

Abstract
PNPLA6 mutations, known to be associated with the development of motor neuron phenotypes, have recently been identified in families with Boucher–Neuhäuser syndrome. Boucher–Neuhäuser is a rare autosomal recessive syndrome characterized by the co-occurrence of cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy. Gait ataxia in Boucher–Neuhäuser usually manifests before early adulthood, although onset in the third or fourth decade has also been reported. However, given the recent identification of PNPLA6 mutations as the cause of this condition, the determining factors of age of symptom onset still need to be established. Here, we have identified a sporadic Boucher–Neuhäuser case with late-onset gait ataxia and relatively milder retinal changes due to compound heterozygous PNPLA6 mutations. Compound heterozygosity was confirmed by cloning and sequencing the patient’s genomic DNA from coding exons 26–29. Furthermore, both mutations (one novel and one known) fell in the phospholipase esterase domain, where most pathogenic mutations seem to cluster. Taken together, we herein confirm PNPLA6 mutations as the leading cause of Boucher–Neuhäuser syndrome and suggest inquiring about a history of hypogonadism or visual changes in patients presenting with late-onset gait ataxia. We also advocate for neuroophthalmologic evaluation in suspected cases.
Keywords: Autosomal recessive, Boucher–Neuhäuser, Chorioretinal dystrophy, Hypogonadotropic hypogonadism, PNPLA6 mutations, NTE domain
Introduction
Boucher–Neuhäuser (B–N; MIM #215470) is a rare syndrome characterized by the triad of early-onset autosomal recessive cerebellar ataxia (ARCA), hypogonadotropic hypogonadism, and chorioretinal dystrophy [1, 2]. Gait ataxia in Boucher–Neuhäuser has been typically reported between the first and third decades of life; later ages of onset are rare [1, 3–5]. Although sporadic cases without apparent consanguinity have been reported, most reports are among siblings (about 80 %) [6, 7], often of consanguineous parents [3, 6]. It is unclear whether there is a reporting bias away from sporadic cases in this autosomal recessive disorder, but this seems plausible. Its genetic causes have recently been established by the identification of PNPLA6 (patatin-like phospholipase domain containing 6) mutations in both B–N and Gordon–Holmes (G–H) syndromes. PNPLA6 genetic variability was known to be implicated in motor neuron phenotypes including spastic paraplegia [8, 9]. PNPLA6 (MIM #603197; 19p13.2) encodes for a neuropathy target esterase (NTE). Five different transcripts have been identified, with the longest transcript, transcript variant 1 (NM_001166111.1), encoding a protein (NP_001159583.1) of 1,375 amino acids. In this study, we have identified a sporadic case of B–N syndrome with an unusually late age of onset of ataxic symptoms due to compound heterozygous PNPLA6 mutations. Details of her phenotype, including ophthalmologic findings, are described.
Materials and methods
Due to the recent identification of PNPLA6 mutations in B–N syndrome [6, 10], these were investigated in a patient with sporadic B–N syndrome. The ethics committees at both Mount Sinai Beth Israel and Icahn School of Medicine at Mount Sinai approved this study, and written informed consent was obtained from the participant. The participant’s DNA samples were isolated from whole blood using standard procedures. Genomic primers for PCR amplifications covering exons and intron–exon boundaries were designed using a primer design public website (http://ihg.gsf.de/ihg/ExonPrimer.html; primer sequences available upon request). All purified PCR products were then sequenced in both forward and reverse directions with Applied Biosystems BigDye terminator v3.1 sequencing chemistry as per the manufacturer’s instructions. The resulting sequencing reactions were resolved on an ABI3130 genetic analyzer (Applied Biosystems, Foster city, CA, USA) and analyzed using Sequencer 5.2.3 software (Gene Codes Corporation, Ann Arbor, MI, USA). Due to the lack of additional family members, and to verify that both mutations identified in this study were located intrans, the patient’s genomic DNA containing both PNPLA6 mutations was amplified by PCR using forward primer 5′-ATGGAGGCCCGAATTCCTAAGTGCTGCTTGCTCACCC-3′ and reverse primer 5′-GCCGCGGTACCTCGAGCATACCACTCTGGGCTTTAAGTAGC-3′, and Phusion High-Fidelity polymerase (Thermo Fisher Scientific Inc, Waltham, MA, USA). Purified PCR fragment of 2,801 bp was cloned into pCMV-HA vector (Clontech, Mountain View, CA, USA) using In-Fusion® HD following the manufacturer’s protocol. Ten independent colonies were picked randomly and sequenced in both directions as described above. PredictSNP (http://loschmidt.chemi.muni.cz/predictsnp), which is a consensus of six prediction tools [11], and Mutation Taster (http://www.mutationtaster.org) were used for mutation pathogenicity prediction. The HomoloGene database from NCBI web site (http://www.ncbi.nlm.nih.gov/homologene) was also used to examine the conservation of the PNPLA6 p.Ser1173Arg mutation in different species.
Results
This 59-year-old woman, born to non-consanguineous parents from Spain and Italy, had normal development until adolescence, when it was noted that she had primary amenorrhea. Hormone therapy for 1 year in her 20s led to menses, but was poorly tolerated and stopped. First onset of neurological symptoms was at age 43, when she noted mild, but progressive, binocular vertical or oblique diplopia on lateral gaze in either direction. Between ages 50–51 she developed gait imbalance and veering to either side, which slowly worsened. When she was 54–55 years her voice became “raspy”.
At age 58, her symptoms started to worsen rapidly over several months. She started to feel mentally clouded and developed word-finding difficulties, although this did not interfere with everyday function. She also reported anhedonia. Of note, she had no monocular visual symptoms, but had seen ophthalmologists in the past who noted retinal abnormalities, suggesting either macular degeneration or a “burn from the sun”.
On examination, she had excellent recall of short- and long-term events. However, her Montreal Cognitive Assessment (MoCA) score was 19/30 (normal >25), with deficits in attention, visuospatial abilities, and delayed recall. She had a scanning quality to her speech, with mild labial and lingual dysarthria. She had decreased hearing to finger rub on the left.
Visual acuity was 20/25 bilaterally. Color vision (Ishihara) was 10/10 plates correct in both eyes. There was no afferent pupillary defect. She had hypometric vertical and horizontal saccades, saccadic smooth pursuit, gaze-evoked nystagmus and poor visual suppression of vestibular ocular reflexes. Although ocular motor range was full, she had an alternating skew deviation on lateral gaze, which explained her diplopia on lateral gaze. There was also a small esophoria in central position that increased in right and left gaze.
Retinal atrophy was evident on the dilated fundoscopic exam, as is shown in Fig. 1. Spectral mode ocular coherence tomography (OCT) imaging (Heidelberg Spectralis) confirmed these changes (Fig. 2). The phototopic electroretinogram (ERG) was reduced by 50 %, but scotopic function reduction was less severe. Goldmann visual field showed bitemporal central defects and a slight blind spot enlargement bilaterally, as well as a small paracentral scotoma to the I2 isopter on the left (Fig. 3a, b).
Fig. 1.

Fundus photography of the posterior pole of the right eye (a) and the region nasal to the optic disk in the left eye (b) demonstrates a large patch of complete retinal pigment epithelium (RPE) and choriocapillaris atrophy nasally adjacent to the optic disk in each eye. A crescent-shaped patch of partial atrophy temporal to the macula is seen in the right eye, along with pigmentary macular changes. Choroidal vessels are visible in atrophic regions in each eye. There was no pigmentary deposition or clumping. Peripapillary regions demonstrate grayish discoloration. Autofluorescence photographs of the right eye (c) and left eye (d) demonstrate crescent-shaped zones of patchy hyperfluorescence temporal to the macula and in the fovea bilaterally
Fig. 2.

Spectral mode OCT images through central fovea in right eye (a) and left eye (b). Note retinal thinning and loss of the outer retinal ellipsoid line temporally (arrows) corresponding to areas of hyper-autofluorescence. Spectral mode OCT images nasal to the optic disks in the right eye (c) and left eye (d). The retinas are severely thinned, with loss of the layered retinal architecture. The choriocapillaris is absent, and adjacent to the optic disks, the larger choroidal vessels are effaced as well. e Nerve fiber layer OCT shows bilateral nasal nerve fiber layer thinning
Fig. 3.
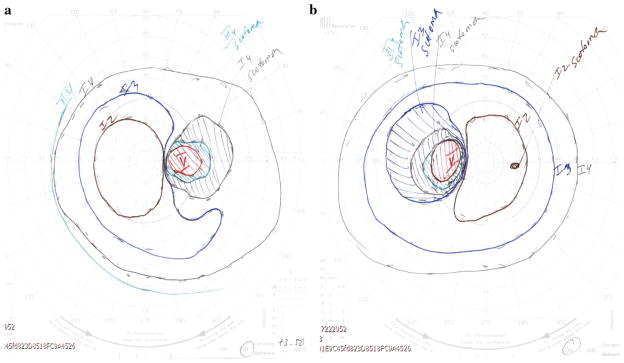
Goldmann visual field in the left eye (a) and right eye (b) shows large temporal defects in each eye and a tiny nasal scotoma in the left eye
She had normal strength, tone and no tremor. Reflexes were normal, except for reduced ankle jerks, and there was mild vibratory loss in her toes. She had dysdiadochokinesia in her arms and legs, dysmetria on heel-to-shin but not on finger-to-nose, and significant overshoot on the finger chase task, bilaterally. Her stance was wide-based, her gait was ataxic with occasional veering to either side, and she was able to tandem walk with much difficulty.
Family history was notable for her mother developing dementia and parkinsonism in her 50s, and death at the age of 77. The patient’s mother also lost eight full-term children within 1 day of their birth, and had three spontaneous abortions of unknown gestation lengths. The patient’s only sibling to survive the perinatal period was her full brother, who is in his 60s.
Brain magnetic resonance imaging (MRI) (Fig. 4a, b) demonstrated superior cerebellar/vermian atrophy and prominent cerebellar folia, with a normal pituitary gland and only minimal, likely age-related periventricular white matter disease. Electromyography and nerve conduction studies (EMG/NCS) showed absent bilateral tibial H reflexes, and absent medial plantar mixed nerve response. The sural and peroneal sensory response amplitudes were in the lower range of normal. Peroneal and tibial F wave minimal latencies were also normal. The peroneal CMAP amplitude and conduction velocity were normal. There were borderline, high-amplitude motor unit potentials and fibrillations in the gastrocnemius muscle. Overall, the study was consistent with a mild distal axonal neuropathy.
Fig. 4.
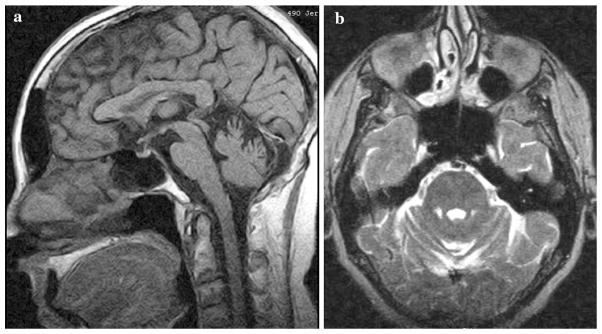
Sagittal T1- (a) and axial T2- (b) weighted brain magnetic resonance images showing cerebellar folia prominence and marked vermian atrophy, respectively
Blood chemistries and thyroid hormones, rheumatologic tests, vitamin E level and paraneoplastic panel were all normal. Luteinizing hormone was low at 0.1 mIU/mL (normal postmenopausal 7.7–58.5), as was follicular stimulating hormone (0.3 mIU/mL, normal postmenopausal 25.8–134.8). Prolactin, estradiol, and random cortisol were within normal ranges (6 ng/mL, 13.23 pg/mL and 3.6 μg/dL, respectively).
Chromosome analysis was performed by Integrated Genetics and revealed a normal female 46, XX karyotype. Mitochondrial DNA mutation and deletion testing for the commercial GeneDx panel of 58 known disease-associated mutations was negative. Subsequent sequencing of the mitochondrial genome showed a m.5780 G>A sequence change in the MT-TC gene, which has previously been associated with sensorineural hearing loss but has also been described as a benign polymorphism [12, 13]. Testing for SCA-7 at Athena Diagnostics, Inc., showed a normal number of 10 CAG repeats for both alleles.
PNPLA6 mutational screening identified compound heterozygous pathogenic mutations, one known (c.3134C>T; p.Ser1045Leu) and one novel (c.3519C>G; p.Ser1173Arg), in coding exons 26 and 29, respectively. The longest transcript, transcript variant 1 (NM_001166111.1), was used for mutation nomenclature. This novel PNPLA6 mutation, which was found to be highly conserved among other orthologs and was predicted to be pathogenic by two computational methods (PredictSNP score: 0.61 and Mutation Taster score: 0.99), fell in the phospholipid esterase domain (Patatin-like domain) at the C-terminal, where the majority of pathogenic PNPLA6 mutations seem to cluster (Fig. 5a, b) [6]. The p.Ser1173Arg mutation was additionally absent in 188 ethnicity-matched, neurologically normal chromosomes tested by direct screening of PNPLA6 exon 29 and other public SNP databases such as dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) and Exome Variant Server of the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) [14]. This, along with the fact that mutations located in the phospholipid esterase domain are prone to abolish the catalytic activity of NTE [6], support the pathogenic role of the novel mutation described here. To further examine whether mutations were transmitted from both parents and were located on separate alleles, the patient’s genomic DNA from coding exon 26 to exon 29 was cloned and sequenced. We observed that five out of ten clones only carried the p.Ser1045Leu mutation, while the other five carried the p.Ser1173Arg mutation alone. This indicates that both mutations were located on separate alleles (one mutation was, therefore, transmitted from the father and one from the mother).
Fig. 5.

a PNPLA6 protein structure showing its predicted functional domains and all described pathogenic mutations. Domains were predicted by SMART (http://smart.embl-heidelberg.de). cNMP stands for cyclic nucleotide-monophosphate binding domain and patatin represents the patatin-like phospholipase domain also known as phospholipid esterase domain. A tyrosine kinase phosphorylation site is also predicted at residues 403–410 (not shown). All mutations identified in BNS are reported above with mutations identified in this study highlighted in bold. The novel PNPLA6 mutation identified in this study is also represented within a rectangle. Below are reported mutations identified in spastic paraplegia (red), spastic ataxia (blue), and GHS (green). b Sequence chromatograms of wild-type and mutant PNPLA6 exon 29, novel mutation (p.Ser1173Arg) is highlighted with a black arrow. Conservation among other species of the novel p.Ser1173Arg mutation (red) is also shown. Of note, conservation scores (GERP++ and PhyloP) were 4.98 and 2.32 for the previously described mutation, and 1.02 and 0.469 for the novel herein described. BT Bos taurus, CL Canis lupus, DR Danio rerio, GG Gallus gallus, HS Homo sapiens, MM Mus musculus. PT Pan troglodytes, RN Rattus norvegicus
Discussion
In this study, we have identified a sporadic B–N case presenting with unusually late-onset gait ataxia due to compound heterozygous PNPLA6 mutations, confirming that genetic variability in PNPLA6 is probably the major cause of Boucher–Neuhäuser syndrome. Our case demonstrated the classic triad of B–N syndrome, which includes cerebellar ataxia, hypopituitary hypogonadism, and chorioretinal dystrophy [1, 2]. Like PLA2G6 (phospholipase A2, Group VI), PNPLA6 contains a patatin-like domain. Interestingly, while PLA2G6 mutations have been reported to cause infantile neuroaxonal dystrophy, atypical parkinsonism, and neurodegeneration with brain iron accumulation, PNPLA6 mutations have been typically related to hereditary spastic paraplegia, and, more recently, ataxia [6, 8, 15, 16]. Although these phenotypic heterogeneities are often seen in genes involved in diseases of the central nervous system [17], it is likely that this broad clinical spectrum relates to the complex role of the patatin-like domain within the brain, which is involved in brain lipid metabolism, neuronal development, intracellular membrane trafficking, and axon maintenance, among others [18, 19]. Given that lipid metabolism is a highly preserved function among species and tissue types, the multisystem effects of PNPLA6 mutations are not surprising. Indeed, PNPLA6 mutations can also cause autosomal recessive spastic paraplegia, spastic ataxia without chorioretinal dystrophy or hypogonadism, and Gordon–Holmes syndrome. G-H differs from B-N in that it lacks chorioretinal dystrophy, often has prominent spasticity [6], and may be due to either mutations in PNPLA6 or RNF216. Dysarthria is usually the presenting neurologic symptom in G-H, but ataxia leading to wheelchair dependency invariably ensues. Dementia is also prominent [20]. Because of phenotypic similarities between RNF216 and PNPLA6 gene mutation carriers, one may speculate that both of these genes are involved in a pathway with common downstream effects. Whether mutations in RNF216 also cause B–N has not been determined.
Whereas the clinical syndrome of B–N was first recognized and reported in 1969 (Table 1), only the cases reported by Synofzik et al. [6, 10] and now ours, have been diagnosed molecularly. In that case series, the age of ataxia onset ranged 6–27 years of age. B–N patients’ ataxic symptoms have been usually reported as subtle “clumsiness” and/or minor balance abnormalities progressing to a gait disorder, with or without dysarthria [6]. Taking into account all clinical B–N reports to date, our case is exceedingly unusual in that the first sign of cerebellar dysfunction (diplopia) appeared at age 43, and gait ataxia did not appear until age 50. Besides ataxia, hormonal dysfunction (resulting from involution of gonadotropic function) is typically not a prominent presenting feature, and it may precede gait ataxia by years. Further, cases may respond to treatment with exogenous gonadotropins, and while our patient did not have children, maintained physiological plasma testosterone concentrations, induced testicular enlargement and induction of spermatogenesis have been reported in G-H syndrome, which is also due to PNPLA6 mutations [21].
Table 1.
Summary of Boucher–Neuhäuser cases reported since 1969
| References | Cases | Consanguinity/ ethnicity |
Age at case description/ last examination |
Mutation (s) | Ataxia onset age |
Age at HH Dx and/ or TX |
Ocular symptom/age of onset or Dx |
Fundoscopic findings/Dx age |
Other neurologic findings |
Imaging or autopsy findings |
|---|---|---|---|---|---|---|---|---|---|---|
| This study | 1 woman | No/mixed Spanish-Italian | 59 | Compound heterozygous [p.Ser1045Leu; p.Ser1173Arg] | 51 | 11 | Diplopia/43 | ChD/58 | CI Hyporreflexia |
CA |
| Synofzik et al. [10] | 1 woman | N/A | 37 | Compound heterozygous [V738Qfs*98; V1110M] | N/A | 14 | VI/37 | ChD/37 | Hyperreflexia and hyporreflexia | CA |
| Synofzik et al. [6] | Family IHG25190 4 siblings: 1 man, 3 women |
Yes/Brazilian | 56 55 53 48 |
Homozygous [p.Thr1058Ile] | 6–7 | N/A | VI/1–3 | ChD | Mild CI | CA Pontine atrophy Small pituitary |
| Family ARCA-05 2 sisters |
No/Italian | 44 42 |
Compound heterozygous [p.Val738Glnfs*98; p.Val1110Met] | 6 (sister with intact vision), 27 (other) | N/A | VI in only 1 sister/12 | ChD in the woman with visual complaints | Mild CI Spasticity |
CA | |
| Family IHG25353 2 brothers |
No/Brazilian | 61 57 |
Compound heterozygous [p.Gly578Trp; p.Phe1066Ser] | 6 | N/A | N/A | ChD | N/A | CA | |
| Family IHG25357 1 man |
No/Venezuelan | 26 | Compound heterozygous [p.Ser1045Leu; p.Pro1122Leu] | 20 | N/A | N/A | ChD | N/A | CA | |
| Kate et al. [7] | Brother and sister | Yes/U | 22 (male proband) N/A |
N/A N/A |
20 18 |
22 N/A |
None N/A |
Mottled retinal pigment epithelium N/A |
CI T CI |
CA ST2WMH CA ST2WMH |
| Ling et al. [3] | 1 man | No/Thai | 45 | N/A | 43 | 43 | VI/39 | Retinal pigment epithelium atrophy choriocapillaris and bone spicule-like clumps of pigment deposits | Right foot dystonia Chorea Titubation |
CA Putaminal and midbrain atrophy |
| Yu et al. [29] | 1 man | U/Asian | 18 | N/A | 16 | 18 | VI and photophobia/12 | Retinal pigment epithelium and choriocapillaris atrophy, with visible choroidal vessels | N | CA |
| Jbour et al. [25] | 3 siblings: 1 man and 2 women | Yes/Arab | 21 (male proband) 17 14 |
N/A | 6 | 20 17 14 |
Astigmatism N/A |
Atrophic PR | CI Bilateral ptosis |
CA |
| Santos et al. [22] | 1 man | Yes/U | 34 | N/A | N/A | N/A | Night blindness | PR | CA ST2WMH |
|
| Rizzi et al. [30] | 2 brothers | No/U | 38 36 |
N/A N/A |
20 24 |
20 18 |
VI/6 VI/24 |
Chorioretinal atrophy with pigmentary changes Bilateral PR with marked choroidal atrophy |
N Bilateral pes cavus N T |
CA CA Frontal cortical atrophy |
| Rump et al. [31] | Brother and sister | No/U | 31 (male proband) 24 |
N/A N/A |
Slight, since childhood | 25 21 |
Night blindness and constricting visual fields/23 Visual field constriction/21 |
Retinal pigment epithelium and choriocapillaris atrophy in the mid-peripheral areas peripapillary atrophy and retinal pigment epithelium alterations of the maculae/23 Atrophic retinal pigment epithelium atrophy Narrow retinal vessels Bone spicule-like clumps of pigment deposition/24 |
Moderate pes cavus Hyporreflexia |
CA (vermian) |
| Salvador et al. [32] | 1 woman | N/A | 39 | N/A | 28 | 17 | Progressive VI and photophobia/37 | Retinal pigment epithelium and choriocapillaris atrophy in the posterior pole and mid-periphery | T Scanning speech |
Diffuse CA |
| Tojo et al. [5] | 2 sisters | Yes/U | 52 57 |
N/A N/A |
20 35 |
28 35 |
“Visual problems”/46 N/A |
ChD/52 ChD/57 |
T Dysarthria Dysarthria |
CA |
| Erdem et al. [23] | 1 man | Yes/U | 27 | N/A | 12 | 27 | VI/12 | Peripheral pigmented macular scars and atypical pigmentary patterns Macular pigment epithelium atrophy |
N Slow pupillary responses Scanning speech |
CA (vermian) Cortical and subcortical atrophy |
| Baroncini et al. [24] | 2 brothers | N/A | 33 31 |
N/A N/A |
25 24 |
20 18 |
VI/6 N/A |
PR/6 Chorioretinal atrophy with macular involvement and pigmentary changes/16 Fine retinal pigmentary changes and coarse macular pigmentation/6 Choroidal atrophy/30 |
T Bilateral pes cavus Brachycephaly T |
Normal CA Mild cerebral atrophy |
| Fok et al. [33] | Brother and sister | No/Chinese | 18 (male proband) 21 |
N/A N/A |
8 6 |
18 21 |
High myopia and astigmatism N/A |
Peripapillary degeneration with chorioretinal atrophy/18 N/A |
N Pendular knee jerks |
Diffuse CA Fourth ventricular dilatation |
| Limber et al. [4] | Family 1 1 woman and 1 man | No/U | 32 (female proband) 25 |
N/A N/A |
27 Early childhood (male brother) |
15 17 |
Trouble reading from a blackboard/20 Scotoma N/A |
Larger choroidal vessels sclerosis atrophic changes of the retinal pigment epithelium with coarse pigmentation PR/15 |
N T N Speech delay |
Chronic cerebellar degeneration (autopsy finding) Slight prominence of cerebellar folia |
| Family 2a 2 sisters and 2 brothers | Yes/mixed Russian– German | 53 (Neuhäuser) and 69 (Limber) (female proband) 58 (proband’s brother) Neuhäuser’s |
N/A N/A |
33–38 12 |
53 N/A |
Scotoma N/A |
Early senile macular degeneration/53 Choroidal atrophy around the disc, macular atrophic lesions/69 N/A |
CI N Ocular dysmetria T |
Normal at age 53 N/A |
|
| Boucher et al. [1] | 2 sisters | No/U | 35 15 |
N/A N/A |
34 U |
35 15 |
VI/4 VI/12 |
ChD ChD |
CI N N Moderate limitation of upgaze Areflexia |
N/A N/A |
CA cerebellar atrophy, ChD chorioretinal dystrophy, CI cognitive impairment, Dx diagnosis, HH hypogonadotropic hypogonadism, N nystagmus, N/A not available, PR pigmentary retinopathy, ST2WMH subcortical T2-weighted white matter hyperintensities, T tremor, Tx treatment, U unknown, VI visual impairment
Previously reported by Neuhäuser and Opitz in 1975. In Neuhäuser’s paper, all four siblings were reported, but only one male and one female were examined. Information on the other two siblings was retrospective
The ocular changes noted in B–N are distinctive in their pattern of progressive RPE and choriocapillaris atrophy, with macular changes in the posterior pole and mid-periphery of each eye. Deposition of brown pigment clumps in the mid-peripheral retina, known as pigmentary clumping, may also be present. Synofzik et al. [6, 10] recently published two separate reports of patients with PNPLA6 mutations including photographic examples of these chorioretinal changes and pigmentary clumping. In one of these reports few visual details were available, but in the other, fundoscopy and OCT findings of a 37-year-old woman with B–N with visual loss and ataxia were shown. OCT demonstrated retinal thinning and atrophy of choroidal vessels in our patient like in Synofzik’s, but the degree of chorioretinal dystrophy was much less severe in ours. Furthermore, our patient lacked pigmentary clumps. We find it interesting that, overall, our patient demonstrated a milder form of the chorioretinal process than previously reported, which is rather counterintuitive given our patient’s older age.
Published B–N case descriptions suggest that additional features may be present, including subcortical T2 white matter hyperintensities [7, 22], basal ganglia [3], midbrain [3], and cortical atrophy [23, 24], pyramidal tract abnormalities and hyperkinetic movements (Table 1). Cognitive decline was noted in our patient and others [1, 4, 7, 25], and we suspect it may constitute a more prominent feature than previously recognized [1]. Although our patient never underwent formal cognitive testing prior to our initial evaluation, she reported normal cognition earlier in her life, and a very clear history of recent cognitive decline. To our knowledge, previous reports of cognitive impairment in B–N patients were present early in the patients’ lives, and progressive decline has not been clearly documented [1, 4, 6, 7, 25].
PNPLA6 encodes for NTE, which is the key protein in the pathogenesis of organophosphorous (OP) compound-induced delayed neuropathy (OPIDN) [26]. In this condition, certain OP esters lead to a degeneration of long axons in the spinal cord and peripheral nerves, either through direct protein inhibition or through generation of neurotoxic complexes. Given the known role of PNPLA6 in the pathogenesis of neuropathy, it is plausible that loss of function mutations may predispose B–N patients to the development of polyneuropathy, which was largely subclinical in our patient as demonstrated only by her EMG/NCS.
NTE is also known to deacetylate intracellular phosphatidylcholine to produce glycerophosphocholine [27]. It has also been reported to avidly hydrolyze a number of lysophospholipids, indicating a role in intracellular membrane trafficking [28]. NTE is predicted to contain a single transmembrane domain, a tyrosine kinase phosphorylation site, and regions of cyclic nucleotide-binding sites (Fig. 5), the latter indicating that it may be regulated by either cAMP or cGMP [28]. Therefore, possible complex interactions between NTE and other molecules (including the transcriptase of RNF216) may give rise to the wide phenotypic spectrum associated with PNPLA6 genetic variability. Genotyping of other B–N cases is necessary to better understand PNPLA6-associated genotype–phenotype correlations.
Our patient’s family history was interesting, but of unclear significance. Since we did not have a sample of our patient’s mother’s DNA, we can only speculate about her possible carrier state. It is plausible that this heterozygous state may have predisposed her for what we can only hypothesize was a dementia with Lewy bodies phenotype. Of course, we also cannot exclude the possibility of a de novo mutation in our patient, which would make her mother’s phenotype completely coincidental.
In sum, we identified a novel B–N case with compound heterozygous PNPLA6 mutations, further confirming the causative role of PNPLA6 in B–N syndrome. Our case shows that sporadic-appearing gait ataxia beginning in the 50s can be due to PNPLA6 mutations, and that it is justified to query for a history of hypogonadism and retinal findings in adult-onset gait ataxia. Furthermore, we recognize that visual symptoms may be absent in patients with mild phenotypes, and suggest that patients in whom B–N is suspected undergo a neuroophthalmologic evaluation. Finally, our case adds weight to the assumption that PNPLA6 mutations are the leading cause of Boucher–Neuhäuser syndrome; since both of this patient’s mutations were located in the phospholipase esterase domain (where most pathogenic mutations seem to cluster, as shown in Fig. 5), we believe the phospholipase esterase domain will be the most appropriate molecular target for the development of novel therapeutic strategies.
Acknowledgments
Authors would like to thank the patient, relatives, and other participants for their contribution to this research. Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institute of Health (NIH) under Award Number R01NS079388 (CPR) and NIH K02NS073836 (RSP).
Footnotes
Ethical Standards The patient described herein has given her informed consent for this manuscript’s publication. No patient-identifying information has been included in this manuscript.
Conflicts of interest Dr. Deik has no conflicts of interest. Ms. Johannes has no conflicts of interest. Dr. Rucker has no conflicts of interest. Dr. Sánchez has no conflict of interest. Dr. Brodie has no conflicts of interest. Dr. Deegan has no conflicts of interest. Ms. Landy has no conflicts of interest. Dr. Kajiwara has no conflict of interest. Dr. Scelsa has no conflicts of interest. Dr. Saunders-Pullman has no conflicts of interest. She is funded in part through NIH K02NS073836. Dr. Paisán-Ruiz has no conflicts of interest.
Contributor Information
A. Deik, Department of Neurology, Parkinson’s Disease and Movement Disorders Center, University of Pennsylvania, 330 S. 9th Street, Philadelphia, PA 19107, USA
B. Johannes, Department of Neurology, Mount Sinai Beth Israel, 10 Union Square East, Suite 5K, New York, NY 10003, USA
J. C. Rucker, Department of Neurology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1137, New York, NY 10029, USA. Department of Ophthalmology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029, USA
E. Sánchez, Department of Neurology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1137, New York, NY 10029, USA
S. E. Brodie, Department of Ophthalmology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029, USA
E. Deegan, Department of Neurology, Mount Sinai Beth Israel, 10 Union Square East, Suite 5K, New York, NY 10003, USA
K. Landy, Icahn School of Medicine at Mount Sinai, The Graduate School of Biomedical Sciences, One Gustave L. Levy Place, New York, NY 10029, USA
Y. Kajiwara, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029, USA
S. Scelsa, Department of Neurology, Mount Sinai Beth Israel, 10 Union Square East, Suite 5K, New York, NY 10003, USA
R. Saunders-Pullman, Email: RSaunder@chpnet.org, Department of Neurology, Mount Sinai Beth Israel, 10 Union Square East, Suite 5K, New York, NY 10003, USA. Department of Neurology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1137, New York, NY 10029, USA
C. Paisán-Ruiz, Email: coro.paisan-ruiz@mssm.edu, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029, USA. Friedman Brain and Mindich Child Health and Development Institutes, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, New York, NY 10029, USA
References
- 1.Boucher BJ, Gibberd FB. Familial ataxia, hypogonadism and retinal degeneration. Acta Neurol Scand. 1969;45:507–510. [PubMed] [Google Scholar]
- 2.Neuhauser G, Opitz JM. Autosomal recessive syndrome of cerebellar ataxia and hypogonadotropic hypogonadism. Clin Genet. 1975;7:426–434. doi: 10.1111/j.1399-0004.1975.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 3.Ling H, Unnwongse K, Bhidayasiri R. Complex movement disorders in a sporadic Boucher–Neuhauser syndrome: phenotypic manifestations beyond the triad. Mov Disord. 2009;24:2304–2306. doi: 10.1002/mds.22831. [DOI] [PubMed] [Google Scholar]
- 4.Limber ER, Bresnick GH, Lebovitz RM, Appen RE, Gilbert-Barness EF, Pauli RM. Spinocerebellar ataxia, hypogonadotropic hypogonadism, and choroidal dystrophy (Boucher–Neuhauser syndrome) Am J Med Genet. 1989;33:409–414. doi: 10.1002/ajmg.1320330325. [DOI] [PubMed] [Google Scholar]
- 5.Tojo K, Ichinose M, Nakayama M, Yamamoto H, Hasegawa T, Kawaguchi Y, Sealfon SC, Sakai O. A new family of Boucher–Neuhauser syndrome: coexistence of Holmes type cerebellar atrophy, hypogonadotropic hypogonadism and retinochoroidal degeneration: case reports and review of literature. Endocr J. 1995;42:367–376. doi: 10.1507/endocrj.42.367. [DOI] [PubMed] [Google Scholar]
- 6.Synofzik M, Gonzalez MA, Lourenco CM, Coutelier M, Haack TB, Rebelo A, Hannequin D, Strom TM, Prokisch H, Kernstock C, Durr A, Schols L, Lima-Martinez MM, Farooq A, Schule R, Stevanin G, Marques W, Jr, Zuchner S. PNPLA6 mutations cause Boucher–Neuhauser and Gordon–Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014;137:69–77. doi: 10.1093/brain/awt326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kate MP, Kesavadas C, Nair M, Krishnan S, Soman M, Singh A. Late-onset Boucher–Neuhauser syndrome (late BNS) associated with white-matter changes: a report of two cases and review of literature. J Neurol Neurosurg Psychiatry. 2011;82:888–891. doi: 10.1136/jnnp.2009.196790. [DOI] [PubMed] [Google Scholar]
- 8.Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, Stevens J, Coon H, Leppert M, Fink JK. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rainier S, Albers JW, Dyck PJ, Eldevik OP, Wilcock S, Richardson RJ, Fink JK. Motor neuron disease due to neuropathy target esterase gene mutation: clinical features of the index families. Muscle Nerve. 2011;43:19–25. doi: 10.1002/mus.21777. [DOI] [PubMed] [Google Scholar]
- 10.Synofzik M, Kernstock C, Haack TB, Schols L. Ataxia meets chorioretinal dystrophy and hypogonadism: Boucher–Neuhauser syndrome due to PNPLA6 mutations. J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2014-307793. [DOI] [PubMed] [Google Scholar]
- 11.Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10:e1003440. doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehtonen MS, Moilanen JS, Majamaa K. Increased variation in mtDNA in patients with familial sensorineural hearing impairment. Hum Genet. 2003;113:220–227. doi: 10.1007/s00439-003-0966-9. [DOI] [PubMed] [Google Scholar]
- 13.Thomas AW, Edwards A, Sherratt EJ, Majid A, Gagg J, Alcolado JC. Molecular scanning of candidate mitochondrial tRNA genes in type 2 (non-insulin dependent) diabetes mellitus. J Med Genet. 1996;33:253–255. doi: 10.1136/jmg.33.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Exome Variant Server. NHLBI Exome Sequencing Project (ESP) Seattle, WA: 2014. [09/2014]. ( http://evs.gs.washington.edu/EVS/) [Google Scholar]
- 15.Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, Coryell JC, Nguyen TM, Nardocci N, Zorzi G, Rodriguez D, Desguerre I, Bertini E, Simonati A, Levinson B, Dias C, Barbot C, Carrilho I, Santos M, Malik I, Gitschier J, Hayflick SJ. Neurodegeneration associated with genetic defects in phospholipase A(2) Neurology. 2008;71:1402–1409. doi: 10.1212/01.wnl.0000327094.67726.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krebs CE, Paisan-Ruiz C. The use of next-generation sequencing in movement disorders. Front Genet. 2012;3:75. doi: 10.3389/fgene.2012.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y, Wang M, Mao F, Shao M, Zhao B, Song Z, Shao C, Gong Y. Knockdown of PNPLA6 protein results in motor neuron defects in zebrafish. Dis Model Mech. 2013;6:404–413. doi: 10.1242/dmm.009688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Read DJ, Li Y, Chao MV, Cavanagh JB, Glynn P. Neuropathy target esterase is required for adult vertebrate axon maintenance. J Neurosci. 2009;29:11594–11600. doi: 10.1523/JNEUROSCI.3007-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margolin DH, Kousi M, Chan YM, Lim ET, Schmahmann JD, Hadjivassiliou M, Hall JE, Adam I, Dwyer A, Plummer L, Aldrin SV, O’Rourke J, Kirby A, Lage K, Milunsky A, Milunsky JM, Chan J, Hedley-Whyte ET, Daly MJ, Katsanis N, Seminara SB. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinton R, Barnett P, Coskeran P, Bouloux PM. Gordon–Holmes spinocerebellar ataxia: a gonadotropin deficiency syndrome resistant to treatment with pulsatile gonadotropin-releasing hormone. Clin Endocrinol (Oxf) 1999;51:525–529. doi: 10.1046/j.1365-2265.1999.00859.x. [DOI] [PubMed] [Google Scholar]
- 22.Santos AV, Saraiva PF, Breia PN. Significance of neuroimaging in the diagnosis of Boucher–Neuhauser syndrome. Acta Med Port. 2003;16:193–195. [PubMed] [Google Scholar]
- 23.Erdem E, Kiratli H, Erbas T, Varli K, Eldem B, Akalin S, Tan E, Topaloglu H, Gedikoglu G. Cerebellar ataxia associated with hypogonadotropic hypogonadism and chorioretinopathy: a poorly recognized association. Clin Neurol Neurosurg. 1994;96:86–91. doi: 10.1016/0303-8467(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 24.Baroncini A, Franco N, Forabosco A. A new family with chorioretinal dystrophy, spinocerebellar ataxia and hypogonadotropic hypogonadism (Boucher–Neuhauser syndrome) Clin Genet. 1991;39:274–277. doi: 10.1111/j.1399-0004.1991.tb03025.x. [DOI] [PubMed] [Google Scholar]
- 25.Jbour AK, Mubaidin AF, Till M, El-Shanti H, Hadidi A, Ajlouni KM. Hypogonadotropic hypogonadism, short stature, cerebellar ataxia, rod-cone retinal dystrophy, and hypersegmented neutrophils: a novel disorder or a new variant of Boucher–Neuhauser syndrome? J Med Genet. 2003;40:e2. doi: 10.1136/jmg.40.1.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emerick GL, Peccinini RG, de Oliveira GH. Organophosphorus-induced delayed neuropathy: a simple and efficient therapeutic strategy. Toxicol Lett. 2010;192:238–244. doi: 10.1016/j.toxlet.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 27.Zaccheo O, Dinsdale D, Meacock PA, Glynn P. Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J Biol Chem. 2004;279:24024–24033. doi: 10.1074/jbc.M400830200. [DOI] [PubMed] [Google Scholar]
- 28.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res. 2006;47:1940–1949. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Yu SI, Kim JL, Lee SG, Kim HW, Kim SJ. Ophthalmologic findings of Boucher–Neuhauser syndrome. Korean J Ophthalmol. 2008;22:263–267. doi: 10.3341/kjo.2008.22.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rizzi R, Carelli V, Monari L, Mochi M, Liguori R, Sensi M, Cocozza S, Filla A, Montagna P. Cerebellar ataxia, hypogonadism and chorioretinopathy: molecular analysis of an Italian family. Ital J Neurol Sci. 1998;19:41–44. doi: 10.1007/BF03028811. [DOI] [PubMed] [Google Scholar]
- 31.Rump P, Hamel BC, Pinckers AJ, van Dop PA. Two sibs with chorioretinal dystrophy, hypogonadotropic hypogonadism, and cerebellar ataxia: Boucher–Neuhauser syndrome. J Med Genet. 1997;34:767–771. doi: 10.1136/jmg.34.9.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvador F, Garcia-Arumi J, Corcostegui B, Minoves T, Tarrus F. Ophthalmologic findings in a patient with cerebellar ataxia, hypogonadotropic hypogonadism, and chorioretinal dystrophy. Am J Ophthalmol. 1995;120:241–244. doi: 10.1016/s0002-9394(14)72612-1. [DOI] [PubMed] [Google Scholar]
- 33.Fok AC, Wong MC, Cheah JS. Syndrome of cerebellar ataxia and hypogonadotropic hypogonadism: evidence for pituitary gonadotrophin deficiency. J Neurol Neurosurg Psychiatry. 1989;52:407–409. doi: 10.1136/jnnp.52.3.407. [DOI] [PMC free article] [PubMed] [Google Scholar]