiments; L.Z., M.X., M.Y., Y.Y., J.D., L.R. and X.L. performed experiments; E.H.B. and M.M. provided critical tissue samples; Y.L., J.L., S.D., W.F., K.F., J.Y.W., Y.Y. and C.W. supervised the study and contributed to data analyses. L.Z. and J.Y.W. wrote the paper.
FUNDING
This work was supported by grants from the National Major Basic Research Program of China (2010CB529603, 2013CB917803, 2011CB932800, 2013CB531002 and 2011CB910503), the National Natural Science Foundation of China (91132710, 91127043, 21273051, 21261130090, 31070657 and 31190062), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA09040300, XDA09030300) and the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-YW-R-154 and KSCX2-EW-J-3). EHB and MM are supported by National Institutes of Health (P30 AG13854). J.Y.W. is supported by ALS Therapy Alliance and NIH (R56NS074763 and RO1AG033004). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of Wu group for helpful discussions and critical reading of the manuscript, Dr M. Shen for expert assistance in molecular dynamic simulation.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Toyama B.H., Weissman J.S. Amyloid structure: conformational diversity and consequences. Annu. Rev. Biochem. 2011;80:557–585. doi: 10.1146/annurev-biochem-090908-120656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bentmann E., Haass C., Dormann D. Stress granules in neurodegeneration—lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 2013;280:4348–4370. doi: 10.1111/febs.12287. [DOI] [PubMed] [Google Scholar]
- 3.Lee E.B., Lee V.M.Y., Trojanowski J.Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo W., Chen Y., Zhou X., Kar A., Ray P., Chen X., Rao E.J., Yang M., Ye H., Zhu L., et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011;18:822–830. doi: 10.1038/nsmb.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cairns N., Bigio E., Mackenzie I.A., Neumann M., Lee V.Y., Hatanpaa K., White C., III, Schneider J., Grinberg L., Halliday G., et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuo P.-H., Doudeva L.G., Wang Y.-T., Shen C.-K.J., Yuan H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009;37:1799–1808. doi: 10.1093/nar/gkp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lukavsky P.J., Daujotyte D., Tollervey J.R., Ule J., Stuani C., Buratti E., Baralle F.E., Damberger F.F., Allain F.H.T. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013;20:1443–1449. doi: 10.1038/nsmb.2698. [DOI] [PubMed] [Google Scholar]
- 8.Chen A.K.H., Lin R.Y.Y., Hsieh E.Z.J., Tu P.-H., Chen R.P.Y., Liao T.-Y., Chen W., Wang C.-H., Huang J.J.T. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 2010;132:1186–1187. doi: 10.1021/ja9066207. [DOI] [PubMed] [Google Scholar]
- 9.Tsao W., Jeong Y.H., Lin S., Ling J., Price D.L., Chiang P.-M., Wong P.C. Rodent models of TDP-43: recent advances. Brain Res. 2012;1462:26–39. doi: 10.1016/j.brainres.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romano M., Feiguin F., Buratti E. Drosophila answers to TDP-43 proteinopathies. J. Amino Acids. 2012;2012:13. doi: 10.1155/2012/356081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inukai Y., Nonaka T., Arai T., Yoshida M., Hashizume Y., Beach T.G., Buratti E., Baralle F.E., Akiyama H., Hisanaga S.-i., et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582:2899–2904. doi: 10.1016/j.febslet.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 12.Cairns N.J., Perrin R.J., Schmidt R.E., Gru A., Green K.G., Carter D., Taylor-Reinwald L., Morris J.C., Gitcho M.A., Baloh R.H. TDP-43 proteinopathy in familial motor neurone disease with TARDBP A315T mutation: a case report. Neuropathol. Appl. Neurobiol. 2010;36:673–679. doi: 10.1111/j.1365-2990.2010.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gitcho M.A., Baloh R.H., Chakraverty S., Mayo K., Norton J.B., Levitch D., Hatanpaa K.J., White C.L., Bigio E.H., Caselli R., et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujita Y., Ikeda M., Yanagisawa T., Senoo Y., Okamoto K. Different clinical and neuropathologic phenotypes of familial ALS with A315E TARDBP mutation. Neurology. 2011;77:1427–1431. doi: 10.1212/WNL.0b013e318232ab87. [DOI] [PubMed] [Google Scholar]
- 15.Robinson J., Geser F., Stieber A., Umoh M., Kwong L., Deerlin V., Lee V.Y., Trojanowski J. TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol. 2013;125:121–131. doi: 10.1007/s00401-012-1055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amador-Ortiz C., Lin W.-L., Ahmed Z., Personett D., Davies P., Duara R., Graff-Radford N.R., Hutton M.L., Dickson D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann. Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arai T., Mackenzie I.A., Hasegawa M., Nonoka T., Niizato K., Tsuchiya K., Iritani S., Onaya M., Akiyama H. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117:125–136. doi: 10.1007/s00401-008-0480-1. [DOI] [PubMed] [Google Scholar]
- 18.Uryu K., Nakashima-Yasuda H., Forman M.S., Kwong L.K., Clark C.M., Grossman M., Miller B.L., Kretzschmar H.A., Lee V.M.-Y., Trojanowski J.Q., et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008;67:555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harper J.D., Wong S.S., Lieber C.M., Lansbury P.T., Jr Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 20.Ranjbar B., Gill P. Circular dichroism techniques: biomolecular and nanostructural analyses—a review. Chem. Biol. Drug Des. 2009;74:101–120. doi: 10.1111/j.1747-0285.2009.00847.x. [DOI] [PubMed] [Google Scholar]
- 21.Sreerama N., Woody R.W. Structural composition of βI- and βII-proteins. Protein Sci. 2003;12:384–388. doi: 10.1110/ps.0235003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Celej M.S., D'Andrea M.G., Campana P.T., Fidelio G.D., Bianconi M.L. Superactivity and conformational changes on alpha-chymotrypsin upon interfacial binding to cationic micelles. Biochem. J. 2004;378:1059–1066. doi: 10.1042/BJ20031536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y.H., Yang J.T., Martinez H.M. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 1972;11:4120–4131. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- 24.Berthomieu C., Hienerwadel R. Fourier transform infrared (FTIR) spectroscopy. Photosynth. Res. 2009;101:157–170. doi: 10.1007/s11120-009-9439-x. [DOI] [PubMed] [Google Scholar]
- 25.Miyazawa T., Blout E.R. Infrared spectra of polypeptides in various conformations—amide I and II bands. J. Am. Chem. Soc. 1961;83:712–719. [Google Scholar]
- 26.Hilbich C., Kisters-Woike B., Reed J., Masters C.L., Beyreuther K. Aggregation and secondary structure of synthetic amyloid βA4 peptides of Alzheimer's disease. J. Mol. Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- 27.Matsuzaki K., Horikiri C. Interactions of amyloid β-peptide (1–40) with ganglioside-containing membranes. Biochemistry. 1999;38:4137–4142. doi: 10.1021/bi982345o. [DOI] [PubMed] [Google Scholar]
- 28.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 29.Li Y., Ray P., Rao E.J., Shi C., Guo W., Chen X., Woodruff E.A., Fushimi K., Wu J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA. 2010;107:3169–3174. doi: 10.1073/pnas.0913602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delisle M., Carpenter S. Neurofibrillary axonal swellings and amyotrophic lateral sclerosis. J. Neurol. Sci. 1984;63:241–250. doi: 10.1016/0022-510x(84)90199-0. [DOI] [PubMed] [Google Scholar]
- 31.Gold B.G. The pathophysiology of proximal neurofilamentous giant axonal swellings: implications for the pathogenesis of amyotrophic lateral sclerosis. Toxicology. 1987;46:125–139. doi: 10.1016/0300-483x(87)90123-5. [DOI] [PubMed] [Google Scholar]
- 32.Tong J., Huang C., Bi F., Wu Q., Huang B., Liu X., Li F., Zhou H., Xia X.-G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013;32:1917–1926. doi: 10.1038/emboj.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y., Yang M., Huang Z., Chen X., Maloney M.T., Zhu L., Liu J., Yang Y., Du S., Jiang X., et al. AxonQuant: a microfluidic chamber culture-coupled algorithm that allows high-throughput quantification of axonal damage. Neurosignals. 2014;22:14–29. doi: 10.1159/000358092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenwald J., Riek R. Biology of amyloid: structure, function, and regulation. Structure. 2010;18:1244–1260. doi: 10.1016/j.str.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Eisenberg D., Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawaya M.R., Sambashivan S., Nelson R., Ivanova M.I., Sievers S.A., Apostol M.I., Thompson M.J., Balbirnie M., Wiltzius J.J.W., McFarlane H.T., et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 37.Wiltzius J.J.W., Landau M., Nelson R., Sawaya M.R., Apostol M.I., Goldschmidt L., Soriaga A.B., Cascio D., Rajashankar K., Eisenberg D. Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol. 2009;16:973–978. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson R., Sawaya M.R., Balbirnie M., Madsen A.O., Riekel C., Grothe R., Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benzinger T.L.S., Gregory D.M., Burkoth T.S., Miller-Auer H., Lynn D.G., Botto R.E., Meredith S.C. Propagating structure of Alzheimer's β-amyloid(10–35) is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA. 1998;95:13407–13412. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang S., Andreasen M., Nielsen J.T., Liu L., Nielsen E.H., Song J., Ji G., Sun F., Skrydstrup T., Besenbacher F., et al. Coexistence of ribbon and helical fibrils originating from hIAPP20–29 revealed by quantitative nanomechanical atomic force microscopy. Proc. Natl. Acad. Sci. USA. 2013;110:2798–2803. doi: 10.1073/pnas.1209955110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petkova A.T., Ishii Y., Balbach J.J., Antzutkin O.N., Leapman R.D., Delaglio F., Tycko R. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petkova A.T., Yau W.-M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2005;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paravastu A.K., Leapman R.D., Yau W.-M., Tycko R. Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmed M., Davis J., Aucoin D., Sato T., Ahuja S., Aimoto S., Elliott J.I., Van Nostrand W.E., Smith S.O. Structural conversion of neurotoxic amyloid-[beta]1-42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C.-W., Giasson B.I., Lewis K.A., Lee V.M., DeMartino G.N., Thomas P.J. A precipitating role for truncated α-synuclein and the proteasome in α-synuclein aggregation: implications for pathogenesis of Parkinson disease. J. Biol. Chem. 2005;280:22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- 47.Colby D.W., Zhang Q., Wang S., Groth D., Legname G., Riesner D., Prusiner S.B. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. USA. 2007;104:20914–20919. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ono K., Condron M.M., Teplow D.B. Structure–neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ono K., Takahashi R., Ikeda T., Yamada M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012;122:883–890. doi: 10.1111/j.1471-4159.2012.07847.x. [DOI] [PubMed] [Google Scholar]
- 50.Xu M., Zhu L., Liu J., Yang Y., Wu J.Y., Wang C. Characterization of β-domains in C-terminal fragments of TDP-43 by scanning tunneling microscopy. J. Struct. Biol. 2013;181:11–16. doi: 10.1016/j.jsb.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang L.-L., Che M.-X., Zhao J., Zhou C.-J., Xie M.-Y., Li H.-Y., He J.-H., Hu H.-Y. Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 2013;288:19614–19624. doi: 10.1074/jbc.M113.463828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim H.J., Kim N.C., Wang Y.-D., Scarborough E.A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A., et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shiina Y., Arima K., Tabunoki H., Satoh J.-i. TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol. 2010;30:641–652. doi: 10.1007/s10571-009-9489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shodai A., Morimura T., Ido A., Uchida T., Ayaki T., Takahashi R., Kitazawa S., Suzuki S., Shirouzu M., Kigawa T., et al. Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43) J. Biol. Chem. 2013;288:14886–14905. doi: 10.1074/jbc.M113.451849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bigio E., Wu J., Deng H.-X., Bit-Ivan E., Mao Q., Ganti R., Peterson M., Siddique N., Geula C., Siddique T., et al. Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol. 2013;125:463–465. doi: 10.1007/s00401-013-1089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nonaka T., Kametani F., Arai T., Akiyama H., Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009;18:3353–3364. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y.-J., Xu Y.-F., Cook C., Gendron T.F., Roettges P., Link C.D., Lin W.-L., Tong J., Castanedes-Casey M., Ash P., et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA. 2009;106:7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barmada S.J., Skibinski G., Korb E., Rao E.J., Wu J.Y., Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 2010;30:639–649. doi: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diaper D.C., Adachi Y., Lazarou L., Greenstein M., Simoes F.A., Di Domenico A., Solomon D.A., Lowe S., Alsubaie R., Cheng D., et al. Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum. Mol. Genet. 2013;22:3883–3893. doi: 10.1093/hmg/ddt243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wuthrich K. NMR of proteins and nucleic acids. New York: John Wiley & Sons; 1986. [Google Scholar]
- 61.Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 62.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 63.Brunger A.T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 64.Koradi R., Billeter M., Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.