

Abstract
Objective
Lung contusion is a major risk factor for the development of acute respiratory distress syndrome. Hypoxia-inducible factor-1α is the primary transcription factor that is responsible for regulating the cellular response to changes in oxygen tension. We set to determine if hypoxia-inducible factor-1α plays a role in the pathogenesis of acute inflammatory response and injury in lung contusion.
Design
Nonlethal closed-chest unilateral lung contusion was induced in a hypoxia reporter mouse model and type 2 cell-specific hypoxia-inducible factor-1α conditional knockout mice. The mice were killed at 5-, 24-, 48-, and 72-hour time points, and the extent of systemic and tissue hypoxia was assessed. In addition, injury and inflammation were assessed by measuring bronchoalveolar lavage cells (flow cytometry and cytospin), albumin (permeability injury), and cytokines (inflammation). Isolated type 2 cells from the hypoxia-inducible factor-1α conditional knockout mice were isolated and evaluated for proinflammatory cytokines following lung contusion. Finally, the role of nuclear factor-κB and interleukin-1β as intermediates in this interaction was studied.
Results
Lung contusion induced profound global hypoxia rapidly. Increased expression of hypoxia-inducible factor-1α from lung samples was observed as early as 60 minutes, following the insult. The extent of lung injury following lung contusion was significantly reduced in conditional knockout mice at all the time points, when compared with the wild-type littermate mice. Release of proinflammatory cytokines, such as interleukin-1β, interleukin-6, macrophage inflammatory protein-2, and keratinocyte chemoattractant, was significantly lower in conditional knockout mice. These actions are in part mediated through nuclear factor-κB. Hypoxia-inducible factor-1α in lung epithelial cells was shown to regulate interleukin-1β promoter activity.
Conclusion
Activation of hypoxia-inducible factor-1α in type 2 cell is a major driver of acute inflammation following lung contusion.
Keywords: cytokines, hypoxia-inducible factor-1α, lung contusion, oxygen-dependent degradation domain luciferase mice, type 2 cells
Lung contusion (LC) is the most common reason for hospital admission following thoracic trauma from motor vehicular accidents and is also a major cause of mortality/morbidity following explosive blast injuries in military and industrial settings (1). In its most severe forms, LC is associated with acute respiratory failure, manifesting as clinical acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), and is also an independent risk factor for ventilator-associated pneumonia (1–3). Our laboratory has previously studied the time course and pathophysiology of isolated LC induced by closed-chest blunt trauma in rodent models (rats and mice) to replicate several clinical features of LC injury (4–11).
Hypoxia, a decrease in available oxygen reaching the tissues of the body, has profound cellular and metabolic consequences. The cellular response to hypoxia is regulated by a family of transcription factors called the hypoxia-inducible factors (HIFs) (12, 13). HIF is composed of two subunits: α subunit (HIF-1α or HIF-2α) and β subunit (HIF-1β). HIF-1α activity is primarily regulated by the abundance of the α subunit. Under hypoxic conditions, HIF-1α is stabilized and translocates into the nucleus where it dimerizes with HIF-1β and activates downstream target genes containing hypoxia-response elements (HREs) within their promoter or enhancer elements (14). These genes include the genes for glycolytic enzymes, sugar transporters, and proangiogenic and inflammatory factors (13, 15–17). HIF-1α has also been shown to modulate inflammation indirectly by influencing the nuclear factor (NF)-κB signaling pathway (18, 19).
Recently, inhibition of HIF-1α was shown to ameliorate lung injury induced by trauma and hemorrhagic shock in rats (20). However, the role of HIF-1α has never been examined or investigated in clinical states characterized by hypoxia, such as LC, ALI, or ARDS.
As hypoxia is an important physiologic attribute of LC, these studies were undertaken to test the hypothesis that hypoxic activation of HIF-1α plays a key role in the generation of acute inflammatory response following LC. The primary role of the alveolar epithelial cells in the initiation and maintenance of ALI remains perplexing. For the longest time, these cells were considered primary targets of injury rather than mechanistically active cells capable of production of key regulatory products including cytokines. Recent reports do suggest that these cells are biologically active (21). However, it is not clear how the AEC regulates the initiation of injury and inflammation. Therefore, we sought to define the specific role of HIF-1α activation in type 2 AEC in defining the inflammatory response.
Materials and Methods
Additional experimental details are available in the online digital supplement (Supplemental Digital Content 1, http://links.lww.com/CCM/A983).
Animals
Male, age-matched (6–8 wk), wild-type (C57BL/6), oxygen-dependent degradation domain (ODD)-luciferase mice and HIF-1α (+/+) and HIF-1α (–/–) mice (Jackson Laboratories, Bar Harbor, ME) were used in this study. All procedures performed were approved by the Institutional Animal Care and Use Committee at the University of Michigan and complied with state, federal, and National Institutes of Health regulations. The number of animals used for different experiments varied from 3 to 14 mice, and a detailed summary is provided for each experimental strategy.
HIF-1α (+/+) and HIF-1α (–/–) Conditional Knockout Mice
Triple transgenic mice were created by mating HIF-1_flox/flox and SP-C-rtTA_/tg/(tetO)7-CMV-Cretg/tg transgenic mice. The generated mice, SP-C-rtTA_/tg/(tetO)7-CMV-Cretg/tg/HIF-1_flox/flox, are capable of respiratory epithelium-specific conditional recombination in the foxed HIF-1α gene upon exposure to doxycycline (22, 23) (a generous gift of John J. LaPres, Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI) (see more details in the Supplementary Method, Supplemental Digital Content 1, http://links.lww.com/CCM/A983).
Murine Model for LC
Male, HIF-1α conditional knockout (cKO) [triple transgenic mice (SP-C-rtTA-/tg/(tetO)7-CMV-Cretg/tg/HIF-1α flox/flox)] and control mice (20–25 g; 6–8 wk; bred in-house) were anesthetized and LC was induced as described by Hoth et al (24) and subsequently modified by our group (9, 10, 25).
Quantitative Analyses of Oxidation Markers by High-Performance Liquid Chromatography Tandem Mass Spectrometry
The protein bound, oxidized amino acids dityrosine, and nitrotyrosine were measured by high-performance liquid chromatography tandem mass spectrometry (HPLC/MS/MS) using a triple quadruple mass spectrometer as described previously (26).
Immunofluorescence Staining
Mice lung sections were prepared and stained as previously described (25) (see more details in the Supplementary Method, Supplemental Digital Content 1, http://links.lww.com/CCM/A983).
Histopathology
Mice lung specimens harvested at the time of death were fixed in 10% formalin, sectioned, and stained with hematoxylin and eosin. Slides were evaluated by an experienced pathologist and graded for the presence of interstitial neutrophilic infiltrate, intra-alveolar hemorrhage, and pulmonary septal edema, as described previously (25).
Preparation and Isolation of Type 2 Alveolar Epithelial Cells From Mice
Crude cell suspensions were prepared from male HIF-1α (–/–) and control mice after LC (27) (see more details in the Supplementary Method, Supplemental Digital Content 1, http://links.lww.com/CCM/A983).
Transfection and Luciferase Assay
Using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), human lung epithelial cell line A549, grown in Dulbecco's modification of eagle's medium/F-12, was cotransfected with 200 ng of pGL3 containing 1KB promoter of human interleukin (IL)-1β or human IL-1β promoter with HRE deleted 50 ng of pcDNA-lacZ and 100 ng oxygen stable HIF-1. Thirty-six hours after transfection, cells were lysed in 200 μL reporter lysis buffer (Promega, Madison, WI), and the luciferase activity was measured in 20 μL of lysate. Luciferase activity was normalized by β-galactosidase activity measured in 5 μL of lysate, as previously described (28, 29).
Statistical Methods
Data are expressed as the mean ± SEM. Statistical significance was estimated using one-way analysis of variance (GraphPad Prism 5.01, La Jolla, CA). Individual intergroup comparisons were analyzed using the two-tailed, unpaired t test with Welch correction. The analyses were run at a significance level of p value of less than 0.05 (25).
Results
ODD-Luciferase Mice Showed Profound Global Hypoxia Following LC
Global hypoxia is a major physiologic attribute of LC and is associated with increased nuclear translocation of HIF-1α. Recently, a mouse model was generated in which a chimeric protein consisting of HIF-1α ODD fused to luciferase was ubiquitously expressed in all tissues. Hypoxic stress leads to the accumulation of ODD-luciferase in the tissues of this mouse model which can be identified by noninvasive bioluminescence measurement. In this study, ODD mice along with uninjured controls were then subjected to an in vivo imaging system (IVIS) to measure the degree of hypoxia 4, 24, and 48 hours after LC. The mice with ODD-linked luciferase showed profound hypoxia especially in the abdominal organs at all-time points after LC (Fig. 1, A and B). In additional experiments using similar animals, LC was induced and the excised organs (lungs, liver, and spleen) were subjected to IVIS. Our results showed that the bioluminescence in the lung regions is lower when compared with the major hypoxic regions of the liver and spleen at all the time points (Fig. 1C).
Figure 1.

A, Lung contusion (LC) is characterized by global hypoxia. The oxygen-dependent degradation domain (ODD)-luciferase mice along with uninjured controls were subjected to in vivo imaging system (IVIS) to measure the degree of hypoxia or hypoxia-inducible factor activation after LC. The mice with ODD-linked luciferase showed increased luminescence at all-time points after LC. Uninjured control, LC 4 hr, LC 24 hr, and LC 48 hr. B, ODD-luciferase mice placed in ventral position at 4 hr after LC demonstrating increased luminescence, especially in the abdomen. C, ODD-luciferase mice were subjected to LC, and the explanted organs were subjected to IVIS to measure the degree of hypoxia after LC. Lung uninjured and LC 24 hr (A), liver uninjured and LC 24 hr (B), and spleen uninjured and LC 24 hr (C).
Lung Homogenates Show Increased Levels of HIF-1α Following LC
Wild-type (C57BL/6) mice were subjected to LC. The lungs of these injured mice along with uninjured controls were harvested at 1, 5, and 24 hours, and HIF-1α expression was determined by Western blot. As seen in Figure 2A, after normalization with Lamin A/B protein, an initial surge in HIF-1α levels was seen at 1 hour after LC which continued to be elevated even at 24-hour time point, compared with the control group.
Figure 2.

Wild-type mice (C57BL/6) showed increased hypoxia-inducible factor (HIF)-1α expression in Western blots following lung contusion (LC). Mice (n = 6) were subjected to LC and the lungs along with uninjured controls were harvested at specified intervals (A). Acriflavine (Acri) administration reduces permeability injury and inflammation after LC. Wild-type (C57 BL/6) mice were pretreated with an intraperitoneal injection of acriflavine at a dose of 10 mg/kg; 48 hr later, the mice were subjected to LC, and permeability injury and inflammation level were determined by enzyme-linked immunosorbent assay as described in Materials and Methods section. Albumin (B), interleukin (IL)-1β (C), IL-6 (D), monocyte chemotactic protein (MCP)-5 (E), nitrotyrosine/tyrosine (F), and dinitrotyrosine/tyrosine (G). BAL = bronchoalveolar lavage.
Acriflavine Administration Inhibits Inflammation and Injury Following LC: the Reduction in Injury Is Not Associated With Increase in Oxidant Generation
Acriflavine, mainly known for its antiseptic properties, inhibits HIF-1α dimerization and prevents its transcriptional activity (30). In this study, we used acriflavine to examine the role of HIF-1α following LC. Wild-type (C57BL/6) mice received acriflavine at a dose of 10 mg/kg 48 hours prior to LC. We first examined the bronchoalveolar lavage (BAL) albumin level at 5- and 24-hour time points by enzyme-linked immunosorbent assay (ELISA) following LC. As expected, there was a significant increase in the BAL albumin level, an indicator of the extent of permeability injury, in LC-induced mice. As shown in Figure 2B, this effect was reversed by acriflavine administration at the 24 hours but not at an earlier time point (5 hr).
To determine if HIF-1α activation has any role in the production of these mediators after LC, we measured the levels of proinflammatory cytokines, such as IL-1β, IL-6, and monocyte chemotactic protein (MCP)-5. The levels of IL-1β were decreased in the 24-hour acriflavine administered animals subjected to LC, when compared with corresponding control animals (Fig. 2C). The increase in the levels of IL-6 was not affected by acriflavine treatment for 5 hours, but it was completely abrogated and brought to the control levels by 24 hours (Fig. 2D). The increased expression of MCP-5 at 24 hours following LC was also significantly inhibited by acriflavine treatment for 24 hours (Fig. 2E). Taken together, these data suggest that HIF-1α is functionally important in the mediation of inflammation and injury following LC.
Our recent study suggests that products of tyrosine oxidation are sensitive markers of oxidative injury and may play an important role in LC (11, 31). To investigate the effect of acriflavine treatment on LC-mediated oxidant production, lung samples were subjected to HPLC/MS/MS, and the levels of nitrotyrosine and dinitrotyrosine, markers of oxidant-mediated lung injury, were measured. However, the results showed that there was no significant difference in the levels of nitrotyrosine and dinitrotyrosine between the acriflavine administered groups and control mice (Fig. 2, F and G).
Confirmation of Inhibition of HIF-1α Following Doxycycline Administration
To specifically characterize the role of HIF-1α in the type 2 AEC, LC was induced in alveolar type 2 cell–specific HIF-1α cKO [HIF-1α (–/–)] [triple transgenic mice (SP-C-rtTA-/tg/(tetO)7-CMV-Cretg/tg/HIF-1α flox/flox)] and control mice. In these mice, feeding doxycycline disrupts HIF-1α from type 2 cells. To confirm if doxycycline will induce recombination within the conditional HIF-1α locus, mice were exposed to doxycycline for 7 weeks starting from postnatal day 1. These mice are referred to as HIF-1α cKO mice. The mice referred to as controls throughout the studies are triple transgenic mice that were maintained on regular food and water. The lungs of control and cKO mice were then removed and analyzed for HIF-1α expression via immunohistochemistry. HIF-1α (+/+) mice showed pronounced HIF-1α expression in the Clara cells lining the bronchiole airway and type 2 cells of the alveoli. In contrast, the cKO mice showed a marked decrease in expression in these cells, with only minor staining visible in the bronchiole airway lining cells and little or no staining in the alveoli (Fig. 3A).
Figure 3.
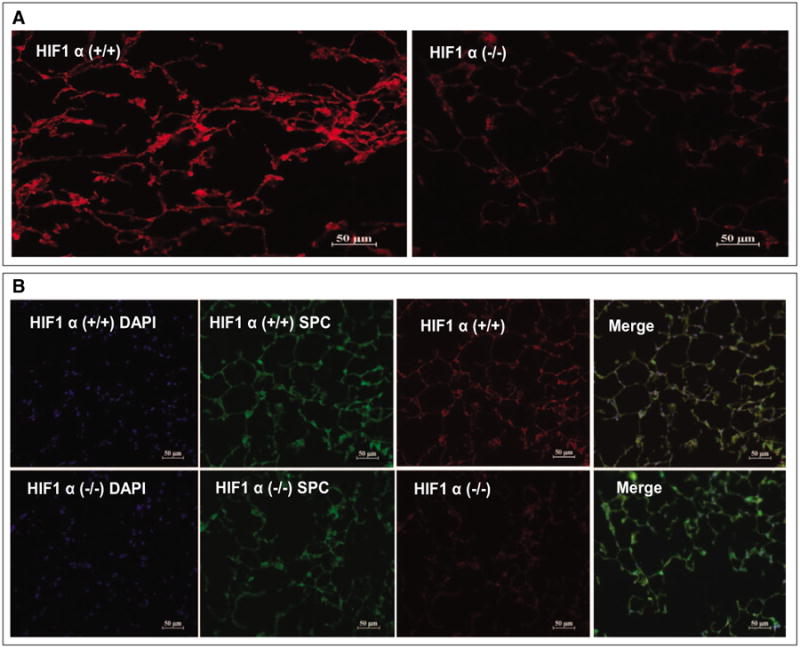
Confirmation of recombination in uninjured hypoxia-inducible factor (HIF)-1α (–/–) mice showed profound inhibition of HIF-1α following doxycycline administration. Immunohistochemistry of lungs samples from HIF-1α (+/+) and HIF-1 (–/–) (A). HIF-1α staining is greatly reduced in the doxycycline-treated animals compared with HIF-1α control. HIF-1α = red, surfactant proteins C (SP-C) = green, and DAPI = blue (B).
Further confirmation using immunohistochemical staining of surfactant proteins C (SP-C) in lung sections from cKO mice shows decreased expression of HIF-1α particularly in the areas of intense staining with SP-C suggesting the specific nature of recombination in type 2 AEC (Fig. 3B). These results confirm that postnatal exposure to doxycycline can induce significant recombination of the HIF-1α locus and that the triple transgenic mouse is a viable model to test the role of HIF-1α following LC.
Downregulation of HIF-1α in Type 2 AEC Results in Reduced Lung Injury and Inflammation Following LC
We have previously shown that the acute inflammatory response in LC is responsible for deficits in oxygenation and increases in quasistatic pulmonary compliance and is associated with severe permeability injury (4, 5, 7, 25). In order to determine the functional aspect of HIF-1α in the extent of mechanical injury following LC, we used cKO mice and the corresponding HIF-1α (+/+) control mice (n = 16). We first examined BAL albumin levels by ELISA at 5-, 24-, 48-, and 72-hour time points following LC. There was a significant decrease in the BAL albumin level, an indicator of the extent of permeability injury due to LC, in the cKO mice when compared with the corresponding HIF-1α (+/+) at all the time points of injury (Fig. 4A).
Figure 4.

Injury and inflammation were reduced in conditional knockout mice following lung contusion (LC). After LC, mice were killed at 5-, 24-, 48-, and 72-hour time points, and albumin and cytokine concentration in the bronchoalveolar lavage (BAL) were determined by enzyme-linked immunosorbent assay as described in Materials and Methods section (mean ± sem, n = 16). Albumin (A), interleukin (IL)-6 (B), IL-1β (C), macrophage inflammatory protein (MIP)-2 (D), keratinocyte chemoattractant (KC) (E), monocyte chemotactic protein (MCP)-5 (F), macrophage (G), and neutrophil (H). Values are represented as mean + sem (n = 16). Statistical analysis was performed on data at each time point, and intergroup comparisons were made with two-tailed unpaired t test with Welch correction. *p value of less than 0.05 hypoxia-inducible factor (HIF)-1α (+/+) vs HIF-1α (–/–). HPF = high power field.
To determine, if HIF-1α activation has any role in the production of pro/anti-inflammatory mediators following LC, we measured the levels of proinflammatory cytokines (IL-1β, IL-6) and MCP-5 chemokines, as well as neutrophilic chemokines macrophage inflammatory protein (MIP)-2 and keratinocyte chemoattractant (KC). The IL-6 levels in the BAL were significantly higher at 5- and 24-hour time points following LC in the control mice compared with the cKO (Fig. 4B). The levels of IL-1β were significantly lower at 5- and 24-hour time points in the cKO mice subjected to LC when compared with corresponding control (Fig. 4C). Levels of MIP-2 and KC were also elevated prominently at 5 hours past LC in the control mice (Fig. 4, D and E) when compared with cKO mice. Levels of macrophage chemokine MCP-5 decreased significantly at 48 and 72 hours in cKO mice compared with control mice (Fig. 4F).
We determined the levels of macrophages and neutrophils in BAL fluid collected from both HIF-1α (–/–) and corresponding wild-type groups at different time intervals using cytospin techniques. The numbers of macrophages were significantly higher at all the time points in the control mice compared with cKO mice (Fig. 4G). We have previously reported that neutrophils are mechanistically important in driving the acute inflammatory response following LC (5, 24, 32, 33). At 48 hours post LC, there was a significant increase in neutrophil levels in the control mice when compared with the cKO (Fig. 4H).
Histological evaluation of the lung tissues suggested that the control mice demonstrated significantly more injury than the cKO mice at all-time points after LC. There was a large area of intra-alveolar hemorrhage in both groups at 5 hours after LC that partially resolved by 24 and 48 hours. However, in the control mice, there were multifocal areas of necrosis and the alveoli were filled with proteinaceous material (fibrin). The alveoli in this area were massively dilated with some loss of alveolar integrity (Fig. 5). On the other hand, histological features suggested considerably less severe injury in the cKO mice. Taken together, these data suggest that activation of HIF-1α specifically in type 2 AEC contributes significantly to lung injury and acute inflammation following LC.
Figure 5.
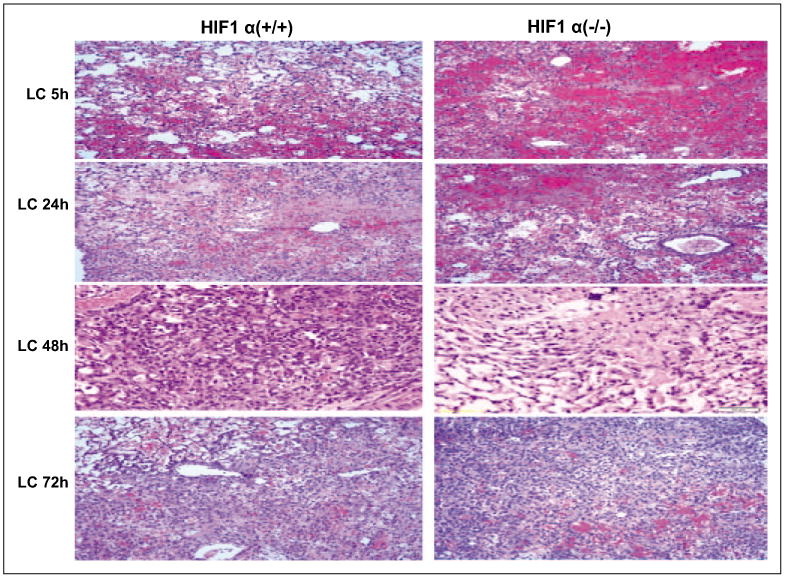
Histological evaluation of hypoxia-inducible factor (HIF)-1α (–/–) mice following lung contusion (LC). Histological evaluations of HIF-1 α (+/+) and HIF-1 α (–/–) mice were conducted as described in Materials and Methods section. The control mice showed signifcantly more injury at 24, 48, and 72 hr. See text for detail.
Alveolar Epithelial Cells Show Increased SP-C Expression in HIF-1α (–/–) Mice Following LC
Surfactant production is an important functional attribute of type 2 AEC. We have previously shown that surfactant dysfunction is responsible for significant physiologic dysfunction in LC (6, 9). It is also well known that surfactant dysfunction worsens lung injury through various mechanisms, including increased atelectasis, inflammation, and alteration of epithelial integrity (9, 34). However, the precise regulation of surfactant production by HIF-1α in type 2 AEC in adult-injured lung is currently not known. We examined the surfactant alteration using immunohistochemical analysis of the SP-C in HIF-1α (+/+) and HIF-1α (–/–) mice, following LC. The lung samples were harvested at the 24- and 48-hour time points and were subjected to immunofluorescence staining with surfactant C for epithelial cells (green), HIF-1α (red), and nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI). The fluorescent images show significantly more surfactant C staining in the alveolar epithelial cells of the HIF-1α (–/–) mice at 48-hour time point compared with the corresponding control mice (Fig. 6). There was no significant difference between these mice at 24 hours after LC. These data strongly indicate that downregulation of HIF-1α in type 2 AEC is associated with significant increase in SP-C levels in injured lungs.
Figure 6.

Hypoxia-inducible factor (HIF)-1α (–/–) mice show increasing surfactant proteins C (SP-C) expression in following lung contusion (LC). Immunohistochemistry of lung samples from HIF-1α (+/+) and HIF-1 (–/–) after LC. Surfactant proteins C (green), HIF-1 (red), and nuclear staining DAPI (blue). *p value of less than 0.05 HIF-1α (+/+) vs HIF-1α (–/–).
HIF-1α in Type 2 AEC Cell Isolates Shows Reduction in Levels of Proinflammatory Cytokines
In a separate experiment, mice were subjected to LC and type 2 epithelial cells were isolated from the lungs of injured animals. Levels of IL-1β, MIP-2, and IL-10 genes were measured by quantitative reverse transcriptase-polymerase chain reaction. These genes play a prominent role in the initiation of acute inflammatory response following LC. We found that the levels of IL-1β after LC was significantly less in the HIF-1α (–/–) mice at all the time points examined, compared with the corresponding HIF-1α (+/+) mice (Fig. 7A). The expression of MIP-2 was found to be significantly elevated in the HIF-1α (+/+) mice following LC, but this phenomenon was not observed in the knockout mice (Fig. 7B). The type 2 cells from HIF-1α (–/–) mice showed significantly increased IL-10 transcript levels compared with the corresponding control mice (Fig. 7C). These results suggest that loss of HIF-1α from alveolar type 2 epithelial and Clara cells of the lungs reduce the inflammatory response following LC.
Figure 7.

RNA from type 2 alveolar epithelial cells induced proinflammatory cytokine production in hypoxia-inducible factor (HIF)-1α (+/+) following lung contusion (LC). RNA isolated from type 2 alveolar epithelial cells collected from HIF-1α (+/+) and HIF-1α (–/–) mice after LC. Then, the levels of proinflammatory mediators interleukin (IL)-1β (A), macrophage inflammatory protein (MIP)-2 (B), and IL-10 (C) were measured. *p value of less than 0.05 HIF-1α (+/+) vs HIF-1α (–/–).
Effects of HIF-1α Are in Part Mediated Through NF-κBp65
Recent evidence has shown that HIF-1α acts through a known secondary transcription factor, NF-κB (35), thus suggesting a cross talk and interdependence between NF-κB and HIF-1α signaling. However, the precise regulation of NF-κB activation on HIF-1α in type 2 AEC in the injured lung is currently not known. We examined NF-κBp65 activation in HIF-1α (+/+) and HIF-1α (–/–) mice subjected to LC, using immunohistochemical analysis and Western blot. The lung samples harvested at 24- and 48-hour time points were subjected to immunofluorescence staining for NF-κB (green) and HIF-1α (red), as well as nuclear staining with DAPI (blue). The fluorescent images show slightly reduced intensity of NF-κB staining in the alveolar epithelial cells of the HIF-1α (–/–) mice at 48-hour time point compared with the HIF-1α (+/+) mice (Fig. 8A). There was no significant difference in NF-κB staining between those mice at 24 hours after LC.
Figure 8.

Hypoxia-inducible factor (HIF)-1α (+/+) induced nuclear factor (NF)-κBp65 expression in alveolar epithelial cells following lung contusion (LC). Immunohistochemistry of lungs samples from HIF-1α (+/+) (A) and HIF-1α (–/–) after LC. A, NF-κBp65 staining (green), HIF-1 staining (red), and nuclear staining DAPI (blue). B, Western blot of NF-κBp65 activation. *p value of less than 0.05 HIF-1α (+/+) vs HIF-1α (–/–).
HIF-1α (+/+) and cKO mice were subjected to LC, and the lungs of these injured mice and uninjured controls were harvested at 24 and 48 hours and assessed for NF-κBp65 activation by Western blot. As seen in Figure 8B, profound expression of NF-κB was seen in HIF-1α (+/+) mice at 48 hours after LC, compared with the control group. Taken together, these results suggest that HIF-1α downregulation of the acute inflammatory response is in part dependent on NF-κBp65.
HIF-1α Activates the Proximal IL-1β Promoter Through a HIF HRE
To explain the HIF-1α-induced interactions at earlier time point of 24 hours, other secondary transcription factors were examined. Sequence analysis revealed the presence of a consensus HRE in the promoter of human IL-1β. In order to investigate whether HIF-1α induces IL-1β expression at the promoter level, we performed luciferase assay in human lung epithelial cell A549, using 1KB of human IL-1β or IL-1β promoter with the HRE mutated. Overexpression of HIF-1α significantly increased the human IL-1β promoter activity, whereas the induction is ablated when HRE is mutated (Fig. 9). These data suggest that HIF-1α induces IL-1β expression by directly binding to its promoter and increases its transcriptional activity.
Figure 9.

Hypoxia-inducible factor (HIF)-1α induces interleukin (IL)-1β expression and increases its transcriptional activity. The experimental methods included transfection of human lung epithelial cell A549 and used a luciferase reporter assay. Statistical analysis was performed on data, and intergroup comparisons were made with two-tailed unpaired t test with Welch correction. HRE = hypoxia-response element
Discussion
There exists a significant gap in the knowledge of factors that are responsible for deterioration of 25–30% patients with LC into severe respiratory failure such as seen with ALI/ARDS. Although hypoxia is a major physiologic consequence of LC, ALI, and ARDS, the precise role of HIF-1α in the pathogenesis of increased permeability or inflammation in the lung remains unresolved. To our knowledge, this study is the first reported study where downregulation of HIF-1α has been studied in animal models of direct lung injury. Specifically, we report that HIF-1α plays a significant role in the progression of lung inflammation and permeability injury following LC. Additionally, the role of type 2 AEC as the primary sentinel cell in the initiation and thereby progression of lung injury has never been fully characterized. There is an evolving concept that these cells are not just innocent bystanders in lung injury but are responsible for production of key regulatory cytokines in addition to surfactant. The current study confirms that downregulation of HIF-1α specifically in type 2 AEC results in significant reduction in lung inflammation and injury.
The critical role of HIF-1α in transcription of various genes involved in cell survival, inflammation, angiogenesis, glycolysis, and iron homeostasis has been well studied (13, 16, 17). HIF-1α signaling in normal development and physiology is underscored by the embryonic lethality observed in mice lacking HIF-1α, HIF-2α, Arnt, and Vhl (15, 36). However, there are few reports suggesting that it is a key regulator in lung injury. In ischemia-reperfusion model of the gut, Feinman et al (37) report that HIF-1α activation is a proximal regulator of gut ischemia-induced lung injury. In addition, the authors using a hemorrhagic shock model reported that HIF-1α activation is necessary for the development of gut injury (37). Additionally, HIF-1α has also been implicated in increased apoptosis of type 2 cells, a feature observed in ALI/ARDS (38). Our data confirm the direct role of HIF-1α in the initiation and maintenance of acute inflammatory response in LC.
It is highly likely that it is the hypoxia that is activating HIF-1α in our model. As seen in the IVIS imaging studies with ODD-luciferase mice, there is global hypoxia. We have also reported that hypoxia is a very prominent and early feature in the rodent model of LC. There are other possible activators of HIF-1α. Oxidants and in particular reactive oxygen species are well known activators of HIF-1α (39). Recently, we have reported that oxidants are responsible for injury and inflammation in LC as well (11). However, the tandem mass spectrometry data of lack of a difference in levels of dityrosine and nitrotyrosine argue against oxidants being the major activator of HIF-1α.
The mechanism of action of how HIF-1α regulates injury downstream likely involves IL-1β-induced aggregation and activation of neutrophils. Nonselective knockdown of HIF-1α with acriflavine results in less inflammation and reduction in levels of IL-1β. Incidentally, the effects of acriflavine are not specific to HIF-1α as some studies have shown that acriflavine additionally inhibits HIF-2α (30). A reduction in the permeability injury and acute inflammatory response were observed when selective knockdown of HIF-1α was performed in type 2 AEC. Our laboratory has shown that acute inflammatory response and injury in LC is neutrophil dependent (7). The histopathological evaluation and reduction in BAL neutrophil numbers suggest that this is an important finding in our study. Furthermore, type 2 AEC isolation experiments suggest that with HIF-1α downregulation, the levels of IL-1β are reduced to negligible levels. Previous studies have clearly reported on the importance of IL-1β in mediation of acute inflammation as a neutrophilic chemokine in various models of injuries (40). Additional studies done in our laboratory suggest that KC and MIP-2 are important neutrophilic chemokines in the evolution of LC (7). Considering that the neutrophil responses in the BAL are only elevated at 48-hour time point even though the neutrophilic chemokines are upregulated at early time points, it is entirely possible that IL-1β is an important mediator of the acute inflammatory response following HIF-1α activation. The transfection experiments with luciferase reporter assay confirm that HIF-1α induces IL-1β expression by directly binding to its promoter and increases its transcriptional activity. These findings are similar to the observed role in linking inflammation and oncogenesis (18). The current study also suggests that the role of HIF-1α at least in part is mediated through NF-κB. This effect is particularly seen at the 48-hour time point following LC. This finding is different from other studies that used cell lines where HIF-1α-mediated NF-κB is specifically elaborated at much earlier time points (35).
One of the primary functions of type 2 AEC is the production of pulmonary surfactant. Active pulmonary surfactant is required to normalize alveolar stability, compliance, and gas exchange (41, 42). Our laboratory has previously reported that, LC in both rat and mouse models, is associated with significant reduction in the quantity as well as change in the quality of pulmonary surfactant (6, 9). The findings presented in this article (Fig. 6) suggest that upregulation of SP-C is observed with knockdown of HIF-1α. There are two possible reasons to explain these findings. One reason would be that HIF-1α directly regulates SP-C levels. The other possibility is that the overall reduction in lung injury and inflammation with the knockdown of HIF-1α results in less surfactant inhibition. Although HREs have been reported in SP-C promoter (43), the precise regulation of and direct interactions of these surfactant proteins by HIF-1α in lung injury is unclear.
In conclusion, the data presented in this article provide clear evidence that hypoxic activation of HIF-1α plays a critical role in the regulation of inflammatory response following LC. Additionally, results indicate a critical role for type 2 AEC in the initiation of acute inflammatory response following LC. Inhibition of HIF-1α apvpears to be a durable target for therapeutic modulation in patients with LC and thereby prevents the progression into ARDS. There are a number of small molecules available for use that specifically inhibit HIF-1α through different mechanisms that include prevention of dimerization and nuclear accumulation (summarized) (44), and some like EZN-2968, an antisense oligonucleotide inhibitor of HIF-1α (ClinicalTrials.gov Identifier: NCT01120288), is currently available in clinical trials.
Supplementary Material
Acknowledgments
We thank Dr. John J. LaPres, Department of Biochemistry and Molecular Biology, Michigan State University, East Lansing, MI, for providing transgenic mice.
Dr. Shah received support for article research from the National Institutes of Health (NIH) (CA148828 and DK095201). Dr. Raghavendran received support for article research from NIH (RO1 HL-102013). He and his institution received grant support from the NIH/National Heart, Lung and Blood Institute.
Footnotes
See also p. 2312).
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (http://journals.lww.com/ccmjournal).
The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Cohn SM. Pulmonary contusion: Review of the clinical entity. J Trauma. 1997;42:973–979. doi: 10.1097/00005373-199705000-00033. [DOI] [PubMed] [Google Scholar]
- 2.Miller PR, Croce MA, Bee TK, et al. ARDS after pulmonary contusion: Accurate measurement of contusion volume identifies high-risk patients. J Trauma. 2001;51:223–228. doi: 10.1097/00005373-200108000-00003. discussion 229-230. [DOI] [PubMed] [Google Scholar]
- 3.Miller PR, Croce MA, Kilgo PD, et al. Acute respiratory distress syndrome in blunt trauma: Identification of independent risk factors. Am Surg. 2002;68:845–850. [PubMed] [Google Scholar]
- 4.Raghavendran K, Davidson BA, Huebschmann JC, et al. Superimposed gastric aspiration increases the severity of inflammation and permeability injury in a rat model of lung contusion. J Surg Research. 2009;155:273–282. doi: 10.1016/j.jss.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raghavendran K, Davidson BA, Helinski JD, et al. A rat model for isolated bilateral lung contusion from blunt chest trauma. Anesth Analg. 2005;101:1482–1489. doi: 10.1213/01.ANE.0000180201.25746.1F. [DOI] [PubMed] [Google Scholar]
- 6.Raghavendran K, Davidson BA, Knight PR, et al. Surfactant dysfunction in lung contusion with and without superimposed gastric aspiration in a rat model. Shock. 2008;30:508–517. doi: 10.1097/SHK.0b013e3181673fc5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raghavendran K, Davidson BA, Woytash JA, et al. The evolution of isolated bilateral lung contusion from blunt chest trauma in rats: Cellular and cytokine responses. Shock. 2005;24:132–138. doi: 10.1097/01.shk.0000169725.80068.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raghavendran K, Notter RH, Davidson BA, et al. Lung contusion: Inflammatory mechanisms and interaction with other injuries. Shock. 2009;32:122–130. doi: 10.1097/SHK.0b013e31819c385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Machado-Aranda D, Wang Z, Yu B, et al. Increased phospholipase A2 and lyso-phosphatidylcholine levels are associated with surfactant dysfunction in lung contusion injury in mice. Surgery. 2013;153:25–35. doi: 10.1016/j.surg.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machado-Aranda DA, Suresh MV, Yu B, et al. Electroporation-mediated in vivo gene delivery of the Na+/K+-ATPase pump reduced lung injury in a mouse model of lung contusion. J Trauma Acute Care Surg. 2012;72:32–39. doi: 10.1097/TA.0b013e31823f0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakshminrusimha S, Suresh MV, Knight PR, et al. Role of pulmonary artery reactivity and nitric oxide in injury and inflammation following lung contusion. Shock. 2013;39:278–285. doi: 10.1097/SHK.0b013e318281d6ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. doi: 10.1152/physrev.1996.76.3.839. [DOI] [PubMed] [Google Scholar]
- 13.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimoda LA, Semenza GL. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. 2011;183:152–156. doi: 10.1164/rccm.201009-1393PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang GL, Jiang BH, Rue EA, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- 18.Jung YJ, Isaacs JS, Lee S, et al. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 19.Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang H, Huang Y, Xu H, et al. Inhibition of hypoxia inducible factor-1 αameliorates lung injury induced by trauma and hemorrhagic shock in rats. Acta Pharmacol Sin. 2012;33:635–643. doi: 10.1038/aps.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhaskaran M, Kolliputi N, Wang Y, et al. Trans-differentiation of alveolar epithelial type II cells to type I cells involves autocrine signaling by transforming growth factor beta 1 through the Smad pathway. J Biol Chem. 2007;282:3968–3976. doi: 10.1074/jbc.M609060200. [DOI] [PubMed] [Google Scholar]
- 22.Saini Y, Greenwood KK, Merrill C, et al. Acute cobalt-induced lung injury and the role of hypoxia-inducible factor 1alpha in modulating inflammation. Toxicol Sci. 2010;116:673–681. doi: 10.1093/toxsci/kfq155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saini Y, Kim KY, Lewandowski R, et al. Role of hypoxia-inducible factor 1{alpha} in modulating cobalt-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L139–L147. doi: 10.1152/ajplung.00252.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoth JJ, Hudson WP, Brownlee NA, et al. Toll-like receptor 2 participates in the response to lung injury in a murine model of pulmonary contusion. Shock. 2007;28:447–452. doi: 10.1097/shk.0b013e318048801a. [DOI] [PubMed] [Google Scholar]
- 25.Suresh MV, Yu B, Machado-Aranda D, et al. Role of macrophage chemoattractant protein-1 in acute inflammation after lung contusion. Am J Respir Cell Mol Biol. 2012;46:797–806. doi: 10.1165/rcmb.2011-0358OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vivekanandan-Giri A, Byun J, Pennathur S. Quantitative analysis of amino acid oxidation markers by tandem mass spectrometry. Methods Enzymol. 2011;491:73–89. doi: 10.1016/B978-0-12-385928-0.00005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol. 1996;14:309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]
- 28.Anderson ER, Xue X, Shah YM. Intestinal hypoxia-inducible factor-2alpha (HIF-2alpha) is critical for efficient erythropoiesis. J Biol Chem. 2011;286:19533–19540. doi: 10.1074/jbc.M111.238667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor M, Qu A, Anderson ER, et al. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140:2044–2055. doi: 10.1053/j.gastro.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee K, Zhang H, Qian DZ, et al. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A. 2009;106:17910–17915. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Suresh MV, Yu B, Lakshminrusimha S, et al. The protective role of MnTBAP in oxidant-mediated injury and inflammation in a rat model of lung contusion. Surgery. 2013;154:980–990. doi: 10.1016/j.surg.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suresh MV, Yu B, Machado-Aranda D, et al. Role of macrophage chemoattractant protein 1 in acute inflammation following lung contusion. Am J Respir Cell Mol Biol. 2012;46:797–806. doi: 10.1165/rcmb.2011-0358OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis CG, Chang K, Osborne D, et al. TLR3 agonist improves survival to secondary pneumonia in a double injury model. J Surg Res. 2013;182:270–276. doi: 10.1016/j.jss.2012.09.039. [DOI] [PubMed] [Google Scholar]
- 34.Holm BA, Notter RH, Siegle J, et al. Pulmonary physiological and surfactant changes during injury and recovery from hyperoxia. J Appl Physiol (1985) 1985;59:1402–1409. doi: 10.1152/jappl.1985.59.5.1402. [DOI] [PubMed] [Google Scholar]
- 35.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412:477–484. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saini Y, Harkema JR, LaPres JJ. HI F1alpha is essential for normal intrauterine differentiation of alveolar epithelium and surfactant production in the newborn lung of mice. J Biol Chem. 2008;283:33650–33657. doi: 10.1074/jbc.M805927200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feinman R, Deitch EA, Watkins AC, et al. HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2010;299:G833–G843. doi: 10.1152/ajpgi.00065.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krick S, Hänze J, Eul B, et al. Hypoxia-driven proliferation of human pulmonary artery fbroblasts: Cross-talk between HIF-1alpha and an autocrine angiotensin system. FASEB J. 2005;19:857–859. doi: 10.1096/fj.04-2890fje. [DOI] [PubMed] [Google Scholar]
- 39.Wang GL, Jiang BH, Semenza GL. Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1995;212:550–556. doi: 10.1006/bbrc.1995.2005. [DOI] [PubMed] [Google Scholar]
- 40.Lappalainen U, Whitsett JA, Wert SE, et al. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 41.Notter RH. Lung Surfactants: Basic Science and Clinical Applications. New York, NY: Marcel Dekker; 2000. [Google Scholar]
- 42.Notter RH, Wang Z. Pulmonary surfactant: Physical chemistry, physiology and replacement. Rev Chem Eng. 1997;13:1–118. [Google Scholar]
- 43.Bryne JC, Valen E, Tang MH, et al. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res. 2008;36:D102–D106. doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powis G, Kirkpatrick L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther. 2004;3:647–654. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.