

Abstract
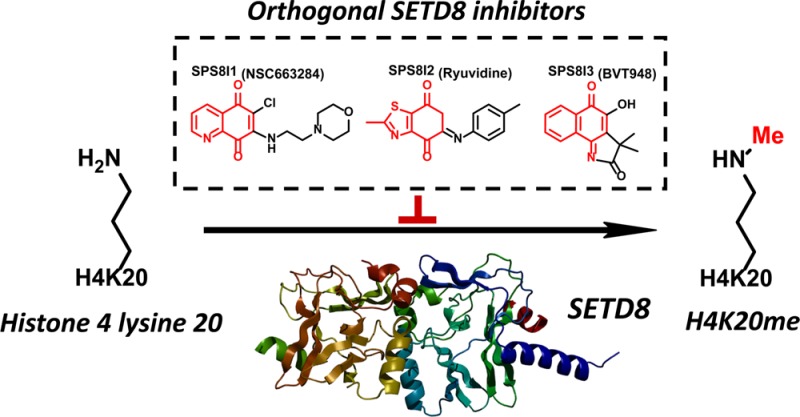
SETD8/SET8/Pr-SET7/KMT5A is the sole protein lysine methyltransferase (PKMT) known to monomethylate lysine 20 of histone H4 in vivo. SETD8’s methyltransferase activity has been implicated in many essential cellular processes including DNA replication, DNA damage response, transcription modulation, and cell cycle regulation. Developing SETD8 inhibitors with cellular activity is a key step toward elucidating the diverse roles of SETD8 via convenient pharmacological perturbation. From the hits of a prior high throughput screen (HTS), SPS8I1–3 (NSC663284, BVT948, and ryuvidine) were validated as potent SETD8 inhibitors. These compounds contain different structural motifs and inhibit SETD8 via distinct modes. More importantly, these compounds show cellular activity by suppressing the H4K20me1 mark of SETD8 and recapitulate characteristic S/G2/M-phase cell cycle defects as observed for RNAi-mediated SETD8 knockdown. The commonality of SPS8I1–3 against SETD8, together with their distinct structures and mechanisms for SETD8 inhibition, argues for the collective application of these compounds as SETD8 inhibitors.
Proteome-wide methylation of lysine residues is an enzymatic posttranslational modification with multifaceted biological outcomes.1,2 This process is catalyzed by protein lysine methyltransferases (PKMTs), which deposit the sulfonium methyl group of the cofactor S-adenosyl-l-methionine (SAM) on diverse histone and nonhistone targets.1,2 Among more than 50 PKMTs encoded by the human genome, SETD8/SET8/Pr-SET7/KMT5A is the sole PKMT known for the in vivo monomethylation of histone H4 lysine 20 (H4K20me1).3,4 The level of the SETD8 protein is finely tuned by multiple E3 ligases throughout a cell cycle with the highest level at G2/M phase and the lowest level at S phase.4−9 Disruption of endogenous SETD8, accompanied by suppression of H4K20 monomethylation, leads to cell cycle defects, chromatin decondensation, and enlarged nuclei, indicating the essential role(s) of SETD8 in DNA replication.10−13 SETD8 also methylates nonhistone targets such as proliferating cell nuclear antigen (PCNA), the tumor suppressor p53, and the p53-stabilizing factor Numb.14−16 Methylation of p53 or Numb results in the downregulation of apoptosis either by antagonizing p53 acetylation, which is required for p53-mediated transcriptional activation, or promoting p53 ubiquitination for degradation.14,15 These findings associate the functions of SETD8 with transcriptional regulation and DNA damage response. Inhibition of SETD8 is thus expected to show a proapoptotic phenotype through the depletion of H4K20 monomethylation, which leads to cell cycle arrest, or p53/Numb-mediated methylation, which results in the upregulation of p53 target genes.14,15 SETD8 has been further implicated in cancer invasiveness and metastasis through its interaction with TWIST,17 a master regulator in epithelial–mesenchymal transition.
The sheer scope of SETD8-associated biology highlights the importance of accessing SETD8 inhibitors, which enable convenient dissection of the functions of SETD8-mediated methylation. Despite such need, few inhibitors of high quality have been reported so far for SETD8 (also see Note),18,19 as well as for other PKMTs implicated in epigenetics and disease.20 Development of PKMT inhibitors aiming at both specificity and potency can be challenging because most PKMTs contain highly similar pockets for binding the SAM cofactor and less-structured regions for binding protein substrates.20 A few examples of potent, selective PKMT inhibitors with demonstrated cellular activities include the chemical probes of G9a/GLP (e.g., UNC0638 and BRD4770), DOT1L (e.g., EPZ000477), and EZH1/2 (e.g., GSK126, EPZ-005687/6438 and EI1).21−26 Prior efforts aimed at SETD8 inhibition have also led to several compounds such as nahuoic acid A18 and bis(bromo/dibromo-methoxylphenol) derivatives19 as in vitro SETD8 inhibitors. However, these compounds have not demonstrated high selectivity or cellular activity against SETD8. The state of the field thus prompted us to explore other small-molecule scaffolds for SETD8 inhibition.
We recently formulated a radioactivity-based scintillation proximity imaging assay (SPA) in a high throughput screening (HTS) format with the purpose of identifying novel SETD8 inhibitors.27 This assay relies on SETD8 to transfer the radioactive [3H-methyl] group from S-adenosyl-l-[3H-methyl]-methionine to a biotinylated H4K20 peptide substrate. The resultant radiolabeled peptide is immobilized onto steptavidin-conjugated SPA imaging polystyrene beads. The proximity between the β-emitting [3H-Me]-labeled peptide and the bead-coated scintillation fluid triggers a luminescence signal, which can be quantified using a LEADseeker system and is suppressed if SETD8 inhibitors are present in the methylation reaction. After screening >5000 compounds from commercial sources (e.g., MicroSource, Prestwick, Tocris, and Sigma), we identified 4 hits, which preferentially suppress the SPA signal of SETD8 but not those of SETD7, SETD2, and GLP (>50% inhibition at a concentration of 10 μM).27 In the present work, we characterize these compounds and demonstrate that NSC663284, BVT948, and ryuvidine (3 out of the 4 HTS hits) inhibit SETD8 via different modes. NSC663284 (SPS8I1), ryuvidine (SPS8I2), and BVT948 (SPS8I3) efficiently and selectively suppress cellular H4K20me1 at doses lower than 5 μM within 24 h (see Figure 1a for the structures of SPS8I1–3). The cells treated with SPS8I1–3 (Small-molecule Pool of SETD8Inhibitor) recapitulate cell-cycle-arrest phenotypes similar to what were reported for knocking down SETD8 by RNAi.11 Given that the three compounds have distinct structures and inhibit SETD8 in different manners, they can be employed collectively as chemical genetic tools to interrogate SETD8-involved methylation.
Figure 1.

Chemical structures, in vitro IC50, and selectivity of SETD8 inhibitors SPS8I1–3. (a) Chemical structures of the three HTS hits with quinonic moieties highlighted in red. SPS8I1 (NSC663284), SPS8I2 (ryuvidine), and SPS8I3 (BVT948) were identified by HTS as potential SETD8 inhibitors and validated in the current work. (b) Dose–response curves of SPS8I1–3. The IC50 values of SPS8I1–3 against SETD8 were measured by the secondary filter paper assay using a low ratio of SAM/peptide/enzyme = 0.75:1.5:1 (see Supporting Information). (c) Selectivity of SPS8I1–3 against a panel of PMTs. The magnitude of IC50 values of SPS8I1–3 is presented against nine phylogenetically related PMTs (their IC50 values are listed in Supplementary Table S1). The diameters of symbols are proportional to the reciprocal values of IC50 and thus higher potency of individual inhibitors. “●” for SPS8I1, “∗L” for SPS8I2 and “+” for SPS8I3.
Among the compounds identified in the SPA-based HTS assays of SETD8, SETD7, SETD2, and GLP, we focused on validating the 4 compounds that were identified solely in the HTS of SETD8.27 The dose–response curves of these compounds against SETD8 were determined by a secondary radiometric filter paper assay.27 Here, the assay parameters including the concentrations of [3H-methyl]-SAM, the H4K20 peptide substrate, and SETD8 (a low ratio of SAM/peptide/enzyme = 0.75:1.5:1) are similar to those used in the primary SPA-based HTS (see Supporting Information). Three compounds (SPS8I1–3) were confirmed as potent inhibitors of SETD8 with apparent IC50 values of 0.21 ± 0.03 μM, 0.5 ± 0.2 μM, and 0.7 ± 0.2 μM, respectively (NSC95397 was triaged because of its high IC50 value of 82 μM) (Figure 1b). The IC50 values largely reflect the interaction between SETD8 and the inhibitors because the concentrations of SAM (0.75 μM) and the H4K20 peptide (1.5 μM) in the assay are far below the values of Km,SAM and Km,H4K20 (24 ± 1 μM and 125 ± 3 μM, respectively, as will be reported elsewhere). The Km,H4K20 value is within 3-fold of that reported previously.28 It is worth noting that the sub-μM IC50 of SPS8I1–3 may not fully reflect their potency because these values fall into the micromolar range of SETD8’s concentration used in the assay and thus represent the lowest values that can be reliably measured under the current settings (see Supporting Information). In addition, the apparent in vitro IC50 values of SPS8I1–3 may alter according to the assay parameters such as the concentrations of reactants and preincubation/reaction time (see discussion later) and the unknown ratio of active versus misfolded SETD8 used in the assay.
To evaluate the selectivity of SPS8I1–3 on SETD8 versus other PMTs, dose–response curves of these compounds were compared among a phylogenic panel of representative human methyltransferases, including 6 PKMTs (SETD2, GLP, G9a, SETD8, SMYD2, and SETD7) and 3 protein arginine methyltransferases (CARM1, PRMT1, and PRMT3) (Figure 1c; Supplmentary Tables S1 and S2). According to the 3 × 9 array of IC50 values, SPS8I1 (see discussion for its non-PMT targets) was identified as the most potent and selective SETD8 inhibitor with an apparent IC50 of 0.21 ± 0.03 μM for SETD8, which is 2.5-fold lower than that of its next hit SMYD2 (0.5 ± 0.2 μM) and >6-fold lower than those of other examined PMTs (from 1.3 to >100 μM) (Figure 1c and Supplementary Table S1). With the 2.5-fold ratio of IC50 values as a threshold, SPS8I2 also demonstrates desired selectivity for SETD8 versus other examined PMTs (Figure 1c and Supplementary Table S1). In contrast, SPS8I3 shows modest selectivity with potential cross-inhibition against PRMT3, SETD2, and CARM1 (Figure 1c and Supplementary Table S1). In a collective view, although the individual SPS8I1–3 might hit multiple PMTs as well as other cellular targets (see discussion later), all three compounds act on SETD8 as the commonly shared PMT target (Figures 1e, 2a).
Figure 2.

Characterization and comparison of SETD8 inhibition by SPS8I1–3. (a) Comparison of SPS8I1–3 as SETD8 inhibitors in vitro and in a cellular setting. The IC50 values of SPS8I1–3 were extracted from Figure 1b. The inhibition modes for SPS8I1–3 (SAM dependence, substrate dependence, slow onset/irreversible characters) were summarized according to the data in panels b–e. Values of kinact and Ki were extracted from Supplementary Figure S1. The EC50 values and cellular phenotypes of SPS8I1–3 are summarized according to the data in Figure 3. The off-target effects of SPS8I1–3 on other PMTs were based on the data in Figure 1c. The bottom line lists the reported non-PMT targets of SPS8I1–3.33−38 (b,c) Characterization of SAM-dependent inhibition of SETD8 by SPS8I1–3. Relative IC50 values were defined as the ratios of IC50 values for each inhibitor (the concentration range of 0.1–100 μM was examined) in the presence of the varied concentrations of the SAM cofactor or substrate in the presence of the lowest concentration of SAM (1 μM) or substrate (5 μM), respectively. These numbers were then plotted against the ratios of [SAM]/Km,SAM or [substrate]/Km,H4K20 (Km,SAM = 24 ± 1 μM and Km,H4K20 or Km,pep = 125 ± 3 μM, as will be reported elsewhere), respectively. The data were arbitrarily fit to 0–3 order exponential curves to show a general trend of IC50 versus the concentrations of SAM or H4K20 peptide. (d) Time-dependent, slow onset inactivation of SETD8 by SPS8I1–3. After incubating with the inhibitors, the relative methyltransferase activity of SETD8 was evaluated by measuring the initial rate and then plotted as the percentage of the loss of the activity versus the DMSO-treated control at different time intervals. (e) Irreversible character of SETD8 inhibition by SPS8I1–3. After preincubating SPS8I1–3 to inactivate SETD8, the mixtures of SETD8 and inhibitors were diluted by 200-fold to lower the concentrations of inhibitors below their IC50 values. The residual methyltransferase activity was monitored and presented as the percentage of the DMSO-treated control.
Given that SPS8I1–3 are structurally distinct except for their quinonic moiety (Figure 1a, highlighted in red), we reasoned that they may act on SETD8 differently (e.g., dependence on cofactor or substrate). Inhibition of SETD8 by SPS8I1–3 was thus examined by measuring relative IC50 values of each inhibitor as a function of the ratios of the concentrations of SAM and the H4K20 peptide substrate versus their respective Km values (Figure 2b,c). SETD8 inhibition mediated by the three compounds (SPS8I1–3) shows a different behavior toward the SAM cofactor with a SAM-dependent character for SPS8I3 (the presence of SAM facilitates SETD8 inhibition) versus a SAM-independent character for SPS8I1 and SPS8I2 (Figure 2b). Concomitantly, SPS8I1 and SPS8I3 are substrate-dependent inhibitors, as shown by the increased IC50 values in the presence of high concentration of substrate, while SPS8I2 is a substrate-independent inhibitor (Figure 2c). Collectively, the three compounds display different characters for SETD8 inhibition within the examined concentrations of SAM and H4K20 peptide, with SPS8I1 favoring the absence of substrate while being insensitive to the presence of SAM, SPS8I3 favoring the absence of substrate and the presence of SAM, and SPS8I2 insensitive to the presence of both SAM and substrate (Figure 2a). Although it is of our interest to explore whether the same trend can be followed within a broader range of concentrations of SAM and the H4K20 peptide (e.g., under saturation conditions), the high Km values (24 μM for SAM and 125 μM for peptide substrate) make it challenging to design such experiments.
Given that SPS8I1–3 share a quinonic motif (Figure 1a) and may act on reactive Cys residues by forming covalent adducts as implicated elsewhere,29 we examined the mechanism of SETD8 inhibition by SPS8I1–3. Incubation of SPS8I1–3 causes a time-dependent inactivation of SETD8’s methyltransferase activity (Figure 2d; Supporting Information). SETD8 inhibition, once achieved by SPS8I1–3, can be sustained over a long period of time (at least 20 h, Figure 2e) even after diluting out free inhibitors (Supporting Information). In contrast, DMSO-treated SETD8 is fully active during the period of incubation (Figure 2d). Given the irreversible character of SPS8I1–3-mediated SETD8 inhibition, we further measured their kinact/Ki values through time-dependent progression. The kinact values of 0.017–0.027 min–1 and the Ki values of 2–17 μM indicate that SPS8I1–3 inhibit SETD8 via an irreversible, slow onset process (Figure 2a, Supplementary Figure S1). As preliminary structure–activity relationship analysis, we also collected three commercial available compounds whose structures contain a quinonic motif and are related to SPS8I1–3 (Supplementary Table S3). All of these compounds are inert to SETD8 inhibition (Supplementary Table S3), suggesting that SPS8I1–3’s special structures rather than the general quinonic motif are essential for SETD8 inhibition.
Structural alignment of the 6 PKMTs examined in the current work (Figure 1c, Supplementary Figure S2a) highlights that SETD8 and SMYD2 contain a distinct Cys residue (Cys270 for SETD8 and Cys181 for SMYD2) around their active sites (Supplementary Figure S2a). Given that SPS8I1 inhibits both SETD8 and SMYD2 (IC50 = 0.21 μM and 0.5 μM, Figure 1c, Supplementary Table S1), we examined whether SETD8’s Cys270 is the target residue to form potential covalent adducts for the observed inhibition. Remarkably, SETD8’s C270S mutant is around 10-fold more resistant to the inhibition of SPS8I1 and SPS8I2 (Supplementary Figure S2b,c) and shows different Hill coefficients in comparison with native SETD8 (Supplementary Figure S2b–d). In contrast, SPS8I3 equally inhibits native SETD8 and the C270S variant (Supplementary Figure S2d). SETD8’s C270 was thus identified as the relevant site for SPS8I1 and SPS8I2 to form covalent adducts, while SPS8I3 may target Cys residues in a more general manner. The different modes of inhibition of SPS8I1–3 may rationalize the modest selectivity of SPS8I3 (SETD8, PRMT3, SETD2, and CARM1 in Figure 1c and Supplementary Table S1) versus the higher specificity of SPS8I1 and SPS8I2 (SETD2 and SMYD2 in Figure 1c and Supplementary Table S1). Here we further performed a Differential Scanning Fluorimetry (DSF) assay to examine inhibitor-bound SETD8. The remarkable thermal shift of 3–6 °C with SYPRO Orange staining for SPS8I1-3 suggests that the SETD8-inhibitor adducts are more susceptible to thermal denaturation and likely adopt less stable, catalytic-inactive conformations (Supplementary Figure S3). In contrast, no such thermal shift was observed for SPS8I1 and SPS8I2 against SETD8’s C270S mutant (Supplementary Figure S3). The collective evidence thus argues that SPS8I1–3 are selective and irreversible inhibitors of SETD8 with distinct structures and at least two modes of interaction (Figure 2a). It remains interesting to solve the structure of the inhibitor-bound SETD8 to further elucidate the mode of interaction.
The irreversible character of SPS8I1–3, together with their different dependence on SAM and substrate for SETD8 inhibition, makes it challenging to evaluate these compounds solely by their in vitro IC50 (or Kd values). We therefore examined the efficiency of SPS8I1–3 by their ability to suppress SETD8’s methylation mark, H4K20me1, under a cellular setting. After treating HEK293T cells with SPS8I1–3, H4K20me1 was rapidly depleted within 24 h, and this effect can be sustained for 3 days with a single -dose of 5 μM SPS8I1, 1 μM SPS8I2, or 5 μM SPS8I3 (Figure 3a; Supplementary Figures S4 and S5). H4K20me1 remained low within 24 h after the treatment with the three inhibitors, indicating that SPS8I1–3 are potent SETD8 inhibitors in the cellular setting. In contrast, no significant change was observed for control marks such as total H4, H4K20me2/3, H3, and H3K9me (Figure 3a; Supplementary Figures S4a, S5). This conclusion was further enforced by quantifying the relative ratios of H4K20me and H4K20me2/3 versus H4 (Supplementary Figure S4b). Because the cells cannot sustain the treatment longer than 3 days (data not shown), we did not further examine other histone marks such as H3K9me2/3, H3K27me2/3, and H3K79me2/3, whose depletion generally takes at least 4 days.23,24 Here the time course of the H4K20me1 depletion is consistent with a previous observation that H4K20me1 is a dynamic histone mark and subject to rapid methylation to H4K20me2/3 by SUV420H1/2.13,30−32 The pool of H4K20me1 can be depleted by blocking its production by SPS8I1–3, coupled with its rapid conversion to H4K20me2/3 by SUV420H1/2. For the cells treated with SPS8I1–3, we also noted a residual level of H4K20me1, which might arise from the H4K20me intermediate during the production of H4K20me2/3 by SUV420H1/2 rather than insufficient inhibition of SETD8 as also observed elsewhere (Figure 3a).13 Collectively, these results demonstrated the efficiency of SPS8I1–3 to inhibit SETD8 under a cellular setting, accompanied by rapid depletion of H4K20me1, at a single dose of 1–5 μM (Figure 2a).
Figure 3.

Cellular inhibition of SETD8 by SPS8I1–3. (a) Western blot of the H4K20me mark upon treatment with SPS8I1–3. The HEK293T cells were treated with varied concentrations of SPS8I1–3 for 3 days (see Supplementary Figure S5 for Day 3). The level of H4K20me was examined as a cellular mark of SETD8’s methyltransferase activity with the levels of H4 and H4K20me2/3 as controls. (b) Cell cycle arrest phenotype associated with SPS8I1–3. HEK293T cells were treated with varied concentrations of SPS8I1–3 for 3 days. The cell cycle distributions of the treated cells were analyzed by flow cytometry. The distributions of cells in S phase and G2/M phase were plotted as their percentage changes in comparison with the DMSO-treated controls. (c) Viability of HEK293T cells treated with SPS8I1–3. HEK293T cells were treated with the varied concentrations of SPS8I1–3 for 3 days. Cell viability was determined by trypan blue staining with the DMSO-treated cells as the controls. Here the cells were treated with 0, 1, and 5 μM of SPS8I1 (left panel); 0 and 1 μM of SPS8I2 (central panel); 0, 1, and 3 μM of SPS8I3 (right panel).
After validating the efficiency of SETD8 inhibition by SPS8I1–3in vitro and in a cellular setting, we further examined the cell-cycle phenotype associated with the treatment of these compounds. Several prior efforts have showed that RNAi-mediated knockdown of SETD8 results in cell-cycle defects at S phase and G2/M phase.11,13 These phenotypes can also be recapitulated by knocking in catalytically dead SETD8 as a dominant-negative mutant.11 After treating HEK293T cells with SPS8I1 at a single subtoxic dose of 5 μM (see viability in Figure 3c), a significant portion of cells were accumulated at S phase after 24 h in comparison with the controls (DMSO or a less effective dose of 1 μM) (Figure 3b, Supplementary Figure S6). The S phase delay was released after 24 h accompanied by increased accumulation of the cells at G2/M phase (Figure 3b, Supplementary Figure S6). A similar phenotype (an increased population of S-phase at 24 h and then an increased population of G2/M phase within 48 h) was also observed upon treating the cells with SPS8I3 at a single dose of 3 μM (Figure 3b, right panel). It is worth noting that the phenotype for SPS8I3 slightly differs from that of SPS8I1 with a more rapid G2/M accumulation for the latter by 24 h. Treating HEK293T cells with SPS8I2 at a single dose of 1 μM also caused S-phase accumulation, although the S-phase arrest was sustained for at least 48 h and did not proceed to G2/M phase as observed for SPS8I1 and SPS8I3 (Figure 3b, middle panel). The slightly different phenotypes of cell cycle defects observed for SPS8I1–3 were likely caused by respective off-target effects (see later discussion). The minimal dosage (1–5 μM SPS8I1–3) and the short treatment period (24 h) required for the manifestation of the cell-cycle defects correlate well with the dose-dependent depletion of SETD8’s methylation mark H4K20me1 (Figure 3a versus b). Such consistency argues that SETD8 inhibition and cell-cycle arrest occur in a comparable time frame upon exposing cells to these inhibitors. The cell-cycle defects were apparently not caused by depleting other histone marks such as H3K9me1/2/3, H3K27me2/3, and H3K79me2/3, a process that generally takes longer than 3 days.23,24SPS8I1–3-associated cell-cycle defects are also unlikely due to general toxicity, given that >50% cells are still viable under these doses (Figure 3c versus Figure 3a,b).
The screening of SPS8I1–3 against the panel of PMTs revealed potential off-target effects on other PMTs (SPS8I1 for SMYD2 and SPS8I3 for PRMT3 and SETD2, Figure 1c). Other cellular targets of SPS8I1–3 were also documented previously in PMT-unrelated settings. For instance, SPS8I1 (NSC663284) was reported as an inhibitor of Cdc25, a dual specificity phosphatase.33−35 The inhibition of Cdc25 by SPS8I1 (NSC663284) was proposed to occur via the production of a covalent adduct or the generation of reactive oxygen species (ROS). For the former mechanism, the catalytic domain of Cdc25 is expected to be inactivated by a 1,4-Michael addition of a serine residue adjacent to the catalytic cysteine. For the latter mechanism, the quinone moiety of this compound may facilitate the oxidation of Cdc25’s catalytic cysteine by the formation of ROS via reductive-oxidative cycling under a cellular context.34 Similarly, SPS8I3 was reported to inhibit several protein tyrosine phosphatases PTPs (e.g., PTP1B) via the production of ROS, such as hydrogen peroxide, and the oxidation of the PTPs’ catalytic cysteines under a cellular setting.36,37SPS8I2 was documented as an in vitro inhibitor of cyclin-dependent kinase 4 and 2 (CDK4/2).38 The off-target effect of SPS8I2 as a CDK2/4 inhibitor may rationalize the severe S-phase defect associated with this compound (Figure 3b).
Nahuoic acid A and bis(bromo/dibromo-methoxylphenol) derivatives were reported previously as SETD8 inhibitors, despite no demonstration of cellular activity for the former and the lack of SETD8 selectivity for the latter.18,19 Interestingly, both of the compounds contain the structural motifs that are amenable for 1,4-Michael addition for reactive Cys residues. Similarly, SPS8I1, SPS8I2, and SPS8I3 contain a quinone moiety, which can also be subject to 1,4-Michael addition of reactive Cys residues such as SET8’s Cys270 for SPS8I1 and SPS8I2 and thus irreversibly inhibit SETD8. The commonality of the reactive functional groups in SPS8I1–3, Nahuoic acid A, and bis(bromo/dibromo-methoxylphenol) derivatives suggests that SETD8 may contain reactive Cys residues such as Cys270 that potentially react with SPS8I1–3 for the observed inhibition. Here we also ruled out the possibility that SPS8I1–3 inhibit SETD8’s methyltransferase activity through their general redox potential for the following reasons: (a) SPS8I1 and SPS8I2 are inert toward SETD8’s C270S mutant, arguing that the inhibitors act on SETD8 by selectively targeting this Cys residue; (b) several structurally related compounds, though containing the redox-active quinonic motif, are inert toward SETD8 (Supplementary Table S3).
SETD8 is the sole PKMT known for H4K20 monomethylation (H4K20me1) in vivo.3,4 H4K20me1 can be either removed by PHF8 or methylated to H4K20me2/3 with a half-lifetime of a few hours.4,13,30−32,39 The dynamic character for H4K20me1 was well reflected by its rapid depletion by SPS8I1–3 within 24 h, in contrast to the more stable histone methylation marks such as H3K9me2/3, H3K27me2/3, and H3K79me2/3.23,24 A residual level of H4K20me1 after the treatment with SPS8I1–3 (Figure 3a) may arise from the incomplete production of H4K20me2/3 by SUV240H1/2.13 In contrast to the dramatic decrease of the H4K20me1 mark upon the treatment with SPS8I1–3, no acute change of the H4K20me2/3 marks was observed, although H4K20me1 has been implicated as SUV420H1/2’s substrate for H4K20me2/3 production.13 This observation suggests that H4K20me2/3, like H3K9me2/3, H3K27me2/3, and H3K79me2/3, are more stable histone methylation marks, and the 1–3-day depletion of H4K20me1 (<5% of the overall methylated H4K20) has a limited impact on overall H4K20me2/3 levels (>95% of the overall methylated H4K20).13,30−32 Besides the H4K20me1 depletion, SPS8I–treated HEK293T cells display cell cycle defects at S phase within 24 h, suggesting that this phenotype is associated with the depletion of H4K20me1 mark but not H4K20me2/3, whose levels remain largely unchanged during the period of treatment. The cell cycle defect can then proceed to G2/M phase within 24–48 h as demonstrated by SPS8I1 and SPS8I3. These results argue that the S phase delay caused by SETD8 inhibition is transient and can eventually advance to G2/M arrest. More importantly, these phenotypes tightly correlate with the H4K20me1 mark and SETD8’s methyltransferase activity. Interestingly, both S-phase and G2/M-phase defects were reported previously as the phenotypes of SETD8 knockdown.11,40 Such discrepancy (S-phase versus G2/M-phase defect) can be rationalized by the different time frames of SETD8 knockdown in individual experimental settings.
In the current work, 3 out of 4 HTS hits were validated in vitro and in a cellular context as potent inhibitors of SETD8. This hit validation rate is remarkable and underscores the robustness of the SPA-based HTS assay for identifying SETD8 inhibitors. The general applicability of the SPA-based HTS assay also highlights its merits for screening other PMTs.27 The 3 compounds were characterized as potent inhibitors of SETD8 with the ability to suppress SETD8’s H4K20me1 mark at single doses of 1–5 μM in a cellular context. Structurally distinct SPS8I1–3 also display different modes of SETD8 inhibition. Such differences also make SPS8I1–3 less likely to act on other common cellular targets besides SETD8. As a result, the shared phenotypes of the 3 compounds are expected to be associated with SETD8 inhibition. More importantly, at the effective doses required for H4K20me1 suppression, the observed cell-cycle-arrest phenotype of SPS8I1–3 is similar to that observed for SETD8 knockdown.11 Strikingly, the phenotypes of the H4K20me1 depletion and cell cycle arrest of SPS8I1–3 can be sustained for up to 3 days at a single dose of <5 μM SPS8I1–3. Such robust inhibition of SETD8 by SPS8I1–3, together with their different off-target effects, argues that these compounds can be used collectively as SETD8 inhibitors to offset off-target effects of individual reagents. At this stage, we envision using all three compounds to examine SETD8 inhibition and then focusing on the phenotypes shared by all of them. SPS8I1 and SPS8I2 were reported to be cytotoxic toward various cancer cell lines.35,38 Given the general cell cycle arrest phenotypes of SPS8I1–3, as well as the specific implication in cancer,14,15,17,35,38 it is of great interest to further examine these compounds and their derivatives for more selective SETD8 inhibitors and novel anticancer reagents.
Methods
Acknowledgments
We thank Drs. Thompson, Min, Frankel, and Trievel for plasmids; the Proteomics Resource Center of the Rockefeller University for preparing peptides; Dr. Dzhekieva for helpful discussion of inhibition experiments; the support of this work by Mr. William H. Goodwin and Mrs. Alice Goodwin (M.L., H.D.), The Commonwealth Foundation for Cancer Research and The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center (M.L., H.D.), NIGMS 1R01GM096056 (M.L.), the NIH Director’s New Innovator Award Program 1DP2-OD007335 (M.L.), NINDS R21NS071520 (M.L.), Starr Cancer Consortium (M.L.) and the Tri-Institutional Training Program in Chemical Biology (G.B.), the William Randolph Hearst Fund in Experimental Therapeutics (H.D.), the Lillian S. Wells Foundation (H.D.), and NIH/NCI Cancer Center Support Grant 5P30 CA008748-44, NIH GM075094 (J.R.) and ACS RSG117619 (J.R.).
Supporting Information Available
Supplementary figures and tables, materials and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Notes
During review of this manuscript, a set of substrate-competitive SETD8 inhibitors were also reported.41
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Luo M. (2012) Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem. Biol. 7, 443–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K.; Rice J. C.; Sarma K.; Erdjument-Bromage H.; Werner J.; Wang Y.; Chuikov S.; Valenzuela P.; Tempst P.; Steward R.; Lis J. T.; Allis C. D.; Reinberg D. (2002) PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213. [DOI] [PubMed] [Google Scholar]
- Wu S.; Rice J. C. (2011) A new regulator of the cell cycle: the PR-Set7 histone methyltransferase. Cell Cycle 10, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S.; Eskildsen M.; Fugger K.; Hansen L.; Larsen M. S.; Kousholt A. N.; Syljuasen R. G.; Trelle M. B.; Jensen O. N.; Helin K.; Sorensen C. S. (2011) SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J. Cell. Biol. 192, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centore R. C.; Havens C. G.; Manning A. L.; Li J. M.; Flynn R. L.; Tse A.; Jin J.; Dyson N. J.; Walter J. C.; Zou L. (2010) CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H.; Hubner M. R.; Beck D. B.; Vermeulen M.; Hurwitz J.; Spector D. L.; Reinberg D. (2010) Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 40, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Wang W.; Kong X.; Congdon L. M.; Yokomori K.; Kirschner M. W.; Rice J. C. (2010) Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 24, 2531–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas T.; Shibata E.; Park J.; Jha S.; Karnani N.; Dutta A. (2010) CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen M. S.; Sy S. M.; van Deursen J. M.; Chen J. (2008) Direct interaction between SET8 and proliferating cell nuclear antigen couples H4-K20 methylation with DNA replication. J. Biol. Chem. 283, 11073–11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston S. I.; McManus K. J.; Adams M. M.; Sims J. K.; Carpenter P. B.; Hendzel M. J.; Rice J. C. (2008) Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J. Biol. Chem. 283, 19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M.; Brustel J.; Kirsh O.; Lefevbre C.; Callanan M.; Sardet C.; Julien E. (2010) The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 12, 1086–1093. [DOI] [PubMed] [Google Scholar]
- Jorgensen S.; Schotta G.; Sorensen C. S. (2013) Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 41, 2797–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Kachirskaia I.; Yamaguchi H.; West L. E.; Wen H.; Wang E. W.; Dutta S.; Appella E.; Gozani O. (2007) Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami G. K.; Liu H.; Galka M.; Voss C.; Wei R.; Muranko K.; Kaneko T.; Cregan S. P.; Li L.; Li S. S. (2013) Dynamic methylation of Numb by Set8 regulates its binding to p53 and apoptosis. Mol. Cell 50, 565–576. [DOI] [PubMed] [Google Scholar]
- Takawa M.; Cho H. S.; Hayami S.; Toyokawa G.; Kogure M.; Yamane Y.; Iwai Y.; Maejima K.; Ueda K.; Masuda A.; Dohmae N.; Field H. I.; Tsunoda T.; Kobayashi T.; Akasu T.; Sugiyama M.; Ohnuma S.; Atomi Y.; Ponder B. A.; Nakamura Y.; Hamamoto R. (2012) Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 72, 3217–3227. [DOI] [PubMed] [Google Scholar]
- Yang F.; Sun L.; Li Q.; Han X.; Lei L.; Zhang H.; Shang Y. (2012) SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 31, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. E.; Dalisay D. S.; Li F.; Amphlett J.; Maneerat W.; Chavez M. A.; Wang Y. A.; Matainaho T.; Yu W.; Brown P. J.; Arrowsmith C. H.; Vedadi M.; Andersen R. J. (2013) Nahuoic acid A produced by a Streptomyces sp. isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org. Lett. 15, 414–417. [DOI] [PubMed] [Google Scholar]
- Valente S.; Lepore I.; Dell’Aversana C.; Tardugno M.; Castellano S.; Sbardella G.; Tomassi S.; Di Maro S.; Novellino E.; Di Santo R.; Costi R.; Altucci L.; Mai A. (2012) Identification of PR-SET7 and EZH2 selective inhibitors inducing cell death in human leukemia U937 cells. Biochimie 94, 2308–2313. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Solomon M. E.; Richon V. M. (2009) Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discovery 8, 724–732. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Wang Q.; Paulk J.; Kubicek S.; Kemp M. M.; Adams D. J.; Shamji A. F.; Wagner B. K.; Schreiber S. L. (2012) A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 7, 1152–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J. C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M. T.; Ott H. M.; Ganji G.; Korenchuk S.; Thompson C.; Van Aller G. S.; Liu Y.; Graves A. P.; Della Pietra A. 3rd; Diaz E.; LaFrance L. V.; Mellinger M.; Duquenne C.; Tian X.; Kruger R. G.; McHugh C. F.; Brandt M.; Miller W. H.; Dhanak D.; Verma S. K.; Tummino P. J.; Creasy C. L. (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. [DOI] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-Lovejoy D.; Liu F.; Rival-Gervier S.; Allali-Hassani A.; Labrie V.; Wigle T. J.; Dimaggio P. A.; Wasney G. A.; Siarheyeva A.; Dong A.; Tempel W.; Wang S. C.; Chen X.; Chau I.; Mangano T. J.; Huang X. P.; Simpson C. D.; Pattenden S. G.; Norris J. L.; Kireev D. B.; Tripathy A.; Edwards A.; Roth B. L.; Janzen W. P.; Garcia B. A.; Petronis A.; Ellis J.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Jin J. (2011) A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Kawano S.; Minoshima Y.; Warholic N. M.; Huang K. C.; Xiao Y.; Kadowaki T.; Uesugi M.; Kuznetsov G.; Kumar N.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Waters N. J.; Smith J. J.; Porter-Scott M.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Uenaka T.; Pollock R.; Kuntz K. W.; Yokoi A.; Keilhack H. (2014) Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854. [DOI] [PubMed] [Google Scholar]
- Qi W.; Chan H.; Teng L.; Li L.; Chuai S.; Zhang R.; Zeng J.; Li M.; Fan H.; Lin Y.; Gu J.; Ardayfio O.; Zhang J. H.; Yan X.; Fang J.; Mi Y.; Zhang M.; Zhou T.; Feng G.; Chen Z.; Li G.; Yang T.; Zhao K.; Liu X.; Yu Z.; Lu C. X.; Atadja P.; Li E. (2012) Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. U.S.A. 109, 21360–21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez G.; Shum D.; Blum G.; Bhinder B.; Radu C.; Antczak C.; Luo M.; Djaballah H. (2012) A high throughput scintillation proximity imaging assay for protein methyltransferases. Comb. Chem. High Throughput Screening 15, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture J. F.; Collazo E.; Brunzelle J. S.; Trievel R. C. (2005) Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 19, 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S.; Lefterov I. M.; Wang M.; Lazo J. S.; Scott C. N.; Wilcox C. S.; Carr B. I. (2003) Binding and inhibition of Cdc25 phosphatases by vitamin K analogues. Biochemistry 42, 10490–10497. [DOI] [PubMed] [Google Scholar]
- Oda H.; Okamoto I.; Murphy N.; Chu J.; Price S. M.; Shen M. M.; Torres-Padilla M. E.; Heard E.; Reinberg D. (2009) Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell. Biol. 29, 2278–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento J. J.; Yang H.; Kelleher N. L.; Mizzen C. A. (2008) Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 28, 468–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee B. M.; Levin R. S.; Dimaggio P. A.; Garcia B. A. (2010) Global turnover of histone post-translational modifications and variants in human cells. Epigenet. Chromatin 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L.; Amoscato A. A.; Bier M. E.; Lazo J. S. (2002) Dual G1 and G2 phase inhibition by a novel, selective Cdc25 inhibitor 6-chloro-7-[corrected](2-morpholin-4-ylethylamino)-quinoline-5,8-dione. J. Biol. Chem. 277, 46877–46885. [DOI] [PubMed] [Google Scholar]
- Brisson M.; Nguyen T.; Wipf P.; Joo B.; Day B. W.; Skoko J. S.; Schreiber E. M.; Foster C.; Bansal P.; Lazo J. S. (2005) Redox regulation of Cdc25B by cell-active quinolinediones. Mol. Pharmacol. 68, 1810–1820. [DOI] [PubMed] [Google Scholar]
- Lazo J. S.; Aslan D. C.; Southwick E. C.; Cooley K. A.; Ducruet A. P.; Joo B.; Vogt A.; Wipf P. (2001) Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J. Med. Chem. 44, 4042–4049. [DOI] [PubMed] [Google Scholar]
- Han Y.; Shen H.; Carr B. I.; Wipf P.; Lazo J. S.; Pan S. S. (2004) NAD(P)H:quinone oxidoreductase-1-dependent and -independent cytotoxicity of potent quinone Cdc25 phosphatase inhibitors. J. Pharmacol. Exp. Ther. 309, 64–70. [DOI] [PubMed] [Google Scholar]
- Liljebris C.; Baranczewski P.; Bjorkstrand E.; Bystrom S.; Lundgren B.; Tjernberg A.; Warolen M.; James S. R. (2004) Oxidation of protein tyrosine phosphatases as a pharmaceutical mechanism of action: a study using 4-hydroxy-3,3-dimethyl-2H-benzo[g]indole-2,5(3H)-dione. J. Pharmacol. Exp. Ther. 309, 711–719. [DOI] [PubMed] [Google Scholar]
- Ryu C. K.; Kang H. Y.; Lee S. K.; Nam K. A.; Hong C. Y.; Ko W. G.; Lee B. H. (2000) 5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents. Bioorg. Med. Chem. Lett. 10, 461–464. [DOI] [PubMed] [Google Scholar]
- Liu W.; Tanasa B.; Tyurina O. V.; Zhou T. Y.; Gassmann R.; Liu W. T.; Ohgi K. A.; Benner C.; Garcia-Bassets I.; Aggarwal A. K.; Desai A.; Dorrestein P. C.; Glass C. K.; Rosenfeld M. G. (2010) PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M.; Murr R.; Herceg Z.; Sardet C.; Julien E. (2007) PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell. Biol. 179, 1413–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A.; Yu W.; Li F.; Bleich R. M.; Herold J. M.; Butler K. V.; Norris J. L.; Korboukh V.; Tripathy A.; Janzen W. P.; Arrowsmith C. H.; Frye S. V.; Vedadi M.; Brown P. J.; Jin J. (2014) Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem. 57, 6822–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.