

Abstract
Developing therapeutics for traumatic brain injury remains a challenge for all stages of recovery. The pathological features of traumatic brain injury are diverse, and it remains an obstacle to be able to target the wide range of pathologies that vary between traumatic brain injured patients and that evolve during recovery. One promising therapeutic avenue is to target the second messengers cAMP and cGMP with phosphodiesterase inhibitors due to their broad effects within the nervous system. Phosphodiesterase inhibitors have the capability to target different injury mechanisms throughout the time course of recovery after brain injury. Inflammation and neuronal death are early targets of phosphodiesterase inhibitors, and synaptic dysfunction and circuitry remodeling are late potential targets of phosphodiesterase inhibitors. This review will discuss how signaling through cyclic nucleotides contributes to the pathology of traumatic brain injury in the acute and chronic stages of recovery. We will review our current knowledge of the successes and challenges of using phosphodiesterase inhibitors for the treatment of traumatic brain injury and conclude with important considerations in developing phosphodiesterase inhibitors as therapeutics for brain trauma.
Keywords: cAMP, cognition, CREB, hippocampus, inflammation, long-term potentiation, phosphodiesterase, protein kinase A, rolipram, synaptic plasticity, traumatic brain injury
1. INTRODUCTION
Traumatic brain injury (TBI) has been hampered in therapeutic development in both the acute and chronic recovery stages [1, 2]. Not only is there a lack of therapeutics adequately addressing the unique consequences of brain trauma, but there is a lack of understanding in the molecular and circuitry changes induced by TBI that need to be targeted with therapeutics. Therapeutic development has also been impeded by the issue that the targets are changing as the TBI survivor recovers and develops coping strategies [3]. An enormous amount of change occurs in the brain in the acute to chronic time windows after brain trauma and even these transitions from acute to subacute to chronic recovery have yet to be identified [4]. Progressive secondary injury coupled with endogenous reparative strategies render the injured brain a moving target for therapeutics. Nevertheless, these are critical issues of importance in understanding the molecular, physiological and circuitry changes that underlie the behavioral problems that develop as a consequence of TBI.
A therapeutic target that is evolving and developing into a key player in multiple recovery mechanisms is phosphodiesterases (PDEs). PDEs subserve a variety of basic functions in the brain. They are involved in synaptic plasticity, homeostasis, and a number of basic behaviors including cognition, anxiety, and mood regulation [5, 6]. PDEs are critically important in neuronal functioning because they are key regulators of the second messenger cyclic nucleotides. Cyclic nucleotides consist of cyclic adenosine 3′-5′-monophosphate (cAMP) and cyclic guanosine 3′-5′-monophosphate (cGMP) and are important second messengers in a variety of cells within the central nervous system (CNS). Cyclic nucleotides are synthesized by adenylyl cyclases (ACs) from ATP and guanylyl cyclases (GCs) from GTP to exert signaling cascades within neurons, microglia, and astrocytes. A key step in control of their activities is hydrolysis to the inactive molecules AMP and GMP. This is performed by the enzymes PDEs, which can be broadly divided into three categories, PDEs that hydrolyze cAMP preferentially, PDEs that hydrolyze cGMP only, and PDEs that hydrolyze both cAMP and cGMP [5].
Eleven PDEs families have been identified with differing specificity towards cAMP and cGMP [5]. These families are numbered 1–11, with individual genes denoted by a letter (e.g., PDE4D being the D gene of the PDE4 family). The PDE4, PDE7, and PDE8 family members are cAMP-specific. The PDE5, PDE6, and PDE9 family members are cGMP-specific. The PDE1, PDE2, PDE3, PDE10, and PDE11 family members hydrolyze both cAMP and cGMP. There are between one to four genes for each PDE family, and within each gene family, numerous splice variants exist and are denoted by a number (e.g., PDE4D3 being splice variant 3 of PDE4D) [5].
Of the 11 PDE families, perhaps the most investigated in the context of brain injury is PDE4. PDE4 is highly specific for cAMP, with a low Km of 1–3 μM [7]. It is not sensitive to cGMP or calcium, but is sensitive to rolipram, a prototypical PDE4 inhibitor. There are four PDE4 genes, 4A, B, C, and D and they are unique within the PDEs by having an upstream conserved region (UCR) which responds to intracellular signals to regulate their activity. The different families are also multiply spliced resulting in different subgroups: long, short, supershort, and dead-short isoforms (Fig. 1). Long isoforms contain both a UCR1 and UCR2, short isoforms lack UCR1 but do contain UCR2, whereas super-short isoforms lack UCR1 and have truncated UCR2 regions; the dead-short isoforms have a truncated catalytic domain and lack both UCR1 and UCR2 [8, 9]. This results in differential regulation because UCR1 can be phosphorylated by protein kinase A (PKA) which activates the enzyme whereas short isoforms can be phosphorylated by phosphoinositide-3-kinase as well as extracellular signal-regulated kinase (ERK) 1/2 in the catalytic domain, resulting in inhibition of the enzyme [10, 11]. The differential regulation of the enzymes suggests that they do not exert redundant functions in the CNS. In fact, isoform-specific knockout mice studies have revealed that PDE4B and PDE4D in particular, subserve very different functions in the brain. PDE4B is involved in anxiety and inflammation, whereas PDE4D is involved in memory formation, adult neurogenesis, and depression [12–17]. Given the diversity of PDEs in the CNS and their various functions, it is important to understand their differential contributions to the pathology of CNS injury and their role in recovery.
Fig. 1.
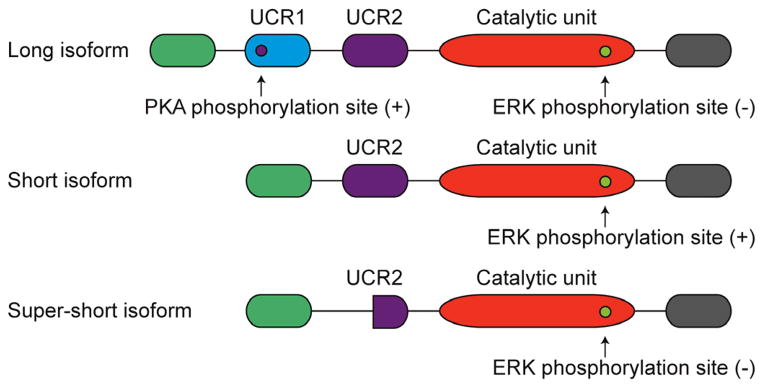
PDE4 isoforms and their post-translational regulation. PDE4 isoforms are classified based on the presence of their upstream conserved regions (UCR) and each isoform is differentially regulated by PKA and ERK 1/2 phosphorylation; these posttranslational modifications result in either an increase (+) or decrease (−) in PDE activity.
This review provides an overview of the regulation of the PDEs and cyclic nucleotide signaling after brain injury, and therapeutic strategies targeting PDEs to improve outcome after CNS injury. Strategies to refine PDE4 inhibitors as therapeutics for brain injury are also discussed.
2. CHANGES IN cAMP SIGNALING AFTER TBI
Many intracellular signaling cascades are activated acutely following TBI, however, the cAMP-PKA signaling pathway is unusual in that this pathway is one of the few that is acutely depressed after TBI [18, 19]. Fluid-percussion brain injury, a brain injury model that results in both focal and diffuse injury within the brain, decreases levels of cAMP within both the injured cortex and hippocampus by 15 minutes after trauma. These changes begin to normalize and within three days, cAMP levels are back to sham, non-injured levels. In contrast to fluid-percussion brain injury, one study has reported no change in cAMP levels after controlled cortical impact, a more focal brain injury model [20]. The depression of cAMP levels parallels decreases in PKA phosphorylation at Thr197 on the catalytic site [18, 21]. Thus, unlike many other signaling molecules such as the calcium/calmodulin-dependent protein kinases, ERK, and protein kinase C which are activated acutely after brain trauma, both cAMP and PKA are transiently downregulated [18, 22–28].
Recently, we found that changes in cAMP levels are sensitive to injury severity. The aforementioned changes were in reference to moderate brain injury severity. In contrast, mild fluid-percussion brain injury has no significant effect on cAMP levels within the hippocampus in young adult animals, although as discussed in Section 4, this does not hold true for aged animals [19]. In addition, cortical changes in cAMP after mild TBI are less significant than what is observed after moderate TBI and independent of age.
An interesting twist on this pathway is the injury-induced changes in the transcription factor cyclic AMP-responsive-element-binding protein (CREB), an important downstream target of cAMP-PKA signaling. Numerous studies have reported that CREB phosphorylation increases within minutes after brain trauma and continues to rise for up to 1 week post-injury [23, 24, 26, 29]. The differential response of CREB versus cAMP and PKA to brain trauma may be due to a number of reasons; however, no study has yet deciphered the cause or meaning of this dissociation. CREB phosphorylation at this site (Ser133) is mediated by numerous protein kinases beyond PKA, including calcium/calmodulin-dependent protein kinase IV and pp90RSK, which are also known to be upregulated after brain trauma [23, 30, 31].
Like TBI, other models of brain injury such as ischemia report similar changes in the cAMP-PKA-CREB signaling pathway. There is a significant increase in CREB phosphorylation after ischemic brain injury [32]. However, the sustained effect of ischemia significantly depresses levels of cAMP, cytosolic PKA catalytic subunit β, nuclear PKA catalytic subunits α and β, and CREB-DNA binding activity in both the cortex and hippocampus [33].
These studies raise interesting questions, how are cAMP levels decreased after brain injury and what are the functional consequences of these decreases? We will attempt to answer these questions in the following sections and discuss the putative mechanisms that contribute to decreases in cAMP levels after TBI (Section 3) and how changes in cAMP and PDE signaling contribute to the pathomechanisms of acute (Section 6) and chronic TBI (Section 7).
3. PHOSPHODIESTERASE REGULATION IN BRAIN TRAUMA
Using the fluid-percussion brain injury model, we have characterized the regulation of a number of PDE isoforms after TBI [34]. After fluid-percussion brain injury, PDE1A protein levels increased rapidly and dramatically within one hour in the injured cerebral cortex, and remained elevated for at least one day thereafter. However, mRNA levels of Pde1A did not change post-injury, suggesting that the increase was not due to gene regulation, but rather was due to translational regulation and/or decreased protein turnover. In the non-injured brain, PDE1A mRNA is most strongly present in the cortex and hippocampus [35]. Similarly after TBI, PDE1A expression was restricted to neurons in the cerebral cortex, particularly in layers IV/V, in both the injured and non-injured brain. Subcellularly, PDE1A localized mainly to the soma and proximal dendrites, and was essentially absent from the nucleus. In contrast, PDE1B levels significantly decreased after TBI, and PDE1C levels did not significantly change after TBI.
Examination of other PDEs within the brain revealed that many were unaltered with brain trauma. Levels of PDE3A, 8A, or 8B were unchanged between 30 minutes to one week post-injury [34]. Further studies are required to investigate whether these particular isoforms contribute to chronic recovery mechanisms in TBI.
In contrast, significant alterations were observed with many of the PDE4 isoforms after TBI [34]. Phosphorylation of PDE4A long isoforms at the PKA phosphorylation site were significantly increased after TBI, remaining high for at least one week. PDE4A mRNA is highly expressed in all cortical regions within the non-injured brain [35]. In both the injured and non-injured parietal cortex, phospho-PDE4A expression was predominantly restricted to neurons throughout all cortical layers. Furthermore, this expression was almost exclusively nuclear. The increased phosphorylation of PDE4A may be responsible, in part, for the decrease in cAMP levels after TBI since PKA phosphorylation of the long isoforms increases PDE4A activity [36]. Interestingly, total PDE4A5 and PDE4A8 (both long isoforms) protein levels decreased over this same time period.
Two isoforms in particular that exhibited dramatic, increased expression within the injured brain were PDE4B2 and PDE4D2 [36]. PDE4B2 levels significantly increased (~10-fold) within one hour after TBI and did not return to sham, non-injured levels until one week post-injury. This paralleled an increase in mRNA expression (~20-fold increase) for this short isoform. In both the injured, ipsilateral parietal cortex as well as the non-injured parietal cortex, PDE4B2 expression was predominantly localized to dendrites. However, PDE4B2 mRNA is most highly expressed in the non-injured hippocampus, although some expression is observed throughout the cortex [37]. Further experiments are needed to evaluate whether hippocampal changes in PDE4B2 are as pronounced as the cortical increases. In contrast to PDE4B2, the levels of the long isoforms PDE4B1, 3 and 4 did not significantly change over this time period.
The other short splice variant that significantly increased after TBI was PDE4D2 [34]. This increase paralleled the changes in PDE4B2, increasing within one hour after TBI, and remaining elevated for up to one week post-injury. Likewise, Pde4D2 mRNA levels increased by ~80-fold. Although PDE4D2 mRNA is found in the cerebral cortex, PDE4D2 protein expression is virtually undetectable in the contralateral and non-injured cortex [34, 38]. In the injured brain, PDE4D2 expression was found throughout the entire ipsilateral cortex, making it an excellent marker for the injured brain. Furthermore, PDE4D2 was absent from neurons and astrocytes. Expression of PDE4D3 and PDE4D4 (long variants) did not significantly change after TBI.
It is significant to note that the genes for both PDE4B2 and PDE4D2, the isoforms that showed the most pronounced changes after TBI, contain cAMP-response elements in their promoters, and increased intracellular cAMP upregulates expression of both isoforms [39–41]. However, with respect to regulation of these genes by cAMP, the activation of CREB during fluid-percussion brain injury is delayed relative to the upregulation seen for PDE4B2 and PDE4D2 [18]. Besides CRE elements, the PDE4B2 promoter also contains an NF-κB canonical site [13, 39]. These isoforms regulate pro-inflammatory cytokine expression within inflammatory cells [13, 40, 42, 43]. The initial increases observed with these isoforms may be due to regulation through the NF-κB pathway within inflammatory cells, whereas the sustained increases may be through the cAMP pathway.
Like PDE1A, PDE4B2, and PDE4D2, PDE10A protein levels significantly increased after TBI within the injured parietal cortex (~2-fold), albeit much more modestly than the others [34]. The increase in PDE10A is likely due to increased gene transcription, since Pde10A mRNA levels were significantly increased by ~10-fold. PDE10 is most strongly expressed within striatum, although it also found at reduced levels within the cerebral cortex [35, 44, 45]. Further work is needed to determine if striatal expression of PDE10A is also regulated by brain injury.
In summary, many but not all, PDE genes and protein expression were upregulated after brain trauma. Most notable include PDE1A, PDE4B2, PDE4D2, and PDE10A, as well as phosphorylation of PDE4A. However, whether these protein changes are the direct cause of the decrease in cAMP levels after TBI has yet to be directly tested. The consequences of these dramatic changes in protein expression are still unknown, although as described in section 6, several may be involved in the inflammatory response to TBI. In addition, whether any of these isoforms are regulated in the chronic phase of recovery after TBI is an area that deserves exploration.
4. INTERACTION OF AGE, TBI AND cAMP
Cognitive dysfunction is one of the most common deteriorating effects of aging in the elderly population. Aging in normal individuals diminishes a variety of cognitive functions, and in particular the consolidation of long-term memories [46]. The hippocampus, a subcortical structure critical for learning and memory, is often implicated in age-related memory deficits [47]. Evidence from human as well as animal studies indicates that aging leads to multiple changes in the hippocampus. These include synaptic dysfunction and deficits in gene expression during memory formation [46]. Together, these studies suggest that age-related cellular signaling changes in the hippocampus likely contribute to memory impairments in the elderly population.
One cell signaling pathway that exhibits several age-related changes is the cAMP-PKA-CREB cascade. Several studies have converged to conclude that basal CREB phosphorylation is lower in pyramidal cells of all subregions of the hippocampus of aged animals as compared to young adult animals [48–53]. The upstream mechanisms that result in this decrease in basal CREB phosphorylation are still being delineated. We and others have shown that basal cAMP levels are not altered in the hippocampi of aged animals [19, 54]. However, there are suggestions that this decrease in basal CREB phosphorylation may be due to increased phosphatase activity since studies have reported age-related increases in phosphatase levels [52, 55].
Additionally, activation of the cAMP-PKA-CREB cascade during learning is deficient in aged animals and this is correlated with age-related cognitive deficits. In a hippocampal-dependent learning paradigm, Monti et al., observed an increase in CREB phosphorylation in the hippocampus 24 hours following learning in young adult animals, but not in aged animals [56]. Although total levels of CREB were unaltered, stimulation of CREB phosphorylation and CREB-mediated gene transcription required for long-term memory formation was dramatically diminished in the aged hippocampus. This deficit in activation of CREB has been suggested to be due to decreases in basal AC expression or regulation [57–59]. Another possibility is dysregulation of PKA since PKA activity levels are depressed with age [60, 61]. Thus, these studies suggest that not only are there alterations in basal CREB phosphorylation levels, but the ability to stimulate CREB phosphorylation and transcription in the aged hippocampus is also deficient during memory formation [53].
Efforts to positively impact the cAMP-PKA-CREB signaling pathway have met with success to improve age-related cognitive deficits. Increasing cAMP levels either directly by cAMP analogs or indirectly by inhibition of PDEs reversed the aged-related memory and synaptic plasticity deficits in aged rodents and in a mouse model of Alzheimer’s disease [62–64]. Rolipram is a PDE4 inhibitor that increases cAMP levels both in vitro and in vivo [65]. Application of a low concentration of rolipram during tetanization converted transient long-term potentiation (LTP) into long-lasting late LTP in hippocampal slices from non-injured aged animals [63, 64, 66]. Administration of a low dose of rolipram to elevate cAMP levels without affecting basal levels improved contextual fear conditioning in aged mice and reversed the age-related deficits in spatial memory in the water maze [63, 64].
Age is one of the most significant prognostic indicators of TBI outcome. A mild TBI that has little or no effect on cognitive functioning in a child or young adult can result in prolonged cognitive disability in aged individuals [3]. There are a number of pathologies that are particularly exacerbated with age after TBI, including intracerebral hemorrhage, elevated glial and macrophage responses, brain tissue damage and disproportionate white matter loss [67, 68]. These pathological changes result in the highest rates of TBI-associated hospitalizations and death in the elderly [69]. TBI is also an epigenetic risk factor for Alzheimer’s and Parkinson’s diseases [70, 71]. Thus, TBI is a significant health problem in the elderly and likely to increase as our population ages.
The detrimental effects of aging on outcome after brain trauma have been recapitulated in animal models. One of the first studies investigated the relationship of calcium dynamics and age since calcium is an important initiating pathomechanism acutely after TBI. Hovda’s group found that TBI results in higher levels of calcium accumulation in the hippocampi of aged animals [72]. This could result in significant dysregulation of key calcium-dependent pathways involved in synaptic function and plasticity, in particular since this study also found that intracellular calcium levels returned to basal levels much more slowly in aged animals after TBI [72]. Later studies investigated other pathological features of TBI in aged animals and several have reported increased oxidative damage and upregulation of pro-inflammatory cytokines [73–78]. In contrast, expression of neuroprotective genes such as hypoxia inducible factor-1α was suppressed following TBI in aged mice [79]. Thus, TBI results in an imbalance between pro-inflammatory cytokines and the expression of neuroprotective genes in aged animals.
Aging results in not only increased inflammation and neuronal death, but also synaptic dysfunction after TBI. Interestingly, these previous studies did not detect any systemic complications due to age since key factors that could contribute to the secondary injury progression such as hypoxia, blood pressure, and weight loss were not altered with age [76, 80, 81]. These studies have also found that the worsened pathology with age corresponds to behavioral deficits. Both cognitive and motor function are more severely impaired in aged animals as compared to young adult animals after TBI [75, 77, 81, 82].
Given the well-known age-related changes in cAMP-mediated signaling in the aged brain, we examined changes in cAMP in aged animals as compared to young adult animals after both mild and moderate TBI [19]. We found that cAMP levels were altered in an injury severity-dependent manner (Fig. 2). Using the fluid-percussion brain injury model as a clinically relevant model, we found that mild TBI resulted in a significant decrease in hippocampal cAMP levels in aged animals, but not in young adult animals. In contrast, moderate TBI significantly lowered cAMP levels in both young adult and aged animals. Using the hippocampal slice preparation, we also investigated the electrophysiological consequences of these differential responses to mild and moderate TBI. At two weeks post-injury, the effects of mild and moderate TBI on basal synaptic transmission and paired-pulse facilitation in area CA1 region of the hippocampus were assessed. Both mild and moderate TBI resulted in hypoexcitability of the Schaffer collateral-evoked field excitatory postsynaptic potential for both age groups as measured by the input-output curve. This depression in basal synaptic transmission was not rescued with rolipram treatment with either age group. Paired-pulse facilitation, a measure of short-term pre-synaptic plasticity, was depressed after moderate TBI, but not mild TBI, in both young adult and aged hippocampal slices. Inhibition of PDE4 with rolipram could not restore this form of short-term plasticity as well. These results indicate that there are similar basal synaptic transmission deficits in both young adult and aged animals after TBI that are therapeutically inaccessible with PDE4 inhibitors.
Fig. 2.

Levels of cAMP are altered in an injury-severity dependent manner. A) Hippocampal cAMP levels were significantly decreased in aged animals, but not in young adult animals following mild TBI. B) Moderate TBI significantly lowers hippocampal cAMP levels in both young adult and aged animals. Adapted from [17].
Hippocampal LTP, a physiological model for learning and memory that requires activation of the cAMP-PKA-CREB pathway, was also investigated [83]. For both age groups, there was a decrease in the ability to maintain the synaptic potentiation, suggestive of a signaling deficit in the expression phase of LTP [19]. Thus, we evaluated whether rolipram could rescue this deficit (Fig. 3). Rolipram rescued LTP in both young adult and aged animals after mild TBI, but rescued LTP only in young adult but not aged animals after moderate TBI [19]. These results suggest that there are differential injury severity consequences of TBI on young adult versus aged animals. Although PDE4 inhibitors may be effective after mild TBI, targeting other pathomechanisms is also required for moderate TBI in the aged population.
Fig. 3.

Differential effects of the PDE4 inhibitor rolipram in rescuing LTP deficits after mild versus moderate TBI. Rolipram improved LTP expression in both young adult and aged animals after mild TBI (A), but rescued LTP only in young adult animals after moderate TBI (B). V, vehicle; R, rolipram. Adapted from [17].
5. ROLE OF cGMP-PDES AND TBI
While the roles played by cAMP-PDEs during brain trauma are by no means completely clear, the roles of cGMP-PDEs are even more poorly defined. However, a growing body of work suggests that inhibition of cGMP-PDEs may be a useful therapeutic intervention after brain injury, albeit with certain caveats.
Very few studies have investigated changes in cGMP and cGMP-specific PDEs after TBI. Povlishock’s group has reported that basal cGMP levels are unaltered after lateral fluid-percussion brain injury [84]. However, there were differential effects on cGMP levels with NMDA stimulation. NMDA induced a decrease in cGMP in the non-injured, contralateral hippocampus, but interestingly, resulted in an increase in cGMP levels in the injured, ipsilateral hippocampus. This effect was only observed in the acute post-injury time period, 60 minutes post-TBI, and stimulated cGMP levels normalized by two weeks post-injury. We have observed analogous transient changes in cGMP-PDEs after fluid-percussion brain injury. PDE1 is a calcium/calmodulin-dependent PDE that hydrolyzes primarily cGMP, although PDE1C also hydrolyzes both cAMP and cGMP. Levels of PDE1A significantly increased within 30 minutes post-TBI within neurons and returned to baseline by one week [34]. Although further work is required in this area, several TBI studies using non-specific PDE1 inhibitors such as caffeine and amantadine have reported improvements in cognitive outcome and reduced inflammation [85, 86].
Another potentially promising therapeutic target is PDE10A. PDE10A is a cGMP-PDE localized predominantly within the striatum [35, 45]. In fluid-percussion brain injury, we observed a significant upregulation of PDE10A in the injured parietal cortex in the acute recovery period between six hours to one day post-injury [34]. Gait deficits are an important deleterious consequence of brain trauma and further work is needed to determine if PDE10A inhibitors, which are currently used for Huntington’s disease, may be a viable therapeutic avenue for TBI [87–89].
Much of the work regarding the cGMP-PDEs in CNS injury has focused on PDE5, the enzyme inhibited by sildenafil (Viagra) and tadalafil (Cialis). Work from Garcia’s group has shown that the non-selective cGMP-PDE inhibitor, zaprinast, reduced neuronal death after cortical cryoinjury and these beneficial effects were essentially mimicked by the PDE5 selective inhibitor, sildenafil [90, 91]. These effects appeared to be multimodal, with enhanced astrogliosis and angiogenesis in the lesioned area and suppression of microglia infiltration. In glia cells around the lesion site, sildenafil significantly upregulated the expression of neuroprotective metal-binding cysteine-rich proteins, metallothioneins I/II [91]. After cerebral ischemia, tadalafil administration was found to facilitate recovery and improve short-term memory deficits by increasing cGMP levels and reducing neuronal apoptosis [92]. In a middle-aged mouse ischemic brain model, sildenafil significantly increased the number of neural stem cells, as well as their neuronal and oligodendrocyte progeny [93]. Furthermore, after experimental subarachnoid hemorrhage (SAH), PDE5 activity, but not expression, increased with a concomitant decrease in cGMP levels [94]. Post-SAH administration of sildenafil significantly decreased PDE5 activity, restored cGMP levels, decreased cerebral vasospasm and neuronal cell death, and improved functional recovery with minimal physiological side effects [94].
However, results from other studies indicate that therapies that target cGMP-PDEs must also take into account how intracellular cGMP will be increased. A number of reports suggest that nitric oxide (NO) activation of the protein kinase G (PKG) pathway may be very detrimental to recovery after brain injury. NO has synaptotoxic actions and NO induction of the GC-PKG pathway has been shown to result in synapse elimination in the CNS, leading to the proposal that it may possibly underlie cognitive deficits in several neurological disorders [95, 96]. For example, the selective serotonin reuptake inhibitor sertraline protects against transient global ischemia-induced behavioral despair, and these beneficial effects are reversed by NO upregulation [97]. Hypothermia therapy also inhibits NO production during cerebral ischemia and upregulation of inducible nitric oxide synthase (iNOS), which might account, at least in part, for the neuroprotective effects of this treatment [98, 99]. In short, it appears that NO activation of the GC-PKG pathway may be deleterious, particularly when it leads to induction of iNOS, and some or most of these deleterious effects may be due to NO itself or its peroxide byproducts driven by iNOS. In support of this hypothesis, the NO donor, ZJM-289, was found to alleviate cerebral ischemic-reperfusion injury through inhibition of neuronal NOS and stimulation of the soluble GC-cGMP pathway [100]. The combination therapy of NO donors and PDE5 inhibitors appears to facilitate neurogenesis, oligodendrogenesis, and angiogenesis after stroke [101]. However, given the complex roles of both iNOS and neuronal NOS in the recovery and pathomechanisms of TBI, this therapeutic route is a challenge for TBI [102, 103].
Guanylyl cyclase activation, and thus cGMP upregulation, can be accomplished by messengers other than NO. One such well-studied activator is atrial natriuretic peptide (ANP), which has been shown to be protective after cerebral ischemia or hemorrhage [104]. One study has shown that stimulation of the cGMP-PKG pathway by ANP, but not by NO, induced morphological changes in microglia through cytoskeleton reorganization [105]. Furthermore, this upregulation of cGMP by ANP did not lead to upregulation of iNOS or tumor necrosis factor-α (TNF-α) expression. In an oxide-injury model, NO and peroxynitrite formation was highly damaging to neurons; however, the PDE inhibitor, propentofylline, could almost entirely block this damage, suggesting PDE inhibitors as therapeutically useful in limiting oxidative damage in neurons [106]. While propentofylline is not selective for cAMP- versus cGMP-PDEs, its beneficial effects may be in this non-selectivity. Indeed, axon regeneration of spinal cord neurons was shown to be improved by a combination of cGMP and cAMP upregulation [107].
It is worth noting that in one study using controlled cortical impact, the continuous administration of a cGMP analogue, 8-bromo-cGMP, significantly worsened injury [108]. In that study, these treatments exacerbated brain edema and worsened blood-brain barrier disruption in the injured hemisphere at 24 hours after injury. However, it is still possible that NO activation may underlie, at least in part, these detrimental effects, as well as activation of the “wrong” cGMP pathway. GC can be divided into two main groups: the membrane-bound or particulate GC, and the cytosolic GC. In the spinal cord, natriuretic peptides activate the particulate GC pool, leading to cGMP accumulation in GABAergic structures [109]. It is thus possible that activation of the particulate GC pool, or isoforms not involved in the NO pathway, may underlie the therapeutic effects.
Relevant to the above is that the dual-specific PDE, PDE3, is strongly expressed in endothelial cells and the PDE3-selective inhibitor, cilostazol, appears to strengthens barrier integrity in brain endothelial cells [110, 111]. Furthermore, in ischemia-reperfusion injury, PDE3B levels increase in neurons and astrocytes, with activation in the latter being stronger [110].
Finally, it is noteworthy that the promoter for PDE5A is responsive to both cAMP and cGMP [112, 113]. Thus, it is possible that treatments that selectively increase cAMP levels may not be useful in all brain injury models, since they could effectively decrease cGMP levels as an unintended consequence. This may explain some of the reported discrepancies in the use of PDE4 inhibitors in brain injury [114, 115].
In summary, there is sufficient data to suggest that increasing intracellular cGMP levels may be therapeutically beneficial after TBI. However, this should be done without increasing NO levels, since depending on the recovery period, this could be deleterious, in particular for the acute time period characterized by lipid oxidation and blood-brain barrier breakdown. Furthermore, there is also evidence for the beneficial effects of specifically targeting PDE5, and that a combination therapy that specifically targets both cGMP- and cAMP-PDEs may be optimal.
6. PDE4 INHIBITORS AS ANTI-INFLAMMTORY AGENTS FOR TBI
Inflammation is an important cause of progressive secondary injury in brain injury [116]. This pathomechanism is a highly dynamic, intricate process regulated by a diverse array of molecules, including cAMP. The ubiquitous nature of cAMP has been exploited to treat a variety of inflammatory diseases, through its effectors PKA and the transcription factor CREB. Elevating cAMP signaling by PDE4 inhibition has been shown to decrease inflammation through attenuation of pro-inflammatory cytokine production in a variety of CNS and systemic inflammatory conditions [117].
After trauma, blood-brain barrier dysfunction is acutely observed as well as activation of inflammatory cells including microglia, invading neutrophils, and macrophages/monocytes [116]. Activation and recruitment of inflammatory cells into the injured brain generates pro-inflammatory cytokines, free radicals and other damaging molecules [116]. Clinical studies have corroborated the increases in pro-inflammatory cytokines and chemokines using biomarkers [118, 119]. Two potent pro-inflammatory cytokines induced by TBI are TNF-α and interleukin-1β (IL-1β) [120–124]. These cytokines are highly cytotoxic, potentiate the inflammatory process in TBI, and are regulated by cAMP signaling.
The benefits of PDE4 inhibition in reducing inflammation and improving histopathology have been well-studied in rodent models of ischemia, multiple sclerosis, stroke, and TBI. After ischemia, PDE4 inhibitors have been found to improve neuronal survival, reduce infarct size, and attenuate inflammation and blood-brain barrier breakdown [125–129]. In a model of multiple sclerosis, experimental autoimmune encephalomyelitis, rolipram and other drug therapies to increase cAMP levels prevents the progression of neurodegeneration and demyelination [130–132]. Together, these studies indicate that targeting PDE4 is effective in reducing neuroinflammation in a variety of disorders.
Contrary to the neuroprotective benefits observed in these models, general PDE4 inhibition has had mixed results for TBI. We initially reported that pre-treatment with rolipram 30 minutes prior to fluid-percussion brain injury reduced inflammation, cortical contusion pathology, and improved neuronal survival [18]. Both TNF-α and IL-1β upregulation were attenuated by rolipram treatment, when given either prior to injury or within 30 minutes post-injury [18, 115]. These results suggested that, like in other CNS injury models, PDE4 inhibitors may be successful at reducing neuroinflammatory signaling in TBI.
However, when rolipram or other structurally distinct PDE4 inhibitors were given in the post-injury recovery period for TBI, cortical contusion volume increased, blood-brain barrier breakdown was aggravated, and hemorrhage in the injured cortex was observed [115]. This was not model-dependent, since this result was observed in two distinct brain injury models, fluid-percussion brain injury and controlled cortical impact [114, 115].
The finding that PDE4 inhibitors increased bleeding in the injured cortex remains unexplained as this has not been reported in other CNS injury models such as ischemia or spinal cord injury [133, 134]. The PDE4D subfamily has been identified in endothelial cells and vascular smooth muscle and may be involved in conferring the vasodilative properties of rolipram [135–139]. In accordance with this, we tested a PDE4 inhibitor that was more selective towards PDE4D as compared to PDE4A or PDE4B and found that this particular inhibitor recapitulated the worsening of pathology seen with the general PDE4 inhibitor rolipram [115]. This raises the possibility that more selective targeting of particular PDE4 isoforms may be a more successful approach in reducing neuroinflammation and improving recovery in the acute phase of TBI.
Given the numerous studies reporting that PDE4 inhibitors reduce expression and secretion of TNF-α and IL-1β from activated microglia, further work is required to determine if more isoform-selective PDE4 inhibitors have the capability of reducing neuroinflammation after TBI [39, 140–145]. In particular, the PDE4B subfamily has been well-studied in systemic inflammation models. It is well-established in other injury and inflammation models that PDE4B, but not PDE4A or PDE4D, induces expression of the pro-inflammatory cytokine TNF-α in circulating monocytes and macrophages [13, 14, 146, 147]. PDE4B2 expression is induced in several inflammatory cell populations, and in particular neutrophils, macrophages, and T-cells [13, 42, 147, 148]. Studies using PDE4B knockout mice have highlighted the role of PDE4B in pro-inflammatory cytokine production. TNF-α production by macrophages in response to an inflammatory stimulus is impaired in macrophages obtained from PDE4B gene-deleted mice [13, 14]. Knockout studies of PDE4B have also discovered that PDE4B is necessary for neutrophil chemotaxis [42]. Recently, it has been reported that the PDE4B subfamily is elevated in activated microglia and macrophages after TBI [149]. Neutrophils are usually the first to infiltrate after TBI; they arrive within hours and peak around 24 hours post-TBI and contribute to edema and apoptotic neuronal death [150, 151]. The temporal dynamics of macrophage, monocyte and lymphocyte infiltration is more variable; the macrophage/monocyte population typically peaks around one to two days and lymphocytes can be identified as soon as two days after injury [116]. The infiltration and activation of these inflammatory cells also contribute to the pathology of TBI and pharmacological targeting of PDE4B in specific inflammatory cell populations may provide a means for reducing inflammation without vascular perturbations and would be greatly advantageous over current inhibitors.
7. PDE4 INHIBITORS AS COGNITIVE ENHANCERS FOR TBI
As discussed in the previous section, the use of PDE inhibitors in the acute phases of TBI requires careful consideration of the isoform targeted and the relevant cell population. In chronic stages of TBI recovery, the pathomechanisms shift to synaptic and axonal dysfunction, circuitry reorganization, depressed neurogenesis, and ongoing neurodegeneration and atrophy. The molecular pathways that underlie these chronic pathomechanisms remain to be elucidated and very little is known how the cAMP-PKA-CREB signaling pathway is regulated chronically after TBI.
To begin to investigate how this pathway is altered chronically after TBI, we first investigated basal changes in these molecules after TBI. Unlike in the acute stages of TBI recovery where CREB phosphorylation undergoes significant upregulation, we found that phospho-CREB levels were significantly reduced at three months post-TBI in the fluid-percussion brain injury model [152]. In addition, in hippocampal slices pharmacologically stimulated with either glutamate or high potassium depolarization, we found that stimulation of phospho-CREB was significantly impaired at three months post-TBI [152]. These results suggest that increasing cAMP levels to boost CREB phosphorylation during learning may improve hippocampal synaptic function.
The effect of PDE4 inhibitors has been extensively investigated in other brain injury models besides TBI. PDE4 inhibition with rolipram ameliorates learning and memory deficits after cerebral ischemia [33, 126, 129]. In addition, administration of rolipram 24 hours before hypoxia protects from permanent neurological dysfunction induced by perinatal hypoxic-ischemic brain injury [153]. Together, and results from other models such as Alzheimer’s disease and psychosis, supports the premise that PDE4 inhibition is a promising therapeutic target for cognitive enhancement [62, 154].
In the context of chronic recovery after brain trauma, TBI-induced cognitive impairments are consistently found in TBI patients from months to years after the initial injury [155]. These deficits have been linked to several memory modalities; however, declarative memory deficits are prevalent among TBI survivors [156]. The unique susceptibility of hippocampus to brain injury, even when not directly damaged results in declarative memory deficits [157]. Rehabilitative strategies, such as neurotransmitter receptor agonists, neurotransmitter reuptake inhibitors or drugs that enhance neurotransmitter release, have been used with limited success in restoring cognitive function after brain injury [1]. To test whether the PDE4 inhibitor rolipram could improve cognitive function after TBI, we administered rolipram to animals at two weeks after fluid-percussion brain injury and tested them on a variety of hippocampal-dependent learning tasks. We found that rolipram improved both cue and contextual fear conditioning, water maze performance, and spatial working memory [158]. These improvements resulted in consolidation of the memories because when we tested animals one month after fear conditioning, TBI animals treated with rolipram still exhibited memory retention. Progressive atrophy of the hippocampus induced by TBI was unaffected by rolipram treatment in this time period, suggesting that rolipram worked to improve cognitive deficits by acting on synaptic signaling pathways rather than cellular loss. To directly test this hypothesis, we analyzed changes in phospho-CREB levels after cue and contextual fear conditioning. In non-injured animals, phosphorylated CREB levels significantly increased within the hippocampus after fear conditioning. However, this increase was not observed in TBI animals. When 1TBI animals were treated with rolipram prior to fear conditioning, the learning-induced increase in CREB phosphorylation was restored, indicating that rolipram boosted cAMP signaling [158]. These results suggest that enhancement of cAMP levels during learning is a viable therapeutic route for the treatment of chronic cognitive impairments for TBI survivors. Whether PDE4 inhibitors can improve other aspects of pathomechanisms such as inflammation, atrophy, and axonal dysfunction in the chronic stages of TBI recovery are important questions remaining to be addressed.
8. CONCLUSIONS AND FUTURE DIRECTIONS
The utility of PDE4 inhibitors to reduce neuroinflammation in the acute recovery stage of TBI remains an important area of research. PDE4B, in particular, is a well-validated target for reducing TNF-α production, an important pathomechanism of TBI [116]. However, it remains to be established whether PDE4B is the critical isoform to target to reduce TNF-α levels and inflammation after TBI. Understanding the critical PDE4 isoforms to target and whether other PDEs such as PDE5 should be considered as TBI therapeutics are areas of ongoing research in the TBI field. In addition, further studies that identify the cell types that upregulate expression of PDE isoforms is important in developing rationally-derived therapies. PDE4 inhibitors are in clinical development for age-related cognitive decline and approved for chronic obstructive pulmonary disorder [159, 160]. Understanding how PDE4 inhibitors could potentially contribute to vascular injury in brain trauma is important, especially in the context of brain injury incidence in the elderly [161].
Developing a PDE4 inhibitor in the chronic recovery stage of TBI appears to be highly promising. However, rolipram results in side effects of nausea and emesis in humans, hampering its development as a cognitive enhancer [162]. In the past few years, it has become clear that PDE4D mediates the emetic effects of rolipram, although PDE4B may contribute as well [163, 164]. Thus, the development of isoform-selective inhibitors that obviate this side-effect but retain the ability for cognitive enhancement is a promising strategy. Although few isoform-selective inhibitors exist to date, structural analyses of PDE4 is leading to the development of more selective PDE4B and PDE4D inhibitors that may begin to address the challenges of potential side effects [165].
TBI is a complicated problem, likely requiring more than just one targeted solution. Targeting PDEs and in particular, PDE4 isoforms, may develop into one aspect of the arsenal against the pathomechanisms of TBI. However, the failure of numerous clinical trials to date in the treatment of TBI suggests that the heterogeneous nature of TBI requires rigorous preclinical studies that address the complexity of TBI pathomechanisms [166]. As our understanding improves of how cyclic nucleotides and PDEs change after TBI and contribute to the pathomechanisms as well as endogenous recovery mechanisms of TBI, so will our understanding of how to harness their power to improve recovery after TBI.
Acknowledgments
This work was supported by a research grant from NIH/NINDS NS069721 to C.M. Atkins.
ABBREVIATIONS
- AC
Adenylyl cyclase
- ANP
Atrial natriuretic peptide
- cAMP
3′,5′-Cyclic adenosine monophosphate
- cGMP
3′,5′-Cyclic guanosine monophosphate
- CNS
Central nervous system
- CREB
Cyclic AMP-responsive-element-binding protein
- ERK
Extracellular signal-regulated kinase
- GC
Guanylyl cyclase
- IL-1β
Interleukin-1 beta
- iNOS
Inducible nitric oxide synthase
- LTP
Long-term potentiation
- NO
Nitric oxide
- PDE
Phosphodiesterase
- PKA
Protein kinase A
- PKG
Protein kinase G
- SAH
Subarachnoid hemorrhage
- TBI
Traumatic brain injury
- TNF-α
Tumor necrosis factor-alpha
- UCR
Upstream conserved region
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors of this paper declare no conflict of interest pertaining to this manuscript.
References
- 1.Wheaton P, Mathias JL, Vink R. Impact of pharmacological treatments on cognitive and behavioral outcome in the postacute stages of adult traumatic brain injury: a meta-analysis. J Clin Psychopharmacol. 2011;31:745–57. doi: 10.1097/JCP.0b013e318235f4ac. [DOI] [PubMed] [Google Scholar]
- 2.Lu J, Gary KW, Neimeier JP, Ward J, Lapane KL. Randomized controlled trials in adult traumatic brain injury. Brain Inj. 2012;26:1523–48. doi: 10.3109/02699052.2012.722257. [DOI] [PubMed] [Google Scholar]
- 3.Himanen L, Portin R, Isoniemi H, Helenius H, Kurki T, Tenovuo O. Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology. 2006;66:187–92. doi: 10.1212/01.wnl.0000194264.60150.d3. [DOI] [PubMed] [Google Scholar]
- 4.Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–40. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 5.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 6.Xu Y, Zhang HT, O’Donnell JM. Phosphodiesterases in the central nervous system: implications in mood and cognitive disorders. Handb Exp Pharmacol. 2011:447–85. doi: 10.1007/978-3-642-17969-3_19. [DOI] [PubMed] [Google Scholar]
- 7.Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10:1503–19. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 8.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Zhang HT. Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr Pharm Des. 2009;15:1688–98. doi: 10.2174/138161209788168092. [DOI] [PubMed] [Google Scholar]
- 10.Sette C, Conti M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem. 1996;271:16526–34. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- 11.Hill EV, Sheppard CL, Cheung YF, Gall I, Krause E, Houslay MD. Oxidative stress employs phosphatidyl inositol 3-kinase and ERK signalling pathways to activate cAMP phosphodiesterase-4D3 (PDE4D3) through multi-site phosphorylation at Ser239 and Ser579. Cell Signal. 2006;18:2056–69. doi: 10.1016/j.cellsig.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 12.Jin SL, Ding SL, Lin SC. Phosphodiesterase 4 and its inhibitors in inflammatory diseases. Chang Gung Med J. 2012;35:197–210. doi: 10.4103/2319-4170.106152. [DOI] [PubMed] [Google Scholar]
- 13.Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-a responses. Proc Natl Acad Sci U S A. 2002;99:7628–33. doi: 10.1073/pnas.122041599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin SL, Lan L, Zoudilova M, Conti M. Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol. 2005;175:1523–31. doi: 10.4049/jimmunol.175.3.1523. [DOI] [PubMed] [Google Scholar]
- 15.Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L, et al. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B) Neuropsychopharm. 2008;33:1611–23. doi: 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li YF, Cheng YF, Huang Y, Conti M, Wilson SP, O’Donnell JM, et al. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J Neurosci. 2011;31:172–83. doi: 10.1523/JNEUROSCI.5236-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, et al. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharm. 2002;27:587–95. doi: 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 18.Atkins CM, Oliva AA, Jr, Alonso OF, Pearse DD, Bramlett HM, Dietrich WD. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol. 2007;208:145–58. doi: 10.1016/j.expneurol.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titus DJ, Furones C, Kang Y, Atkins CM. Age-dependent alterations in cAMP signaling contribute to synaptic plasticity deficits following traumatic brain injury. Neuroscience. 2013;231:182–94. doi: 10.1016/j.neuroscience.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell MJ, Kochanek PM, Carcillo JA, Mi Z, Schiding JK, Wisniewski SR, et al. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat. J Neurotrauma. 1998;15:163–70. doi: 10.1089/neu.1998.15.163. [DOI] [PubMed] [Google Scholar]
- 21.Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem Rev. 2001;101:2243–70. doi: 10.1021/cr000226k. [DOI] [PubMed] [Google Scholar]
- 22.Atkins CM. Decoding hippocampal signaling deficits after traumatic brain injury. Trans Stroke Res. 2011;2:546–55. doi: 10.1007/s12975-011-0123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atkins CM, Oliva AA, Jr, Alonso OF, Chen S, Bramlett HM, Hu BR, et al. Hypothermia treatment potentiates ERK1/2 activation after traumatic brain injury. Eur J Neurosci. 2007;26:810–9. doi: 10.1111/j.1460-9568.2007.05720.x. [DOI] [PubMed] [Google Scholar]
- 24.Dash PK, Moore AN, Dixon CE. Spatial memory deficits, increased phosphorylation of the transcription factor CREB, and induction of the AP-1 complex following experimental brain injury. J Neurosci. 1995;15:2030–9. doi: 10.1523/JNEUROSCI.15-03-02030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Folkerts MM, Parks EA, Dedman JR, Kaetzel MA, Lyeth BG, Berman RF. Phosphorylation of calcium calmodulin-dependent protein kinase II following lateral fluid percussion brain injury in rats. J Neurotrauma. 2007;24:638–50. doi: 10.1089/neu.2006.0188. [DOI] [PubMed] [Google Scholar]
- 26.Hu B, Liu C, Bramlett H, Sick TJ, Alonso OF, Chen S, et al. Changes in trkB-ERK1/2-CREB/Elk-1 pathways in hippocampal mossy fiber organization after traumatic brain injury. J Cereb Blood Flow Metab. 2004;24:934–43. doi: 10.1097/01.WCB.0000125888.56462.A1. [DOI] [PubMed] [Google Scholar]
- 27.Otani N, Nawashiro H, Fukui S, Nomura N, Yano A, Miyazawa T, et al. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22:327–34. doi: 10.1097/00004647-200203000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Yang K, Taft WC, Dixon CE, Todaro CA, Yu RK, Hayes RL. Alterations of protein kinase C in rat hippocampus following traumatic brain injury. J Neurotrauma. 1993;10:287–95. doi: 10.1089/neu.1993.10.287. [DOI] [PubMed] [Google Scholar]
- 29.Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Voluntary exercise following traumatic brain injury: brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience. 2004;125:129–39. doi: 10.1016/j.neuroscience.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 30.Shaywitz AJ, Greenberg ME. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 31.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–14. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka K, Nogawa S, Nagata E, Ito D, Suzuki S, Dembo T, et al. Persistent CREB phosphorylation with protection of hippocampal CA1 pyramidal neurons following temporary occlusion of the middle cerebral artery in the rat. Exp Neurol. 2000;161:462–71. doi: 10.1006/exnr.1999.7313. [DOI] [PubMed] [Google Scholar]
- 33.Nagakura A, Niimura M, Takeo S. Effects of a phosphodiesterase IV inhibitor rolipram on microsphere embolism-induced defects in memory function and cerebral cyclic AMP signal transduction system in rats. Br J Pharmacol. 2002;135:1783–93. doi: 10.1038/sj.bjp.0704629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliva AA, Jr, Kang Y, Furones C, Alonso OF, Bruno O, Dietrich WD, et al. Phosphodiesterase isoform-specific expression induced by traumatic brain injury. J Neurochem. 2012;123:1019–29. doi: 10.1111/jnc.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharm. 2010;59:367–74. doi: 10.1016/j.neuropharm.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 36.MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, McSorley T, et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1) Br J Pharmacol. 2002;136:421–33. doi: 10.1038/sj.bjp.0704743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reyes-Irisarri E, Perez-Torres S, Miro X, Martinez E, Puigdomenech P, Palacios JM, et al. Differential distribution of PDE4B splice variant mRNAs in rat brain and the effects of systemic administration of LPS in their expression. Synapse. 2008;62:74–9. doi: 10.1002/syn.20459. [DOI] [PubMed] [Google Scholar]
- 38.Richter W, Jin SL, Conti M. Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem J. 2005;388:803–11. doi: 10.1042/BJ20050030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verghese MW, McConnell RT, Strickland AB, Gooding RC, Stimpson SA, Yarnall DP, et al. Differential regulation of human monocyte-derived TNFa and IL-1b by type IV cAMP-phosphodiesterase (cAMP-PDE) inhibitors. J Pharmacol Exp Ther. 1995;272:1313–20. [PubMed] [Google Scholar]
- 40.Manning CD, Burman M, Christensen SB, Cieslinski LB, Essayan DM, Grous M, et al. Suppression of human inflammatory cell function by subtype-selective PDE4 inhibitors correlates with inhibition of PDE4A and PDE4B. Br J Pharmacol. 1999;128:1393–8. doi: 10.1038/sj.bjp.0702911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.D’Sa C, Tolbert LM, Conti M, Duman RS. Regulation of cAMP-specific phosphodiesterases type 4B and 4D (PDE4) splice variants by cAMP signaling in primary cortical neurons. J Neurochem. 2002;81:745–57. doi: 10.1046/j.1471-4159.2002.00878.x. [DOI] [PubMed] [Google Scholar]
- 42.Ariga M, Neitzert B, Nakae S, Mottin G, Bertrand C, Pruniaux MP, et al. Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol. 2004;173:7531–8. doi: 10.4049/jimmunol.173.12.7531. [DOI] [PubMed] [Google Scholar]
- 43.Reyes-Irisarri E, Sanchez AJ, Garcia-Merino JA, Mengod G. Selective induction of cAMP phosphodiesterase PDE4B2 expression in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2007;66:923–31. doi: 10.1097/nen.0b013e3181567c31. [DOI] [PubMed] [Google Scholar]
- 44.Coskran TM, Morton D, Menniti FS, Adamowicz WO, Kleiman RJ, Ryan AM, et al. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem. 2006;54:1205–13. doi: 10.1369/jhc.6A6930.2006. [DOI] [PubMed] [Google Scholar]
- 45.Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113–26. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- 46.Samson RD, Barnes CA. Impact of aging brain circuits on cognition. Eur J Neurosci. 2013;37:1903–15. doi: 10.1111/ejn.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geinisman Y, Detoledo-Morrell L, Morrell F, Heller RE. Hippocampal markers of age-related memory dysfunction: behavioral, electrophysiological and morphological perspectives. Prog Neurobiol. 1995;45:223–52. doi: 10.1016/0301-0082(94)00047-l. [DOI] [PubMed] [Google Scholar]
- 48.Williams CM, El Mohsen MA, Vauzour D, Rendeiro C, Butler LT, Ellis JA, et al. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radic Biol Med. 2008;45:295–305. doi: 10.1016/j.freeradbiomed.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 49.Countryman RA, Gold PE. Rapid forgetting of social transmission of food preferences in aged rats: relationship to hippocampal CREB activation. Learn Mem. 2007;14:350–8. doi: 10.1101/lm.524907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brightwell JJ, Gallagher M, Colombo PJ. Hippocampal CREB1 but not CREB2 is decreased in aged rats with spatial memory impairments. Neurobiol Learn Mem. 2004;81:19–26. doi: 10.1016/j.nlm.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 51.Porte Y, Buhot MC, Mons N. Alteration of CREB phosphorylation and spatial memory deficits in aged 129T2/Sv mice. Neurobiol Aging. 2008;29:1533–46. doi: 10.1016/j.neurobiolaging.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 52.Hsu KS, Huang CC, Liang YC, Wu HM, Chen YL, Lo SW, et al. Alterations in the balance of protein kinase and phosphatase activities and age-related impairments of synaptic transmission and long-term potentiation. Hippocampus. 2002;12:787–802. doi: 10.1002/hipo.10032. [DOI] [PubMed] [Google Scholar]
- 53.Hattiangady B, Rao MS, Shetty GA, Shetty AK. Brain-derived neurotrophic factor, phosphorylated cyclic AMP response element binding protein and neuropeptide Y decline as early as middle age in the dentate gyrus and CA1 and CA3 subfields of the hippocampus. Exp Neurol. 2005;195:353–71. doi: 10.1016/j.expneurol.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 54.Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1. 2 during aging. Proc Natl Acad Sci U S A. 2003;100:16018–23. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci. 2001;21:4066–73. doi: 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monti B, Berteotti C, Contestabile A. Dysregulation of memory-related proteins in the hippocampus of aged rats and their relation with cognitive impairment. Hippocampus. 2005;15:1041–9. doi: 10.1002/hipo.20099. [DOI] [PubMed] [Google Scholar]
- 57.Mons N, Segu L, Nogues X, Buhot MC. Effects of age and spatial learning on adenylyl cyclase mRNA expression in the mouse hippocampus. Neurobiol Aging. 2004;25:1095–106. doi: 10.1016/j.neurobiolaging.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 58.Reis GF, Lee MB, Huang AS, Parfitt KD. Adenylate cyclase-mediated forms of neuronal plasticity in hippocampal area CA1 are reduced with aging. J Neurophysiol. 2005;93:3381–9. doi: 10.1152/jn.00827.2003. [DOI] [PubMed] [Google Scholar]
- 59.Amenta F, Cavallotti C, de Michele M, Ricci A, Vega JA. Changes of dopamine-sensitive cyclic AMP-generating system in the rat hippocampus as a function of age. Arch Gerontol Geriatr. 1990;10:279–85. doi: 10.1016/0167-4943(90)90029-6. [DOI] [PubMed] [Google Scholar]
- 60.Karege F, Lambercy C, Schwald M, Steimer T, Cisse M. Differential changes of cAMP-dependent protein kinase activity and 3H-cAMP binding sites in rat hippocampus during maturation and aging. Neurosci Lett. 2001;315:89–92. doi: 10.1016/s0304-3940(01)02358-8. [DOI] [PubMed] [Google Scholar]
- 61.Karege F, Schwald M, Lambercy C, Murama JJ, Cisse M, Malafosse A. A non-radioactive assay for the cAMP-dependent protein kinase activity in rat brain homogenates and age-related changes in hippocampus and cortex. Brain Res. 2001;903:86–93. doi: 10.1016/s0006-8993(01)02409-x. [DOI] [PubMed] [Google Scholar]
- 62.Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–34. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci U S A. 1998;95:15020–5. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, et al. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci U S A. 1999;96:5280–5. doi: 10.1073/pnas.96.9.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giorgi M, Modica A, Pompili A, Pacitti C, Gasbarri A. The induction of cyclic nucleotide phosphodiesterase 4 gene (PDE4D) impairs memory in a water maze task. Behav Brain Res. 2004;154:99–106. doi: 10.1016/j.bbr.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 66.Navakkode S, Sajikumar S, Frey JU. The type IV-specific phosphodiesterase inhibitor rolipram and its effect on hippocampal long-term potentiation and synaptic tagging. J Neurosci. 2004;24:7740–4. doi: 10.1523/JNEUROSCI.1796-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bigler ED, Anderson CV, Blatter DD. Temporal lobe morphology in normal aging and traumatic brain injury. Am J Neuroradiol. 2002;23:255–66. [PMC free article] [PubMed] [Google Scholar]
- 68.Lee JC, Cho GS, Choi BO, Kim HC, Kim WK. Aging exacerbates intracerebral hemorrhage-induced brain injury. J Neurotrauma. 2009;26:1567–76. doi: 10.1089/neu.2008.0630. [DOI] [PubMed] [Google Scholar]
- 69.Susman M, DiRusso SM, Sullivan T, Risucci D, Nealon P, Cuff S, et al. Traumatic brain injury in the elderly: increased mortality and worse functional outcome at discharge despite lower injury severity. J Trauma. 2002;53:219–23. doi: 10.1097/00005373-200208000-00004. [DOI] [PubMed] [Google Scholar]
- 70.Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci Biobehav Rev. 2012;36:1376–81. doi: 10.1016/j.neubiorev.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 71.Stern MB. Head trauma as a risk factor for Parkinson’s disease. Mov Disord. 1991;6:95–7. doi: 10.1002/mds.870060202. [DOI] [PubMed] [Google Scholar]
- 72.Osteen CL, Moore AH, Prins ML, Hovda DA. Age-dependency of 45calcium accumulation following lateral fluid percussion: acute and delayed patterns. J Neurotrauma. 2001;18:141–62. doi: 10.1089/08977150150502587. [DOI] [PubMed] [Google Scholar]
- 73.Sandhir R, Puri V, Klein RM, Berman NE. Differential expression of cytokines and chemokines during secondary neuron death following brain injury in old and young mice. Neurosci Lett. 2004;369:28–32. doi: 10.1016/j.neulet.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 74.Shah SA, Prough DS, Garcia JM, DeWitt DS, Hellmich HL. Molecular correlates of age-specific responses to traumatic brain injury in mice. Exp Gerontol. 2006;41:1201–5. doi: 10.1016/j.exger.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 75.Onyszchuk G, He YY, Berman NE, Brooks WM. Detrimental effects of aging on outcome from traumatic brain injury: a behavioral, magnetic resonance imaging, and histological study in mice. J Neurotrauma. 2008;25:153–71. doi: 10.1089/neu.2007.0430. [DOI] [PubMed] [Google Scholar]
- 76.Gilmer LK, Ansari MA, Roberts KN, Scheff SW. Age-related mitochondrial changes after traumatic brain injury. J Neurotrauma. 2010;27:939–50. doi: 10.1089/neu.2009.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Itoh T, Imano M, Nishida S, Tsubaki M, Mizuguchi N, Hashimoto S, et al. Increased apoptotic neuronal cell death and cognitive impairment at early phase after traumatic brain injury in aged rats. Brain Struct Funct. 2013;218:209–20. doi: 10.1007/s00429-012-0394-5. [DOI] [PubMed] [Google Scholar]
- 78.Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic Biol Med. 2006;41:77–85. doi: 10.1016/j.freeradbiomed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 79.Anderson J, Sandhir R, Hamilton ES, Berman NE. Impaired expression of neuroprotective molecules in the HIF-1a pathway following traumatic brain injury in aged mice. J Neurotrauma. 2009;26:1557–66. doi: 10.1089/neu.2008.0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hamm RJ, Jenkins LW, Lyeth BG, White-Gbadebo DM, Hayes RL. The effect of age on outcome following traumatic brain injury in rats. J Neurosurg. 1991;75:916–21. doi: 10.3171/jns.1991.75.6.0916. [DOI] [PubMed] [Google Scholar]
- 81.Hamm RJ, White-Gbadebo DM, Lyeth BG, Jenkins LW, Hayes RL. The effect of age on motor and cognitive deficits after traumatic brain injury in rats. Neurosurg. 1992;31:1072–7. doi: 10.1227/00006123-199212000-00013. [DOI] [PubMed] [Google Scholar]
- 82.Maughan PH, Scholten KJ, Schmidt RH. Recovery of water maze performance in aged versus young rats after brain injury with the impact acceleration model. J Neurotrauma. 2000;17:1141–53. doi: 10.1089/neu.2000.17.1141. [DOI] [PubMed] [Google Scholar]
- 83.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–26. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 84.Temple MD, Delahunty TM, Hamm RJ, Phillips LL, Lyeth BG, Povlishock JT. Subtle alterations in NMDA-stimulated cyclic GMP levels following lateral fluid percussion brain injury. J Neurotrauma. 2001;18:47–55. doi: 10.1089/089771501750055767. [DOI] [PubMed] [Google Scholar]
- 85.Dixon CE, Kraus MF, Kline AE, Ma X, Yan HQ, Griffith RG, et al. Amantadine improves water maze performance without affecting motor behavior following traumatic brain injury in rats. Restor Neurol Neurosci. 1999;14:285–94. [PubMed] [Google Scholar]
- 86.Li W, Dai S, An J, Li P, Chen X, Xiong R, et al. Chronic but not acute treatment with caffeine attenuates traumatic brain injury in the mouse cortical impact model. Neuroscience. 2008;151:1198–207. doi: 10.1016/j.neuroscience.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 87.Williams G, Morris ME, Schache A, McCrory PR. Incidence of gait abnormalities after traumatic brain injury. Arch Phys Med Rehabil. 2009;90:587–93. doi: 10.1016/j.apmr.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 88.Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shin SS, Bray ER, Zhang CQ, Dixon CE. Traumatic brain injury reduces striatal tyrosine hydroxylase activity and potassium-evoked dopamine release in rats. Brain Res. 2011;1369:208–15. doi: 10.1016/j.brainres.2010.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pifarre P, Prado J, Giralt M, Molinero A, Hidalgo J, Garcia A. Cyclic GMP phosphodiesterase inhibition alters the glial inflammatory response, reduces oxidative stress and cell death and increases angiogenesis following focal brain injury. J Neurochem. 2010;112:807–17. doi: 10.1111/j.1471-4159.2009.06518.x. [DOI] [PubMed] [Google Scholar]
- 91.Prado J, Pifarre P, Giralt M, Hidalgo J, Garcia A. Metallothioneins I/II are involved in the neuroprotective effect of sildenafil in focal brain injury. Neurochem Int. 2013;62:70–8. doi: 10.1016/j.neuint.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 92.Ko IG, Shin MS, Kim BK, Kim SE, Sung YH, Kim TS, et al. Tadalafil improves short-term memory by suppressing ischemia-induced apoptosis of hippocampal neuronal cells in gerbils. Pharmacol Biochem Behav. 2009;91:629–35. doi: 10.1016/j.pbb.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 93.Zhang RL, Chopp M, Roberts C, Wei M, Wang X, Liu X, et al. Sildenafil enhances neurogenesis and oligodendrogenesis in ischemic brain of middle-aged mouse. PLoS One. 2012;7:e48141. doi: 10.1371/journal.pone.0048141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Han BH, Vellimana AK, Zhou ML, Milner E, Zipfel GJ. Phosphodiesterase 5 inhibition attenuates cerebral vasospasm and improves functional recovery after experimental subarachnoid hemorrhage. Neurosurg. 2012;70:178–86. doi: 10.1227/NEU.0b013e31822ec2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sunico CR, Gonzalez-Forero D, Dominguez G, Garcia-Verdugo JM, Moreno-Lopez B. Nitric oxide induces pathological synapse loss by a protein kinase G-, Rho kinase-dependent mechanism preceded by myosin light chain phosphorylation. J Neurosci. 2010;30:973–84. doi: 10.1523/JNEUROSCI.3911-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moreno-Lopez B, Gonzalez-Forero D. Nitric oxide and synaptic dynamics in the adult brain: physiopathological aspects. Rev Neurosci. 2006;17:309–57. doi: 10.1515/revneuro.2006.17.3.309. [DOI] [PubMed] [Google Scholar]
- 97.Gaur V, Kumar A. Behavioral, biochemical and cellular correlates in the protective effect of sertraline against transient global ischemia induced behavioral despair: possible involvement of nitric oxide-cyclic guanosine monophosphate study pathway. Brain Res Bull. 2010;82:57–64. doi: 10.1016/j.brainresbull.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 98.Chatzipanteli K, Wada K, Busto R, Dietrich WD. Effects of moderate hypothermia on constitutive and inducible nitric oxide synthase activities after traumatic brain injury in the rat. J Neurochem. 1999;72:2047–52. doi: 10.1046/j.1471-4159.1999.0722047.x. [DOI] [PubMed] [Google Scholar]
- 99.Kader A, Frazzini VI, Baker CJ, Solomon RA, Trifiletti RR. Effect of mild hypothermia on nitric oxide synthesis during focal cerebral ischemia. Neurosurg. 1994;35:272–7. doi: 10.1227/00006123-199408000-00013. [DOI] [PubMed] [Google Scholar]
- 100.Zhuang P, Ji H, Zhang YH, Min ZL, Ni QG, You R. ZJM-289, a novel nitric oxide donor, alleviates the cerebral ischaemic-reperfusion injury in rats. Clin Exp Pharmacol Physiol. 2010;37:e121–7. doi: 10.1111/j.1440-1681.2010.05353.x. [DOI] [PubMed] [Google Scholar]
- 101.Chen J, Chopp M. Neurorestorative treatment of stroke: cell and pharmacological approaches. NeuroRx. 2006;3:466–73. doi: 10.1016/j.nurx.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Foley LM, Hitchens TK, Melick JA, Bayir H, Ho C, Kochanek PM. Effect of inducible nitric oxide synthase on cerebral blood flow after experimental traumatic brain injury in mice. J Neurotrauma. 2008;25:299–310. doi: 10.1089/neu.2007.0471. [DOI] [PubMed] [Google Scholar]
- 103.Bayir H, Kagan VE, Clark RS, Janesko-Feldman K, Rafikov R, Huang Z, et al. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem. 2007;101:168–81. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- 104.Naruse S, Aoki Y, Takei R, Horikawa Y, Ueda S. Effects of atrial natriuretic peptide on ischemic brain edema in rats evaluated by proton magnetic resonance method. Stroke. 1991;22:61–5. doi: 10.1161/01.str.22.1.61. [DOI] [PubMed] [Google Scholar]
- 105.Boran MS, Baltrons MA, Garcia A. The ANP-cGMP-protein kinase G pathway induces a phagocytic phenotype but decreases inflammatory gene expression in microglial cells. Glia. 2008;56:394–411. doi: 10.1002/glia.20618. [DOI] [PubMed] [Google Scholar]
- 106.Kohgami S, Ogata T, Morino T, Yamamoto H, Schubert P. Pharmacological shift of the ambiguous nitric oxide action from neurotoxicity to cyclic GMP-mediated protection. Neurol Res. 2010;32:938–44. doi: 10.1179/016164110X12681290831243. [DOI] [PubMed] [Google Scholar]
- 107.Murray AJ, Peace AG, Shewan DA. cGMP promotes neurite outgrowth and growth cone turning and improves axon regeneration on spinal cord tissue in combination with cAMP. Brain Res. 2009;1294:12–21. doi: 10.1016/j.brainres.2009.07.071. [DOI] [PubMed] [Google Scholar]
- 108.Fukui S, Fazzina G, Amorini AM, Dunbar JG, Marmarou A. Differential effects of atrial natriuretic peptide on the brain water and sodium after experimental cortical contusion in the rat. J Cereb Blood Flow Metab. 2003;23:1212–8. doi: 10.1097/01.WCB.0000088762.02615.30. [DOI] [PubMed] [Google Scholar]
- 109.Marsala J, Lukacova N, Kolesar D, Sulla I, Galik J, Marsala M. The distribution of primary nitric oxide synthase- and parvalbumin- immunoreactive afferents in the dorsal funiculus of the lumbosacral spinal cord in a dog. Cell Mol Neurobiol. 2007;27:475–504. doi: 10.1007/s10571-007-9140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mitome-Mishima Y, Miyamoto N, Tanaka R, Oishi H, Arai H, Hattori N, et al. Differences in phosphodiesterase 3A and 3B expression after ischemic insult. Neurosci Res. 2013;75:340–8. doi: 10.1016/j.neures.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 111.Horai S, Nakagawa S, Tanaka K, Morofuji Y, Couraud PO, Deli MA, et al. Cilostazol strengthens barrier integrity in brain endothelial cells. Cell Mol Neurobiol. 2013;33:291–307. doi: 10.1007/s10571-012-9896-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lin CS. Tissue expression, distribution, and regulation of PDE5. Int J Impot Res. 2004;16 (Suppl 1):S8–S10. doi: 10.1038/sj.ijir.3901207. [DOI] [PubMed] [Google Scholar]
- 113.Lin CS, Lin G, Xin ZC, Lue TF. Expression, distribution and regulation of phosphodiesterase 5. Curr Pharm Des. 2006;12:3439–57. doi: 10.2174/138161206778343064. [DOI] [PubMed] [Google Scholar]
- 114.Atkins CM, Cepero ML, Kang Y, Liebl DJ, Dietrich WD. Effects of early rolipram treatment on histopathological outcome after controlled cortical impact injury in mice. Neurosci Lett. 2013;532:1–6. doi: 10.1016/j.neulet.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Atkins CM, Kang Y, Furones C, Truettner JS, Alonso OF, Dietrich WD. Postinjury treatment with rolipram increases hemorrhage after traumatic brain injury. J Neurosci Res. 2012;90:1861–71. doi: 10.1002/jnr.23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 117.Dal Piaz V, Giovannoni MP. Phosphodiesterase 4 inhibitors, structurally unrelated to rolipram, as promising agents for the treatment of asthma and other pathologies. Eur J Med Chem. 2000;35:463–80. doi: 10.1016/s0223-5234(00)00179-3. [DOI] [PubMed] [Google Scholar]
- 118.Goodman JC, Robertson CS, Grossman RG, Narayan RK. Elevation of tumor necrosis factor in head injury. J Neuroimmunol. 1990;30:213–7. doi: 10.1016/0165-5728(90)90105-v. [DOI] [PubMed] [Google Scholar]
- 119.Ross SA, Halliday MI, Campbell GC, Byrnes DP, Rowlands BJ. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br J Neurosurg. 1994;8:419–25. doi: 10.3109/02688699408995109. [DOI] [PubMed] [Google Scholar]
- 120.Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF a and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–9. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 121.Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42:177–85. doi: 10.1016/0165-5728(93)90008-m. [DOI] [PubMed] [Google Scholar]
- 122.Yan HQ, Banos MA, Herregodts P, Hooghe R, Hooghe-Peters EL. Expression of interleukin (IL)-1 b, IL-6 and their respective receptors in the normal rat brain and after injury. Eur J Immunol. 1992;22:2963–71. doi: 10.1002/eji.1830221131. [DOI] [PubMed] [Google Scholar]
- 123.Vitarbo EA, Chatzipanteli K, Kinoshita K, Truettner JS, Alonso OF, Dietrich WD. Tumor necrosis factor a expression and protein levels after fluid percussion injury in rats: The effect of injury severity and brain temperature. Neurosurg. 2004;55:416–24. doi: 10.1227/01.neu.0000130036.52521.2c. [DOI] [PubMed] [Google Scholar]
- 124.Kinoshita K, Chatzipanteli K, Vitarbo E, Truettner JS, Alonso OF, Dietrich WD. Interleukin-1b messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: importance of injury severity and brain temperature. Neurosurg. 2002;51:195–203. doi: 10.1097/00006123-200207000-00027. [DOI] [PubMed] [Google Scholar]
- 125.Kato H, Araki T, Itoyama Y, Kogure K. Rolipram, a cyclic AMP-selective phosphodiesterase inhibitor, reduces neuronal damage following cerebral ischemia in the gerbil. Eur J Pharmacol. 1995;272:107–10. doi: 10.1016/0014-2999(94)00694-3. [DOI] [PubMed] [Google Scholar]
- 126.Imanishi T, Sawa A, Ichimaru Y, Miyashiro M, Kato S, Yamamoto T, et al. Ameliorating effects of rolipram on experimentally induced impairments of learning and memory in rodents. Eur J Pharmacol. 1997;321:273–8. doi: 10.1016/s0014-2999(96)00969-7. [DOI] [PubMed] [Google Scholar]
- 127.Block F, Tondar A, Schmidt W, Schwarz M. Delayed treatment with rolipram protects against neuronal damage following global ischemia in rats. Neuroreport. 1997;8:3829–32. doi: 10.1097/00001756-199712010-00033. [DOI] [PubMed] [Google Scholar]
- 128.Block F, Schmidt W, Nolden-Koch M, Schwarz M. Rolipram reduces excitotoxic neuronal damage. Neuroreport. 2001;12:1507–11. doi: 10.1097/00001756-200105250-00041. [DOI] [PubMed] [Google Scholar]
- 129.Li LX, Cheng YF, Lin HB, Wang C, Xu JP, Zhang HT. Prevention of cerebral ischemia-induced memory deficits by inhibition of phosphodiesterase-4 in rats. Metab Brain Dis. 2011;26:37–47. doi: 10.1007/s11011-011-9235-0. [DOI] [PubMed] [Google Scholar]
- 130.Paintlia AS, Paintlia MK, Singh I, Skoff RB, Singh AK. Combination therapy of lovastatin and rolipram provides neuroprotection and promotes neurorepair in inflammatory demyelination model of multiple sclerosis. Glia. 2009;57:182–93. doi: 10.1002/glia.20745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abbas N, Zou LP, Pelidou SH, Winblad B, Zhu J. Protective effect of rolipram in experimental autoimmune neuritis: protection is associated with down-regulation of IFN-g and inflammatory chemokines as well as up-regulation of IL-4 in peripheral nervous system. Autoimmunity. 2000;32:93–9. doi: 10.3109/08916930008994078. [DOI] [PubMed] [Google Scholar]
- 132.Sommer N, Loschmann PA, Northoff GH, Weller M, Steinbrecher A, Steinbach JP, et al. The antidepressant rolipram suppresses cytokine production and prevents autoimmune encephalomyelitis. Nat Med. 1995;1:244–8. doi: 10.1038/nm0395-244. [DOI] [PubMed] [Google Scholar]
- 133.Souza DG, Cassali GD, Poole S, Teixeira MM. Effects of inhibition of PDE4 and TNF-alpha on local and remote injuries following ischaemia and reperfusion injury. Br J Pharmacol. 2001;134:985–94. doi: 10.1038/sj.bjp.0704336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schaal SM, Garg MS, Ghosh M, Lovera L, Lopez M, Patel M, et al. The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury. PLoS One. 2012;7:e43634. doi: 10.1371/journal.pone.0043634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Birk S, Edvinsson L, Olesen J, Kruuse C. Analysis of the effects of phosphodiesterase type 3 and 4 inhibitors in cerebral arteries. Eur J Pharmacol. 2004;489:93–100. doi: 10.1016/j.ejphar.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 136.Willette RN, Shiloh AO, Sauermelch CF, Sulpizio A, Michell MP, Cieslinski LB, et al. Identification, characterization, and functional role of phosphodiesterase type IV in cerebral vessels: effects of selective phosphodiesterase inhibitors. J Cereb Blood Flow Metab. 1997;17:210–9. doi: 10.1097/00004647-199702000-00011. [DOI] [PubMed] [Google Scholar]
- 137.Mehats C, Jin SL, Wahlstrom J, Law E, Umetsu DT, Conti M. PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J. 2003;17:1831–41. doi: 10.1096/fj.03-0274com. [DOI] [PubMed] [Google Scholar]
- 138.Miwa T, Mori A, Nakahara T, Ishii K. Intravenously administered phosphodiesterase 4 inhibitors dilate retinal blood vessels in rats. Eur J Pharmacol. 2009;602:112–6. doi: 10.1016/j.ejphar.2008.10.060. [DOI] [PubMed] [Google Scholar]
- 139.Tilley DG, Maurice DH. Vascular smooth muscle cell phosphodiesterase (PDE) 3 and PDE4 activities and levels are regulated by cyclic AMP in vivo. Mol Pharmacol. 2002;62:497–506. doi: 10.1124/mol.62.3.497. [DOI] [PubMed] [Google Scholar]
- 140.Si Q, Nakamura Y, Ogata T, Kataoka K, Schubert P. Differential regulation of microglial activation by propentofylline via cAMP signaling. Brain Res. 1998;812:97–104. doi: 10.1016/s0006-8993(98)00954-8. [DOI] [PubMed] [Google Scholar]
- 141.Zhang B, Yang L, Konishi Y, Maeda N, Sakanaka M, Tanaka J. Suppressive effects of phosphodiesterase type IV inhibitors on rat cultured microglial cells: Comparison with other types of cAMP-elevating agents. Neuropharm. 2002;42:262–9. doi: 10.1016/s0028-3908(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 142.Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFa production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999;837:203–12. doi: 10.1016/s0006-8993(99)01666-2. [DOI] [PubMed] [Google Scholar]
- 143.Yoshikawa M, Suzumura A, Tamaru T, Takayanagi T, Sawada M. Effects of phosphodiesterase inhibitors on cytokine production by microglia. Mult Scler. 1999;5:126–33. doi: 10.1177/135245859900500210. [DOI] [PubMed] [Google Scholar]
- 144.Prabhakar U, Lipshutz D, Bartus JO, Slivjak MJ, Smith EF, 3rd, Lee JC, et al. Characterization of cAMP-dependent inhibition of LPS-induced TNF alpha production by rolipram, a specific phosphodiesterase IV (PDE IV) inhibitor. Int J Immunopharmacol. 1994;16:805–16. doi: 10.1016/0192-0561(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 145.Foey AD, Field S, Ahmed S, Jain A, Feldmann M, Brennan FM, et al. Impact of VIP and cAMP on the regulation of TNF-a and IL-10 production: Implications for rheumatoid arthritis. Arthritis Res Ther. 2003;5:R317–28. doi: 10.1186/ar999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hansen G, Jin S, Umetsu DT, Conti M. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc Natl Acad Sci U S A. 2000;97:6751–6. doi: 10.1073/pnas.97.12.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jin SL, Goya S, Nakae S, Wang D, Bruss M, Hou C, et al. Phosphodiesterase 4B is essential for T(H)2-cell function and development of airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol. 2010;126:1252–9. e12. doi: 10.1016/j.jaci.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang P, Wu P, Ohleth KM, Egan RW, Billah MM. Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophils. Mol Pharmacol. 1999;56:170–4. doi: 10.1124/mol.56.1.170. [DOI] [PubMed] [Google Scholar]
- 149.Ghosh M, Garcia-Castillo D, Aguirre V, Golshani R, Atkins CM, Bramlett HM, et al. Proinflammatory cytokine regulation of cyclic AMP-phosphodiesterase 4 signaling in microglia in vitro and following CNS injury. Glia. 2012;60:1839–59. doi: 10.1002/glia.22401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Keeling KL, Hicks RR, Mahesh J, Billings BB, Kotwal GJ. Local neutrophil influx following lateral fluid-percussion brain injury in rats is associated with accumulation of complement activation fragments of the third component (C3) of the complement system. J Neuroimmunol. 2000;105:20–30. doi: 10.1016/s0165-5728(00)00183-1. [DOI] [PubMed] [Google Scholar]
- 151.Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflamm. 2012;9:17. doi: 10.1186/1742-2094-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Atkins CM, Falo MC, Alonso OF, Bramlett HM, Dietrich WD. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett. 2009;459:52–6. doi: 10.1016/j.neulet.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee HT, Chang YC, Wang LY, Wang ST, Huang CC, Ho CJ. cAMP response element-binding protein activation in ligation preconditioning in neonatal brain. Ann Neurol. 2004;56:611–23. doi: 10.1002/ana.20259. [DOI] [PubMed] [Google Scholar]
- 154.Wiescholleck V, Manahan-Vaughan D. PDE4 inhibition enhances hippocampal synaptic plasticity in vivo and rescues MK801-induced impairment of long-term potentiation and object recognition memory in an animal model of psychosis. Transl Psychiatry. 2012;2:e89. doi: 10.1038/tp.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Christensen BK, Colella B, Inness E, Hebert D, Monette G, Bayley M, et al. Recovery of cognitive function after traumatic brain injury: a multilevel modeling analysis of Canadian outcomes. Arch Phys Med Rehabil. 2008;89:S3–15. doi: 10.1016/j.apmr.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 156.Mathias JL, Mansfield KM. Prospective and declarative memory problems following moderate and severe traumatic brain injury. Brain Inj. 2005;19:271–82. doi: 10.1080/02699050400005028. [DOI] [PubMed] [Google Scholar]
- 157.Serra-Grabulosa JM, Junque C, Verger K, Salgado-Pineda P, Maneru C, Mercader JM. Cerebral correlates of declarative memory dysfunctions in early traumatic brain injury. J Neurol Neurosurg Psychiatry. 2005;76:129–31. doi: 10.1136/jnnp.2004.027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Titus DJ, Sakurai A, Kang Y, Furones C, Jergova S, Santos R, et al. Phosphodiesterase inhibition rescues chronic cognitive deficits induced by traumatic brain injury. J Neurosci. 2013;33:5216–26. doi: 10.1523/JNEUROSCI.5133-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lopez-Campos JL, Calero Acuna C. What is in the guidelines about the pharmacological treatment of chronic obstructive pulmonary disease? Expert Rev Respir Med. 2013;7:43–51. doi: 10.1586/ers.13.17. [DOI] [PubMed] [Google Scholar]
- 160.Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharm (Berl) 2009;202:419–43. doi: 10.1007/s00213-008-1273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta:GA: 2010. [Google Scholar]
- 162.Bielekova B, Richert N, Howard T, Packer AN, Blevins G, Ohayon J, et al. Treatment with the phosphodiesterase type-4 inhibitor rolipram fails to inhibit blood-brain barrier disruption in multiple sclerosis. Mult Scler. 2009;15:1206–14. doi: 10.1177/1352458509345903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Robichaud A, Stamatiou PB, Jin SL, Lachance N, MacDonald D, Laliberte F, et al. Deletion of phosphodiesterase 4D in mice shortens a(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest. 2002;110:1045–52. doi: 10.1172/JCI15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gurney ME, Burgin AB, Magnusson OT, Stewart LJ. Small molecule allosteric modulators of phosphodiesterase 4. Handb Exp Pharmacol. 2011:167–92. doi: 10.1007/978-3-642-17969-3_7. [DOI] [PubMed] [Google Scholar]
- 165.Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010;28:63–70. doi: 10.1038/nbt.1598. [DOI] [PubMed] [Google Scholar]
- 166.Kochanek PM, Bramlett H, Dietrich WD, Dixon CE, Hayes RL, Povlishock J, et al. A novel multicenter preclinical drug screening and biomarker consortium for experimental traumatic brain injury: operation brain trauma therapy. J Trauma. 2011;71:S15–24. doi: 10.1097/TA.0b013e31822117fe. [DOI] [PubMed] [Google Scholar]