

Abstract
The aim of this study is to investigate the potential of nanostructured lipid carriers (NLCs) in improving the oral bioavailability of a lipid lowering agent, fenofibrate (FEN). FEN-loaded NLCs (FEN-NLCs) were prepared by hot homogenization followed by an ultrasonication method using Compritol 888 ATO as a solid lipid, Labrafil M 1944CS as a liquid lipid, and soya lecithin and Tween 80 as emulsifiers. NLCs were characterized in terms of particle size and zeta pote\ntial, surface morphology, encapsulation efficiency, and physical state properties. Bioavailability studies were carried out in rats by oral administration of FEN-NLC. NLCs exhibited a spherical shape with a small particle size (84.9 ± 4.9 nm). The drug entrapment efficiency was 99% with a loading capacity of 9.93 ± 0.01% (w/w). Biphasic drug release manner with a burst release initially, followed by prolonged release was depicted for in vitro drug release studies. After oral administration of the FEN-NLC, drug concentration in plasma and AUCt-∞ was fourfold higher, respectively, compared to the free FEN suspension. According to these results, FEN-NLC could be a potential delivery system for improvement of loading capacity and control of drug release, thus prolonging drug action time in the body and enhancing the bioavailability.
KEY WORDS: bioavailability, fenofibrate, nanoparticles, nanostructured lipid carriers
Introduction
Fenofibrate (FEN), a isopropyl ester of 2-(4-[4-chloro-benzoyl] phenoxy)-2-methylpropanoic acid been used regularly to reduce elevated levels of cholesterol, low-density lipoprotein cholesterol, apolipoprotein B, total triglycerides, and triglyceride-rich very low-density lipoprotein (1,2). Specifically, it is used alone or in combination with statins in treatment of hypercholesterolemia and hypertriglyceridemia. FEN belongs to BCS class II drug (low aqueous solubility and high permeability) with high lipophilicity (log p = 5.24) (3). This leads to low aqueous solubility and poor gastrointestinal absorption, resulting in low, erratic, and highly variable inter- and intra-subject bioavailability.
Several approaches have been investigated in an effort to improve the dissolution and bioavailability profile of FEN, including nanocrystals, SMEDDS, nanosuspension, and solid lipid nanoparticles (SLNs) (4–7). Among them, physiological lipid nanocarriers, such as SLNs, have been a subject of great interest within the scientific community due to their ability to evade the hepatic first-pass metabolism and enhance intestinal permeability (8,9). SLNs combine the unique advantages of other colloidal carriers, including physical stability, protection of incorporated sensitive drug molecules from degradation, and longer blood circulation time (10). Despite of several above advantages, SLNs have to face some challenger such as relatively low drug loading capacity, associating with gelation, drug leakage, and possible expulsion of the drug during storage (11). These problems were resolved with introduction of nanostructured lipid carriers (NLCs), a second generation of SLNs. NLCs have a solid matrix blended with a liquid lipid (oil) to form an unstructured matrix that improved the drug loading capacity of nanoparticles and reduced drug expulsion from the matrix during storage (12,13). With more imperfection of the crystal structure and lack of polymorphisms compared to SLNs, NLCs have found wide applications in oral and parenteral delivery systems. In addition, passage of NLCs through multiple biological barriers and efficient transport the therapeutic moiety via the lymphatic pathway for enhancement of the oral bioavailability has been demonstrated (14).
Therefore, the aim of this work was to evaluate NLC as a tool for enhancement of the oral bioavailability of a poorly water-soluble compound, FEN. To validate this aim, Compritol 888 ATO and Labrafil M 1944 CS were chosen as respective solid and liquid lipids for formulation of FEN-loaded NLCs (FEN-NLCs). Because nanoparticle size and surface properties are limiting factors in transport across the intestinal barrier, we have studied the influence of Compritol/Labrafil ratios in terms of size, polydispersity, and charge. In addition, this investigation reports on the morphological analysis, solid state characterization, drug release kinetics, and in vivo pharmacokinetic study in order to establish FEN-NLC as a suitable oral delivery system.
Materials and Methods
Materials
Fenofibrate and fenofibric acid were a generous gift from Dong-A Pharm. Co. (Yongin, South Korea). Compritol 888 ATO (Compritol) and Labrafil M 1944 CS (Labrafil) were obtained from Gattefosse (Saint-Priest Cedex, France). Polysorbate 80 (Tween 80) was purchased from DC Chemical Co. (Seoul, South Korea). All other chemicals were of analytical grade and used without further purification.
Preparation of Nanostructured Lipid Carriers (NLCs)
FEN-loaded nanostructured lipid carriers (FEN-NLCs) were prepared by hot homogenization followed by an ultrasonication method (8). The oil phase, consisting of Compritol, Labrafil, and Lecithin, was heated to 80°C. The aqueous phase, including Tween 80, was heated to the same temperature and then added to the lipid phase and mixed using an Ultra-Turrax® T-25 homogenizer (IKA®-Werke, Staufen, Germany) at 13,500 rpm for 3 min. Then, the coarse emulsion was ultra-sonicatedusing a probe sonicator (Vibracell VCX130; Sonics, USA). The resulting suspensions were cooled down in ice bath. The compositions of the NLCs are shown in Table I.
Table I.
Composition of NLC Formulations
| Formulations | Fenofibarte (mg) | Compritol (mg) | Labrafil (mg) | Lecithin (mg) | Tween 80 (mg) | Distilled water (mL) |
|---|---|---|---|---|---|---|
| N0 | − | 500 | − | 500 | 500 | 15 |
| N10 | − | 450 | 50 | 500 | 500 | 15 |
| N20 | − | 400 | 100 | 500 | 500 | 15 |
| N30 | − | 350 | 150 | 500 | 500 | 15 |
| N40 | − | 300 | 200 | 500 | 500 | 15 |
| N50 | − | 250 | 250 | 500 | 500 | 15 |
| dN0 | 50 | 500 | − | 500 | 500 | 15 |
| dN10 | 50 | 450 | 50 | 500 | 500 | 15 |
| dN20 | 50 | 400 | 100 | 500 | 500 | 15 |
| dN30 | 50 | 350 | 150 | 500 | 500 | 15 |
| dN40 | 50 | 300 | 200 | 500 | 500 | 15 |
| dN50 | 50 | 250 | 250 | 500 | 500 | 15 |
Measurements of Particle Size and Zeta Potential
Particle size (nm), polydispersity index (PDI), and ζ-potential (ZP, mV) of NLCs were determined at 25°C by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) equipped with a He–Ne laser operated at a wavelength of 635 nm. Measurements were performed at a fixed scattering angle of 90°. Analysis of particle size, polydispersity index, and ζ-potential was performed using manufacturer-supplied software (Nano DTS software, version 6.34) that employs cumulant analysis. After performing measurements at least three times, we calculated means for each variable.
Encapsulation Efficiency and Loading Capacity
The unbound FEN was separated by ultrafiltration centrifugation. Briefly, 1 mL of FEN-NLC dispersion was placed in the upper chamber of a centrifuge tube matched with an ultrafilter (Amicon ultra, MWCO 10 kDa, Millipore Co., USA) and centrifuged for 15 min at 5,000 rpm. The filtrate was analyzed using the HPLC method. HPLC conditions were as follows: the mobile phase, a mixture of acetonitrile and phosphate buffer (90:10, v/v), pH 2.5, was filtered through a 0.22-μm membrane filter and eluted at 286 nm with a flow rate of 1.0 mL/min. The drug loading content was the ratio of incorporated drug to lipid (w/w). Drug loading capacity (LC) and entrapment efficiency (EE) were calculated using equations:
Where mdrug in NLC = minitial drug − mfree drug is the mass of FEN incorporated in NLC, minitial drug is the mass of total FEN added, and mlipid is the mass of lipid added to make NLC.
Morphological Analysis
The morphology of FEN-NLC was examined by transmission electron microscopy (H7600, Hitachi, Japan). Samples were prepared by negative staining with a drop of 1% aqueous solution of acid phosphotungstic for contrast enhancement and then a drop of that was placed onto a copper grid and air-dried. The morphology was further confirmed by scanning electron microscopy (SEM; S-4100, Hitachi, Japan). The lyophilized powders were fixed on a brass stub using double-sided adhesive tape, and a platinum coating (6 nm/min) was applied using Hitachi Ion Sputter (E-1030) for 120 s at 15 mA at a vacuum of 6 Pa.
Solid State Characterization of Lyophilized FEN-NLC
Differential scanning calorimetry analysis was performed using a DSC-Q200 differential scanning calorimeter (TA Instruments, USA). All formulation ingredients and freeze-dried FEN-NLC were placed in aluminum pans, and the temperature was increased from 40°C to 120°C at a rate of 10°C/min under a dynamic nitrogen atmosphere with a flow rate of 50 mL/min. An empty pan was used as a reference.
In addition, crystalline structure of the lyophilized FEN-NLC was investigated using an X-ray diffractometer (X’Pert PRO MPD diffractometer, Almelo, the Netherlands) with a copper anode (Cu Kα radiation, 40 kV, 30 mA, λ = 0.15418). The data used were typically collected with a step width of 0.04° with a detector resolution in 2θ (diffraction angle) between 10°C and 60°C at an ambient temperature.
Fourier-transform infrared spectroscopy was conducted in order to obtain conformational information on the lipid molecules. The spectra were recorded using a Thermo Scientific Nicolet Nexus 670 FT-IR Spectrometer and Smart iTR with a diamond window (Thermo Fisher Scientific Inc., Waltham, MA). The spectra were employed at 550–4,000 cm−1 and obtained at a resolution of 4 cm−1 and at 32 scans per sample. Background scanning and correction were performed before each measurement, and all measurements were performed thrice at room temperature.
In Vitro Drug Release
In vitro release study was performed on NLCs dispersion within 24 h of preparation. One milliliter of dispersion was poured to a dialysis tube (molecular weight cutoff 3,500, Membra-Cel®, Viskase, USA), and the sealed tube was introduced into a vial containing 35 mL of distilled water. Samples were horizontally shaken in a shaker (HST-205 SW, Hanbaek ST Co., South Korea) at 37 ± 1°C and 100 rpm. At predetermined time intervals (0.5, 1, 2, 4, 6, 12, and 24 h), a 0.5-mL sample of the medium was taken and replaced with the same amount of fresh medium. The amount of FEN released from the NLC was evaluated using the HPLC method described above.
Lyophilization of FEN-NLC
Lyophilization of FEN-NLC was performed using trehalose as a cryoprotectant. The dispersion was pre-frozen (−80°C) for 12 h and subsequently lyophilized at a temperature of −25°C for 24 h followed by a secondary drying phase of 12 h at 20°C.
Stability Study
Optimized formulation was stored at three different conditions (4°C, room temperature and after lyophilization) for 3 months. Physical stability was determined on the day of production and on the 90th day.
Pharmacokinetic Study
Twelve Sprague–Dawley (SD) rats (250 ± 50 g) were randomly divided into groups; each group included six rats. The animals were quarantined in an animal house maintained at 25 ± 2°C and 50–60% RH, and fasted for 12 h prior to conduct of the experiments. The protocols for the animal studies were approved by the Institutional Animal Ethical Committee, Yeungnam University, South Korea. FEN-NLC and FEN suspension (1% PVP-K30) were administrated at a dose of 25 mg/kg as FEN via oral route. Blood samples were taken from the right femoral artery at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 6, 12, 18, and 24 h after administration and transferred to heparinized micro tubes. Then, the heparinized blood was centrifuged for 10 min at 10,000 rpm, and the plasma obtained was stored frozen at −20°C until analysis. Drug concentration in plasma was evaluated using the HPLC method via analysis of fenofibric acid—active form and major metabolite of fenofibrate after oral administration. Briefly, drug was extracted with acetonitrile solution then injected to HPLC using mobile phase was composed of a mixture of 0.02 M phosphoric acid and acetonitrile (45:55%, v/v), pH 3.0 at wavelength 287 nm with a flow rate of 1.2 mL/min (1). Standard non-compartmental analysis was performed for determination of the pharmacokinetic parameters, including maximum plasma concentration (Cmax), time taken for its occurrence (Tmax), half-life (t1/2), area under the curve from 0 to infinity (AUC0-∞), and the elimination rate constant (Kel) for each rat using the standard edition of WinNonlinTM, version 2.1 (Pharsight Corp., Mountain View, CA, USA). Student t tests were performed for evaluation of significant differences between the two formulations. Values are reported as mean ± S.D. and the data were considered statistically significant at p < 0.05.
Results and Discussion
Preparation of Fenofibrate-Loaded NLCs
FEN-NLCs were prepared by hot homogenization followed by an ultrasonication method. Different blends of solid lipid and liquid lipid (Labrafil) were optimized in order to obtain weight proportion with a uniform nano-sized particle. The average diameters, ζ-potential, and polydispersity index of resulting NLCs are shown in Fig. 1. As can be seen, nanoparticle size was increased with the increasing amount of Labrafil, followed by a decrease in size when the proportion of liquid lipid exceeded 10% w/w. It can be assumed that the solid lipid particles can encapsulate up to 10% w/w of liquid lipid, and the initial increase in size was due to the swollen core of the particles loaded with liquid oil. Afterward, the particle size showed a dramatic decrease and remained constant for the further addition of Labrafil up to 50% w/w. This could be due to the fact that excess oil was excluded during the lipid crystallization process. In addition, reduction in viscosity and surface tension level with the higher Labrafil content was responsible for the smaller particle sizes. A similar phenomenon was reported during the formation of drug-loaded NLCs (15,16). Trends similar to those of blank NP were observed for FEN-NLC, and addition of FEN had a negligible effect on the size and PDI of blank NP. This could be due to the better dispersity of FEN in solid lipid as well as liquid lipid, which resulted in unchanged particle sizes.
Fig. 1.
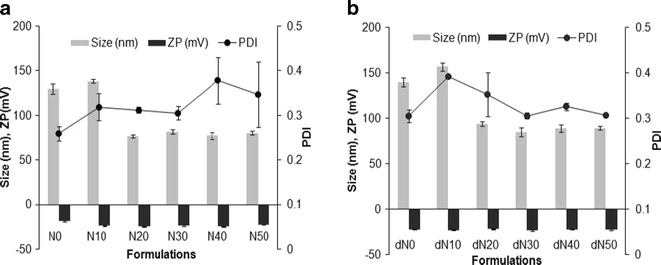
Particle size, polydispersity index and ζ-potential of various NLC formulations. Data represent mean ± standard deviation (n = 3)
An important aspect of nanoparticles as ideal drug delivery carriers is their drug loading capacity. In this regard, effects of Labrafil concentration on drug entrapment efficiency were investigated and are shown in Table II. It is clear that the entrapment efficiency of nanoparticles increased from ∼90% to ∼100% upon increasing the percentage of Labrafil from 0% to 50% w/w. Thus, incorporation of Labrafil in lipid matrix resulted in higher EE, as compared to the one without liquid lipid. This could be due to the high lipophilic nature (log p ∼ 5.24) of FEN, which could be easily incorporated into the lipid matrix. Compritol itself is a long chain fatty acid, facilitating the entrapment of FEN in NLC structure (8,17). In addition, among various liquid lipids, Labrafil showed the highest solubility for FEN (1). Based on these results, dN30 was selected as an optimized formulation, which consisted of 50 mg FEN incorporated into 350 mg Compritol, 150 mg Labrafil as a lipid core, and lipid nanoparticles were emulsified by 500 mg lecithin—a lipophilic surfactant and 500 mg Tween 80—a hydrophilic surfactant in15 mL water.
Table II.
Entrapment Efficiency (EE) and Loading Capacity (LC) of NLCs
| Formulations | EE (%) | LC (%) |
|---|---|---|
| dNLC-0 | 92.73 ± 1.17 | 9.27 ± 0.12 |
| dNLC-10 | 94.68 ± 0.83 | 9.47 ± 0.08 |
| dNLC-20 | 98.00 ± 0.33 | 9.80 ± 0.03 |
| dNLC-30 | 99.31 ± 0.09 | 9.93 ± 0.01 |
| dNLC-40 | 99.51 ± 0.03 | 9.95 ± 0.01 |
| dNLC-50 | 99.60 ± 0.06 | 9.96 ± 0.01 |
Morphological Analysis
Figures 2 and 3 show TEM and SEM images of the FEN-NLC. As indicated in these figures, particle diameters were consistent with the results obtained by DLS characterization and depicted a mono-dispersed spheroid-like appearance with a distinct boundary between each particle. The mean diameters of lipid particles were around ∼100 nm, with no visible aggregation of particles. SEM image depicted large particles of even-surfaced rectangular crystals for pure FEN powder, corresponding to the crystalline nature (Fig. 3a). However, the morphology of the FEN-NLC showed a uniform and spherical shape with a clear absence of crystalline nature of free drug (Fig. 3b).
Fig. 2.

TEM image of FEN-NLC. Composition of FEN-NLC was dN30
Fig. 3.

SEM images of a FEN powder and b Lyophilized FEN-NLC. Composition of FEN-NLC was dN30
Solid State Characterization
DSC thermograms of pure FEN, Compritol, blank NLC, and FEN-NLC are presented in Fig. 4. Crystalline FEN showed a sharp endothermic peak at 81°C, corresponding to its melting point (4). However, the thermograms of FEN-NLC did not show any peak for FEN, indicating that the drug is present in either an amorphous state or a molecularly dispersed state. On the other hand, due to colloidal size range of the lipid particles, a remarkably lower endothermic peak of lipid was observed in blank NLC and FEN-NLC compared with that of pure Compritol (72°C). In addition, shift in melting point of NLC may be attributed to the interaction of solid lipid with the liquid lipid during the preparation process. A less ordered crystal or amorphous lipid matrix would be favorable for encapsulating greater amounts of drug (18). Figure 5 shows the XRD patterns of Compritol, FEN, blank NLC, and FEN-NLC formulation. Consistent with the DSC thermogram, XRD patterns showed numerous diffraction peaks for pure FEN, corresponding to its crystalline structure. Such characteristic patterns were absent in the diffraction peaks of FEN-NLC, indicating the presence of drug in the amorphous form, withstanding the DSC data.
Fig. 4.
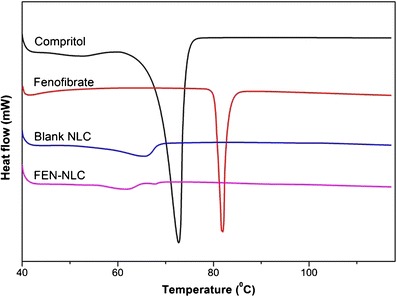
Differential scanning calorimetric thermograms of Compritol, FEN, blank NLC, and FEN-NLC. Composition of FEN-NLC was dN30
Fig. 5.
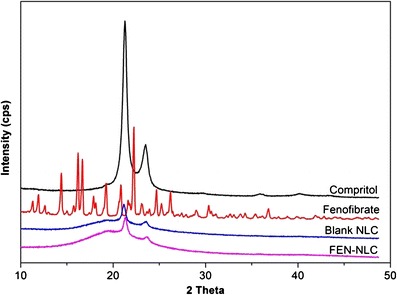
X-ray diffraction patterns of Compritol, FEN, blank NLC, and FEN-NLC. Composition of FEN-NLC was dN30
FTIR studies were performed in order to determine any physical or chemical interaction between FEN and Compritol (Fig. 6). FTIR spectrum of FEN showed typical absorption bands at 1,725.57, 1,648.43, 1,300.80, and 2,983.46 cm−1 for C=O stretching of the ester group, ketone group, ether group, and aromatic stretching, respectively (19). Similarly, Compritol showed absorption bands of C–H and C=O stretching at 2,815 and 1,738 cm−1, respectively. FEN-NLC showed peaks at 1,727.16, 1,749.54, and 3,000.25 cm−1 for ester, ketone, and aromatic stretching, respectively. These typical absorption bands clearly indicated the absence of any physical or chemical interaction between Compritol and the drug.
Fig. 6.
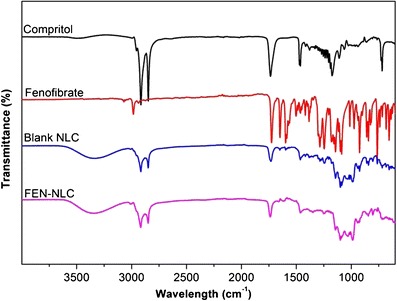
Fourier-transform infrared spectroscopy spectra of Compritol, FEN, blank NLC, and FEN-NLC. Composition of FEN-NLC was dN30
In Vitro Release
The in vitro release profile of FEN-NLC and FEN suspension is displayed in Fig. 7. As can be seen in case of FEN-NLC, a bi-phasic release pattern was observed with an initial burst release of drug, followed by sustained release of drug at a constant rate. Approximately ∼60% of drug was released from formulations in the first 4 h, followed by relatively slower release up to 24 h. The initial burst release might be attributed to a drug enriched shell model of incorporation of FEN into the NLC system. A significant proportion of active drug remains in the outer shell during the supersaturation step, leading to a short diffusion path of the drug to release media. In addition, a high amount of surfactant used in the formulation accelerates the release of drug in the media (20). In the second stage, FEN is released from the deeper lipid matrix/core in a sustained manner culminating in diffusion and erosion mechanisms (16,21). Furthermore, FEN has no ionizable group so its solubility and dissolution is pH-independent (22). On the other hand, FEN suspension showed dissolution rate very low due to high crystalline state of plain drug that consistent with DSC and XRD data.
Fig. 7.
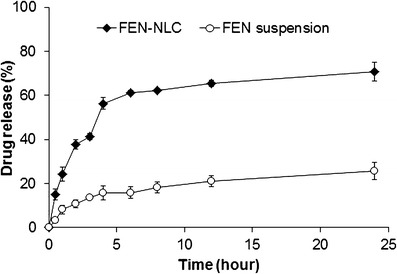
In vitro drug release of FEN from FEN-NLC and FEN suspension in distilled water. Composition of FEN-NLC was dN30. Each value represents the mean ± standard deviation (n = 3)
Stability Study
The stability of NLC formulations was ascertained by monitoring the particle size, PDI, and surface charge under different conditions. FEN-NLC maintained its colloidal stability under both temperature conditions and the particle size remained the same. Similarly, all of the formulations exhibited good storage stability for at least 3 months (data not shown). In addition, lyophilized FEN-NLC (85.4 ± 3.5 nm, −22.6 ± 1.7 mV) maintained its physical properties and maintained size and charge characteristics similar to those of the freshly prepared one (84.9 ± 4.9 nm, −23.4 ± 0.8 mV). Trehalose was used as a cryoprotectant in order to shield the nanoparticles during the freeze-drying process. These results indicate the tremendous storage stability of NLC formulations.
Oral Bioavailability
The plasma concentration–time profiles of FEN suspension and FEN-NLC after oral administration to rats are shown in Fig. 8. At all points, significantly higher FEN plasma concentrations were observed (p < 0.05) for rats applied with FEN-NLC than for those applied with FEN suspension. Table III shows the respective pharmacokinetic parameters (Cmax, Tmax, and AUC0–∞). As can be seen, Cmax (21.72 μg/mL) and AUC0-∞ (400.03 μg · h/mL) of FEN-NLCs were four times higher than those of the FEN suspension. These results clearly suggest that incorporation of FEN into NLCs resulted in augmentation orally absorption of FEN. The improved bioavailability by the NLC-modified formulation could be attributed to the efficient uptake of nanoparticles through the GI tract, increased GI permeability by surfactants and higher dissolution rate, reduced degradation. In addition, particle size of NLC played an important role in the absorption rate (23). Due to their small particle size, NLCs might have exhibited enhanced adhesion to the GI wall or entered the inter-villar spaces and prolong interaction time on the GI tract and enhanced bioavailability (24). In the present study, average particle size of nanoparticles was maintained below the optimum range of 200 nm, which led to bypassing of liver first pass metabolism. Moreover, Tween 80 and lecithin in the NLC preparation act as surfactants might account for the increased permeability of the intestinal membrane (25). During the NLC formation, a greater proportion of FEN accumulates in the core lipid matrix, which not only reduces enzymatic degradation during the absorption process but also offers long circulation time in vivo (26). In addition, owing to the high hydrophobicity of glyceride fatty acid, lymphatic uptake of FEN-NLC could be expected (17,27). Therefore, the in vivo pharmacokinetic profile unambiguously demonstrated that the NLC could be a suitable prospect for improvement of the oral bioavailability of FEN.
Fig. 8.
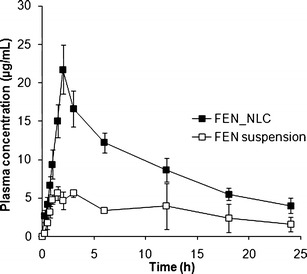
Plasma concentration–time profile of FEN after oral administration of FEN suspension (square) and FEN-NLC (filled square) at a dose to 25 mg/kg to rats. Composition of FEN-NLC was dN30. Data are expressed as the mean ± standard deviation (n = 6)
Table III.
Pharmacokinetic Parameters of FEN After Oral Administration of FEN Suspension and FEN-NLC to Rats
| Parameters | FEN suspensiona | FEN-NLC |
|---|---|---|
| C max (μg/mL) | 5.38 ± 0.45 | 21.72 ± 2.53* |
| T max (h) | 2.8 ± 0.5 | 2.0 ± 0.0 |
| t 1/2 (h) | 10.4 ± 2.9 | 30.96 ± 7.29 |
| K el (h−1) | 0.056 ± 0.018 | 0.023 ± 0.005 |
| AUC0-∞ (h.μg/mL) | 97.10 ± 18.82 | 400.03 ± 93.15* |
Each value represents the mean ± S.D. (n = 6)
aModified from reference (1)
*p < 0.05, compared with the drug suspension
Conclusions
In this study, FEN-NLCs were successfully prepared and achieved for their potential to increase drug permeability across the gastrointestinal barrier. Solid lipid to liquid lipid ratio was investigated to optimize the suitable NLC, in terms of particle size, PDI, and ζ-potential. The optimized FEN-NLC presented particles in nanometric size range with good physical stability and high encapsulation capacity. Solid state characterizations were performed to confirm the presence of drug in the amorphous and molecularly dispersed state, which would in turn enhance the in vivo performance. The higher Cmax levels, along with higher AUC (fourfold) of NLC formulations in plasma, clearly proved distinct enhancement in rate and extent of drug bioavailability. This remarkable augmentation in bioavailability would lately result in a progress in the intensity of the therapeutic effect of FEN. This study can be potentially explored for development of appropriate platform technologies for improvement of the bioavailability of other BCS class II drugs, which correspond to the undergoing extensive hepatic first-pass effect, P-gp efflux, and/or restricted permeation.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (No. 2012R1A2A2A02044997 and No. 2012R1A1A1039059).
Contributor Information
Chul Soon Yong, Phone: +82-53-810-2812, Email: csyong@yu.ac.kr.
Jong Oh Kim, Phone: +82-53-810-2813, Email: jongohkim@yu.ac.kr.
References
- 1.Lee DW, Marasini N, Poudel BK, Kim JH, Cho HJ, Moon BK, et al. Application of Box-Behnken design in the preparation and optimization of fenofibrate-loaded self-microemulsifying drug delivery system (SMEDDS) J Microencapsul. 2014;31(1):31–40. doi: 10.3109/02652048.2013.805837. [DOI] [PubMed] [Google Scholar]
- 2.Zuo B, Sun Y, Li H, Liu X, Zhai Y, Sun J, et al. Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals. Int J Pharm. 2013;455(1):267–75. doi: 10.1016/j.ijpharm.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 3.Niu X, Wan L, Hou Z, Wang T, Sun C, Sun J, et al. Mesoporous carbon as a novel drug carrier of Fenofibrate for enhancement of the dissolution and oral bioavailability. Int J Pharm. 2013;452(1–2):382–9. doi: 10.1016/j.ijpharm.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 4.Kim GG, Poudel BK, Marasini N, Lee DW, Hiep TT, Yang KY, et al. Enhancement of oral bioavailability of fenofibrate by solid self-microemulsifying drug delivery systems. Drug Dev Ind Pharm. 2013;39(9):1431–8. doi: 10.3109/03639045.2012.719903. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Gu L, Xu Y, Wang Y. Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behavior in rats. Drug Dev Ind Pharm. 2009;35(7):827–33. doi: 10.1080/03639040802623941. [DOI] [PubMed] [Google Scholar]
- 6.Hanafy A, Spahn-Langguth H, Vergnault G, Grenier P, Tubic Grozdanis M, Lenhardt T, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59(6):419–26. doi: 10.1016/j.addr.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 7.He H, Yang R, Tang X. in vitro and in vivo evaluation of fenofibrate solid dispersion prepared by hot-melt extrusion. Drug Dev Ind Pharm. 2010;36(6):681–7. doi: 10.3109/03639040903449720. [DOI] [PubMed] [Google Scholar]
- 8.Tran TH, Ramasamy T, Cho HJ, Kim YI, Poudel BK, Choi HG, et al. Formulation and optimization of raloxifene-loaded solid lipid nanoparticles to enhance oral bioavailability. J Nanosci Nanotechnol. 2014;14(7):4820–31. [DOI] [PubMed]
- 9.Ramasamy T, Khandasami US, Ruttala H, Shanmugam S. Development of solid lipid nanoparticles enriched hydrogels for topical delivery of anti-fungal agent. Macromol Res. 2012;20(7):682–92. doi: 10.1007/s13233-012-0107-1. [DOI] [Google Scholar]
- 10.Subedi RK, Kang KW, Choi H-K. Preparation and characterization of solid lipid nanoparticles loaded with doxorubicin. Eur J Pharm Sci. 2009;37(3):508–13. doi: 10.1016/j.ejps.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Ramasamy T, Tran TH, Choi JY, Cho HJ, Kim JH, Choi HG, et al. Layer-by-layer coated lipid-polymer hybrid nanoparticles designed for use in anticancer drug delivery. Carbohydr Polym. 2014;102:653–61. doi: 10.1016/j.carbpol.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Beloqui A, Solinís MÁ, Gascón AR, del Pozo-Rodríguez A, des Rieux A, Préat V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release. 2013;166(2):115–23. doi: 10.1016/j.jconrel.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 13.Zhuang C-Y, Li N, Wang M, Zhang X-N, Pan W-S, Peng J-J, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394(1):179–85. doi: 10.1016/j.ijpharm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Khan AA, Mudassir J, Mohtar N, Darwis Y. Advanced drug delivery to the lymphatic system: lipid-based nanoformulations. Int J Nanomedicine. 2013;8:2733–44. doi: 10.2147/IJN.S41521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agrawal Y, Petkar KC, Sawant KK. Development, evaluation and clinical studies of Acitretin loaded nanostructured lipid carriers for topical treatment of psoriasis. Int J Pharm. 2010;401(1):93–102. doi: 10.1016/j.ijpharm.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 16.Hu F-Q, Jiang S-P, Du Y-Z, Yuan H, Ye Y-Q, Zeng S. Preparation and characterization of stearic acid nanostructured lipid carriers by solvent diffusion method in an aqueous system. Colloids Surf B: Biointerfaces. 2005;45(3):167–73. doi: 10.1016/j.colsurfb.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Aji Alex M, Chacko A, Jose S, Souto E. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42(1):11–8. doi: 10.1016/j.ejps.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Lin X, Li X, Zheng L, Yu L, Zhang Q, Liu W. Preparation and characterization of monocaprate nanostructured lipid carriers. Colloids Surf A Physicochem Eng Asp. 2007;311(1):106–11. doi: 10.1016/j.colsurfa.2007.06.003. [DOI] [Google Scholar]
- 19.Ige PP, Baria RK, Gattani SG. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf B: Biointerfaces. 2013;108:366–73. doi: 10.1016/j.colsurfb.2013.02.043. [DOI] [PubMed] [Google Scholar]
- 20.Huang X, Chen Y-J, Peng D-Y, Li Q-L, Wang X-S, Wang D-L, et al. Solid lipid nanoparticles as delivery systems for Gambogenic acid. Colloids Surf B: Biointerfaces. 2013;102:391–7. doi: 10.1016/j.colsurfb.2012.08.058. [DOI] [PubMed] [Google Scholar]
- 21.Abdelbary G, Fahmy RH. Diazepam-loaded solid lipid nanoparticles: design and characterization. AAPS PharmSciTech. 2009;10(1):211–9. doi: 10.1208/s12249-009-9197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (Self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9(3):E344–52. doi: 10.1208/aapsj0903041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hussain N, Jaitley V, Florence AT. Recent advances in the understanding of uptake of microparticulates across the gastrointestinal lymphatics. Adv Drug Deliv Rev. 2001;50(1):107–42. doi: 10.1016/S0169-409X(01)00152-1. [DOI] [PubMed] [Google Scholar]
- 24.Vasir JK, Tambwekar K, Garg S. Bioadhesive microspheres as a controlled drug delivery system. Int J Pharm. 2003;255(1):13–32. doi: 10.1016/S0378-5173(03)00087-5. [DOI] [PubMed] [Google Scholar]
- 25.Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114(1):53–9. doi: 10.1016/j.jconrel.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Zhang T, Chen J, Zhang Y, Shen Q, Pan W. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur J Pharm Sci. 2011;43(3):174–9. doi: 10.1016/j.ejps.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Kumar VV, Chandrasekar D, Ramakrishna S, Kishan V, Rao YM, Diwan PV. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335(1):167–75. doi: 10.1016/j.ijpharm.2006.11.004. [DOI] [PubMed] [Google Scholar]