

Abstract
Introduction
No head-to-head trials have compared the efficacy of the oral therapies, fingolimod, dimethyl fumarate and teriflunomide, in multiple sclerosis. Statistical modeling approaches, which control for differences in patient characteristics, can improve indirect comparisons of the efficacy of these therapies.
Methods
No evidence of disease activity (NEDA) was evaluated as the proportion of patients free from relapses and 3-month confirmed disability progression (clinical composite), free from gadolinium-enhancing T1 lesions and new or newly enlarged T2 lesions (magnetic resonance imaging composite), or free from all disease measures (overall composite). For each measure, the efficacy of fingolimod was estimated by analyzing individual patient data from fingolimod phase 3 trials using methodologies from studies of other oral therapies. These data were then used to build binomial regression models, which adjusted for differences in baseline characteristics between the studies. Models predicted the indirect relative risk of achieving NEDA status for fingolimod versus dimethyl fumarate or teriflunomide in an average patient from their respective phase 3 trials.
Results
The estimated relative risks of achieving NEDA status for fingolimod versus placebo in a pooled fingolimod trial population were numerically greater (i.e., fingolimod more efficacious) than the estimated relative risks for dimethyl fumarate or teriflunomide versus placebo in each respective trial population. In indirect comparisons, the predicted relative risks for all composite measures were better for fingolimod than comparator when tested against the trial populations of those treated with dimethyl fumarate (relative risk, clinical: 1.21 [95% confidence interval 1.06–1.39]; overall: 1.67 [1.08–2.57]), teriflunomide 7 mg (clinical: 1.22 [1.02–1.46]; overall: 2.01 [1.38–2.93]) and teriflunomide 14 mg (clinical: 1.14 [0.96–1.36]; overall: 1.61 [1.12–2.31]).
Conclusion
Our modeling approach suggests that fingolimod therapy results in a higher probability of NEDA than dimethyl fumarate and teriflunomide therapy when phase 3 trial data are indirectly compared and differences between trials are adjusted for.
Electronic supplementary material
The online version of this article (doi:10.1007/s12325-014-0167-z) contains supplementary material, which is available to authorized users.
Keywords: Disease-modifying therapies, Fingolimod, Indirect comparison, Multiple sclerosis, Neurology, Oral therapies
Introduction
Multiple sclerosis (MS) is a chronic, neurodegenerative disease in which putative auto-inflammatory responses attack myelinated axons of the central nervous system (CNS), causing the formation of scar tissue and disruption of nerve impulses traveling to and from the brain. This damage can result in a wide range of possible physical and mental symptoms [1]. Relapsing–remitting MS (RRMS), the type of MS that is the first diagnosis in 80–85% of patients, is characterized by episodes of neurological dysfunction, known as relapses, followed by periods of remission. Disease-modifying therapies (DMTs) form the mainstay of first-line treatment for RRMS. Until recently, most approved DMTs required administration by injection (interferon beta and glatiramer acetate) or intravenous infusion (natalizumab). Injectable agents are, however, associated with injection site reactions, as well as other tolerability issues (such as influenza-like symptoms), poor patient adherence and moderate efficacy [2, 3]. Three new oral therapies with different mechanisms of action have recently been approved for the treatment of MS. Fingolimod was the first oral therapy approved for the treatment of relapsing MS. It was approved as a first-line treatment in the USA in September 2010, and was recommended in the EU in March 2011 for the treatment of patients with high disease activity despite previous treatment with at least one other DMT and individuals with rapidly evolving severe RRMS [4]. Subsequently, teriflunomide was approved in the USA in September 2012 and in Europe in March 2013 [5, 6]. Dimethyl fumarate (DMF; BG-12) was approved in the USA in March 2013 and recently in Europe as well [7, 8].
DMTs aim to reduce the frequency and severity of relapses, extend the time intervals between relapses and slow progression to permanent disability [2]. To assess these treatment goals, annualized relapse rates (ARRs) or time to first relapse and disability progression, as measured by the expanded disability status scale (EDSS), are the primary clinical endpoints of phase 3 studies of therapies for RRMS, with magnetic resonance imaging (MRI) measures of disease activity and burden (CNS lesions) as secondary endpoints. Oral therapies have been shown to offer benefits with regard to these clinical and MRI outcomes when compared with placebo in phase 3 trials [9–13]. The clinical efficacy of these therapies over traditional injectable DMTs has been demonstrated for fingolimod in the trial assessing injectable interferon versus FTY720 oral in RRMS (TRANSFORMS) [14], and for the 7 mg dose (but not the 14 mg dose) of teriflunomide in the teriflunomide and Rebif (TENERE) trial [15]. Findings of these phase 3 trials indicate that most doses of oral therapies may represent an advance in the treatment of MS because they offer effective treatment options that are often better tolerated and more convenient than the traditional injectable DMTs.
In response to this therapeutic progress, treatment expectations and goals have evolved to encompass potential remission from the progressive symptoms of MS, known as freedom from disease activity or no evidence of disease activity (NEDA) [16]. Several exploratory analyses have investigated the efficacy of oral DMTs versus placebo on achieving NEDA status, defined as an absence of relapses, disability progression lasting at least 3 months and no new MRI lesions [17–22]. Post hoc analyses of the 2-year, placebo-controlled, phase 3 FTY720 research evaluating effects of daily oral therapy in multiple sclerosis (FREEDOMS) trial demonstrated that a significantly higher proportion of patients treated with fingolimod 0.5 mg achieved NEDA status than those treated with placebo (33% vs. 13%; P < 0.001) [21]. In an integrated post hoc analysis of the phase 3 determination of the efficacy and safety of oral fumarate in relapsing–remitting multiple sclerosis (DEFINE) and comparator and an oral fumarate in relapsing–remitting multiple sclerosis (CONFIRM) trials, the proportion of individuals free from disease activity over 2 years was higher for the DMF 240 mg twice daily group than for the placebo group (23% vs. 11%; P < 0.0001) [20]. In a post hoc analysis of teriflunomide multiple sclerosis oral (TEMSO), a greater proportion of patients treated with teriflunomide 7 or 14 mg were free from disease activity than individuals receiving placebo (18% and 23% vs. 14%; P = 0.0293 and P = 0.0002, respectively) [23].
There are no head-to-head controlled trials comparing the efficacy of the different oral DMTs. This is an area of much interest to neurologists and healthcare decision makers; therefore, several indirect treatment comparisons have recently been performed. Of these, two studies have compared fingolimod with teriflunomide [24, 25]. A network meta-analysis (NMA) found a significantly lower ARR with fingolimod than with teriflunomide 14 mg, but no significant difference in the proportion of patients with 3-month confirmed disability progression [24]. A separate NMA study found no statistically significant differences between fingolimod and teriflunomide 7 or 14 mg on measures of freedom from relapse and disease progression [25]. A recent study has additionally compared fingolimod with DMF using an NMA approach and found no significant differences in ARR or in the proportion of patients with disability progression lasting at least 3 months [26].
Standard NMA methods may be susceptible to bias because of differences in trial populations and methodologies. The placebo-controlled trials of these oral MS therapies are not sufficiently similar and differences between the trials, including differences in patient populations, endpoint definitions and methods for dealing with non-completers, have not been taken into account in any of the NMAs of these therapies performed to date. Subgroup and post hoc analyses of the phase 3 trials of DMTs have demonstrated that differences in patient baseline characteristics influence the observed effect of DMTs on ARRs and disability progression [14, 27], and that the application of different definitions of disability progression has a large impact on disability outcomes [28]. Therefore, it is important to adjust for these potentially confounding factors when assessing the comparative efficacy of these oral DMTs. Limited methodology exists to perform this type of adjusted comparison. Therefore, we developed a statistical modeling approach to compare treatment effects that adjusted for differences in patient characteristics and methodologies across the MS trials and allowed for the use of a combination of individual patient- and population-level data, thus permitting the utilization of all available data for these treatments [29–32]. Here, we have compared the effectiveness of oral therapies for MS (fingolimod 0.5 mg, DMF 240 mg twice daily and teriflunomide 7 or 14 mg) for achieving NEDA status. Our modeling approach uses all publicly available data for oral therapies and individual patient-level data from the phase 3 placebo-controlled trials of fingolimod.
Methods
Clinical Trials
The methodological details of the five double-blind, randomized, controlled, phase 3 trials for fingolimod (FREEDOMS and FREEDOMS II), DMF (DEFINE and CONFIRM) and teriflunomide (TEMSO) are described elsewhere [9–13]. This analysis used data for the placebo groups of these trials and the following treatment groups: fingolimod 0.5 mg, DMF 240 mg twice daily and teriflunomide 7 and 14 mg. Comparisons with DMF 240 mg three times daily were also performed. The number of patients randomized to each group and the differences in inclusion and exclusion criteria among trials are described in Supplementary Material S1. As data for this study were obtained from these trials and do not involve any new studies of human or animal subjects, ethical approval or participant’s informed consent was not required. All studies assessed ARR or time to first relapse as the primary endpoint and time to 3-month confirmed disability progression as a key secondary endpoint. Definitions of 3-month confirmed disability progression differed across the trials. In FREEDOMS, FREEDOMS II and TEMSO, confirmed disability progression was considered to be an increase of 1 EDSS point for patients with a baseline score of 0–5.0, and of 0.5 points for individuals with a baseline EDSS score of 5.5 (FREEDOMS) or greater than 5.5 (TEMSO). In DEFINE and CONFIRM, confirmed disability progression was defined as an increase of 1 point in individuals with an EDSS score of 1.0–5.0, and of at least 1.5 points in patients with a baseline EDSS score of 0.
NEDA Outcomes
NEDA was evaluated as the proportion of patients free from relapses, free from 3-month confirmed disability progression, free from gadolinium (Gd)-enhancing T1 lesions and free from new or newly enlarged T2 lesions. Using a similar methodology to the post hoc analyses of the placebo-controlled, phase 3 trial of natalizumab, AFFIRM [19], these individual components were combined to assess NEDA in three composite measures. The clinical composite of NEDA measured freedom from relapses and 3-month confirmed disability progression. The MRI composite of NEDA measured freedom from Gd-enhancing T1 lesions and new or newly enlarged T2 lesions. The overall composite or overall NEDA measured freedom from all of these disease outcomes.
In the FREEDOMS trials, if patients did not complete the trial and were disease free at their last study visit, they were counted as having achieved NEDA status [21]. This method was also assumed for the TEMSO trial where all patients who were randomized were included in the analysis, so we assumed that a disease-free non-completer was counted as having achieved NEDA status. In the DEFINE and CONFIRM trials, it was assumed that non-completers were removed from the analysis if they were disease free because these analyses were performed by the same investigators as the original AFFIRM analyses, which excluded these patients from analyses [21]. In the absence of published information from the DEFINE, CONFIRM and TEMSO trials it was assumed that all patient visits (i.e., both scheduled and unscheduled) were assessed for presence of disease activity.
Statistical Modeling
Models were built to estimate the efficacy of fingolimod in improving the probability (and thereby relative risk [RR] compared with placebo) of achieving NEDA status, and to compare the efficacy with that of other DMTs. Individual patient data from the pooled fingolimod phase 3 trials, FREEDOMS and FREEDOMS II, were used to build binomial regression models to estimate the proportion of patients achieving NEDA status. Data from FREEDOMS and FREEDOMS II were pooled by including a study-level stratifying variable. For each component and composite measure, the efficacy of fingolimod was estimated by re-analyzing the individual patient data from the fingolimod phase 3 trials using methodologies from studies of other oral therapies (adjusted only for endpoint definitions and how trial non-completers contributed to the analyses). Owing to differences in definitions and methodologies between the trials, two slightly different sets of models, termed ‘estimated’ models, were constructed; one for fingolimod versus DMF and another for fingolimod versus teriflunomide. Models for the DMF comparisons were based on the same definitions of disability progression used in the DEFINE and CONFIRM trials for patients with an EDSS score of 0 at baseline (i.e., 1.5-point change), whereas models for teriflunomide comparisons utilized the same definition as originally used in the FREEDOMS study for patients with an EDSS score of 0 at baseline (i.e., 1-point change). The outcomes in the models also took into account differences in the methods of dealing with non-completers across the various trials, with disease-free patients in FREEDOMS excluded from the models for the DMF comparisons if they did not complete the trial (as assumed in the DEFINE and CONFIRM trials). Thus, these estimates took into account methodological differences between trials and were termed the ‘estimated’ RRs of achieving NEDA status. The RR of achieving NEDA status for fingolimod versus placebo and for DMF or teriflunomide versus placebo was combined using the method proposed by Bucher et al. [33] to assess the RR of achieving NEDA status for fingolimod versus DMF or teriflunomide. The need for different adjustments to compare fingolimod with DMF and teriflunomide prevented the use of an NMA approach [34], and separate indirect comparisons are needed to indirectly compare the estimated RRs of achieving NEDA status for fingolimod versus DMF and teriflunomide.
Because application of the indirect comparison method proposed by Bucher et al. [33] to the treatment effect estimates requires the assumption that patient characteristics do not influence the treatment effect, we extended the method by building further models, based on the estimated models, to adjust for possible differences in baseline characteristics between the studies. In each set of estimated models, which accounted for differences in methodologies across trials, two models were constructed for each component and composite measure; an initial and a final model, in which individual patient data from the FREEDOMS trials were used to estimate the contribution of baseline characteristics to measures of NEDA. The prediction method for these initial and final models is described in Supplementary Material S2. Initial models were built by including pre-specified baseline covariates as main and treatment interaction (i.e., potential treatment modifier) effects. Covariates likely to modify the treatment effect were selected based on the results of previous subgroup analyses of FREEDOMS [35] and AFFIRM [36], as well as clinical expert opinion. Final models were developed by selecting the baseline covariates that were most predictive of the respective outcomes using a backward stepwise algorithm that used the Akaike information criterion (AIC) as the metric to retain the best, but simplest, model. This method avoids over-parameterizing the model. Goodness-of-fit was assessed by a Hosmer–Lemeshow grouping method (Supplementary Material S3). Initial models for the DMF comparisons included the following eight pre-specified baseline covariates (continuous variables were centered about their means): age, sex, previous DMT use, duration of MS, number of relapses in the past year, EDSS score at baseline (0–1.5, 2–2.5, ≥3), number of Gd-enhancing T1 lesions and cube root of the total volume of T2 lesions. Owing to unavailability of data, the initial models for the teriflunomide comparisons excluded the cube root of total volume of T2 lesions. The EDSS score was split into two categories (≤3.5 and >3.5) based on the stratification of randomized patients in the TEMSO trial.
An indirect comparison of the oral therapies was performed in three steps (Fig. 1). First, models were used to predict the RR of achieving NEDA status for fingolimod versus placebo in an average patient in a pooled DEFINE and CONFIRM population, and in the TEMSO population (termed ‘predicted’ models and ‘predicted’ RRs, respectively). Second, the estimated RR of achieving NEDA status for DMF versus placebo in the pooled DEFINE and CONFIRM population was calculated using a fixed-effect inverse variance-weighted method of the RRs from each study, a standard method for pooling outcomes from studies that provides a weighted average of estimates. The RRs from each study were found from data in Havrdova et al. [20] reporting the probabilities of patients achieving NEDA status in each arm, with the variance of these probabilities calculated from the sample size in each arm, excluding disease-free patients who did not complete the study. Because this number is not reported, we estimated it assuming that non-completers had the same likelihood of being disease free as those who completed the trial. This is likely to be a conservative assumption; in the FREEDOMS study, non-completers were less likely to be disease free than completers, leading to the sample size being reduced too much and an inflated variance of the pooled RR estimate. Similar calculations for estimating the RR of achieving NEDA status were performed for teriflunomide versus placebo in the TEMSO population using results from Freedman et al. [23]. Third, the estimated RR of achieving NEDA status for fingolimod versus placebo in the pooled FREEDOMS population (from the ‘estimated’ models) and the predicted RRs for fingolimod versus placebo in comparator trial populations (from the ‘predicted’ models) were compared with those calculated for DMF and teriflunomide in their respective trials. An indirect comparison of the efficacy of fingolimod and DMF or teriflunomide was performed by comparing the estimated RRs of achieving NEDA status for each treatment versus placebo using the Bucher et al. [33] method. Results are expressed as the RR (95% confidence interval [CI]) of achieving NEDA status, with an RR greater than 1.0 indicating an improved outcome; the higher the RR, the better the outcome for the patient.
Fig. 1.
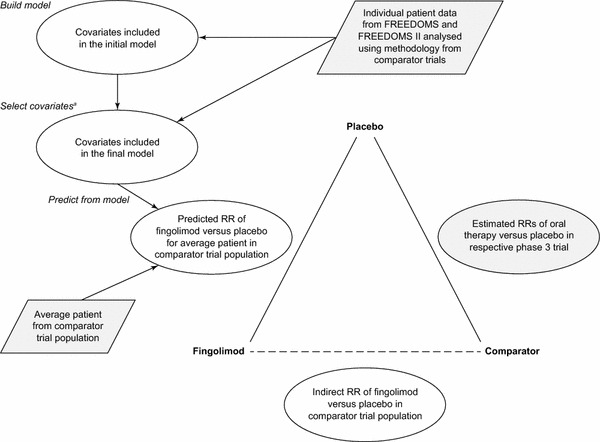
Schematic of the modeling approach. aFinal models selected baseline characteristics that were most predictive of the outcome using a stepwise algorithm that used the Akaike information criterion as the metric to retain the best model. FREEDOMS FTY720 research evaluating effects of daily oral therapy in multiple sclerosis, RR relative risk
Compliance With Ethics Guidelines
The analysis in this article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Results
Patient Baseline Characteristics
Patient baseline demographics and disease characteristics in the pooled FREEDOMS and FREEDOMS II, pooled DEFINE and CONFIRM, and TEMSO populations are compared in Table 1. In general, patient demographics and the mean number of relapses in the past year were similar across the trials, but there were notable differences between the populations regarding previous DMT use, the number of Gd-enhancing lesions and the mean volume of T2 lesions. In particular, more patients in the pooled FREEDOMS population had previously used DMTs (51.0%) than in the other trial populations (27.0–35.4%).
Table 1.
Baseline demographics and disease characteristics for patients in FREEDOMS and FREEDOMS II, DEFINE and CONFIRM, and TEMSO
| Characteristics | FREEDOMS and FREEDOMS IIa [9, 10] (N = 1,556) | DEFINE and CONFIRMb [11, 12] (N = 2,301) | TEMSO [13] (N = 1,088) |
|---|---|---|---|
| Demographics | |||
| Mean age, years | 38.6 | 38.0 | 37.9 |
| Sex, % female | 74.3 | 71.9 | 72.2 |
| Previous therapy with any approved DMT, % | 51.0 | 35.4 | 27.0 |
| Clinical | |||
| Mean disease duration, years | 9.2 | 8.1 [42] | 8.7 |
| Number of relapses in the past year, mean | 1.5 | 1.3 | 1.37 |
| EDSS score, % | |||
| 0 | 7.9 | 5.1 | NA |
| 1.0–1.5 | 24.2 | 24.6 | NA |
| 2.0–2.5 | 31.9 | 29.3 | NA |
| 3.0–3.5 | 19.6 | 24.7 | 77.1c [43] |
| 4.0–4.5 | 11.8 | 12.8 | 22.9d [43] |
| ≥5 | 4.7 | 3.6 | NA |
| Mean | 2.4 | NR | 2.7 |
| MRI measures | |||
| Gd-enhancing T1-weighted lesions, mean | 1.4 | 1.9 [11, 44] | 1.7 |
| Mean volume of T2-weighted lesions, mm3 | 5,858 | 10,766 [44] | NR |
CONFIRM comparator and an oral fumarate in relapsing–remitting multiple sclerosis, DEFINE determination of the efficacy and safety of oral fumarate in relapsing–remitting multiple sclerosis, DMF dimethyl fumarate, DMT disease-modifying therapy, EDSS expanded disability status scale, FREEDOMS FTY720 research evaluating effects of daily oral therapy in multiple sclerosis, Gd gadolinium, MRI magnetic resonance imaging, NA not applicable, NR not reported, TEMSO teriflunomide multiple sclerosis oral
aFingolimod 0.5 mg and placebo groups only
bDMF twice daily, DMF three times daily and placebo groups
cPatients with EDSS score ≤3.5. The proportions of patients with EDSS scores of ≤3.5 are 83.6% in FREEDOMS and FREEDOMS II, and 84.8% in DEFINE and CONFIRM
dPatients with EDSS score >3.5. The proportions of patients with EDSS scores of >3.5 are 16.4% in FREEDOMS and FREEDOMS II, and 15.2% in DEFINE and CONFIRM
Comparisons Without Adjustment for Baseline Characteristics
When the efficacy of fingolimod was estimated by analyzing patient data from the FREEDOMS trials using methodologies from studies of other oral therapies, the estimated RRs for fingolimod versus placebo in the pooled FREEDOMS population were consistently greater than the estimated RRs for DMF versus placebo in the pooled DEFINE and CONFIRM population, and for teriflunomide versus placebo in TEMSO for all composite measures of NEDA (Fig. 2). Using the methodology in the DMF trials, the estimated RR (95% CI) of the overall composite, or overall NEDA, for fingolimod versus placebo in the pooled FREEDOMS population (3.25 [2.51–4.21]) was greater than that for DMF versus placebo in the pooled DEFINE and CONFIRM population (2.05 [1.41–3.00]) (Fig. 2, rows 1 and 2). Using the methodology in the teriflunomide trials, the estimated RR of the overall NEDA for fingolimod versus placebo in the pooled FREEDOMS population (2.78 [2.22–3.49]) was significantly greater than that for teriflunomide 7 mg versus placebo (1.28 [0.92–1.78]), and teriflunomide 14 mg versus placebo (1.59 [1.16–2.19]) in the TEMSO population (Fig. 2, rows 4 and 5 for 7 mg dose and rows 4 and 6 for 14 mg dose). Similar trends were seen for the clinical and MRI composite measures (Fig. 2).
Fig. 2.
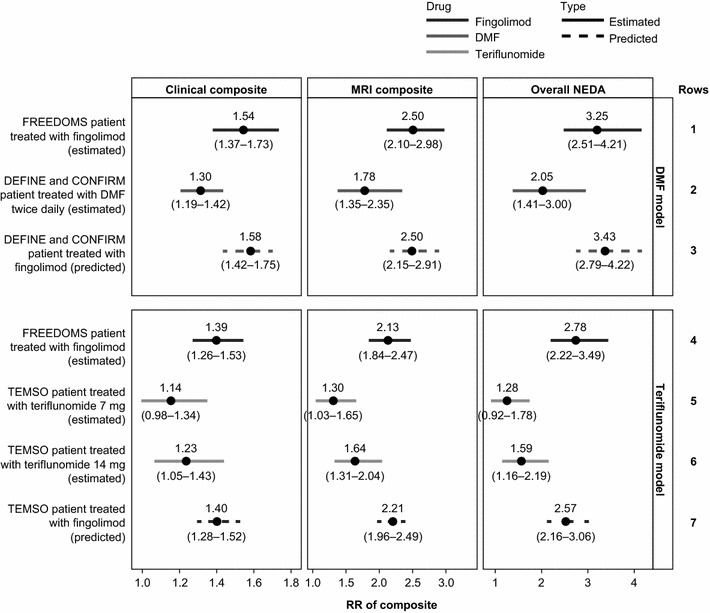
RRs of achieving NEDA status for fingolimod, DMF and teriflunomide versus placebo. Estimated RRs for the pooled FREEDOMS population, pooled DEFINE and CONFIRM population, and TEMSO populations are shown as solid lines as indicated (estimated). Dashed lines represent the predicted RRs for fingolimod versus placebo in alternative trial populations using the final models (predicted). An RR above 1.0 indicates an improved outcome for treatment relative to placebo. CONFIRM comparator and an oral fumarate in relapsing–remitting multiple sclerosis, DEFINE determination of the efficacy and safety of oral fumarate in relapsing–remitting multiple sclerosis, DMF dimethyl fumarate, FREEDOMS FTY720 research evaluating effects of daily oral therapy in multiple sclerosis, MRI magnetic resonance imaging, NEDA no evidence of disease activity, RR relative risk, TEMSO teriflunomide multiple sclerosis oral
Baseline Covariates Selected for Inclusion in the Final Models
The effect of each covariate included in the initial models on the predicted clinical, MRI and overall composite measures for fingolimod versus placebo was explored by changing them one at a time. To demonstrate the effect of covariates on predicting the efficacy of fingolimod, the clinical composite measure is used as an example. In models for the DMF comparisons, age and previous DMT use were found to be the best predictors of no evidence of clinical disease activity using AIC selection, whereas age was found to be the only predictor of no evidence of clinical disease activity in the models for the teriflunomide comparison. The covariates included in the initial and final models for each component measure are shown in Supplementary Material S4 and Fig. 3, respectively.
Fig. 3.
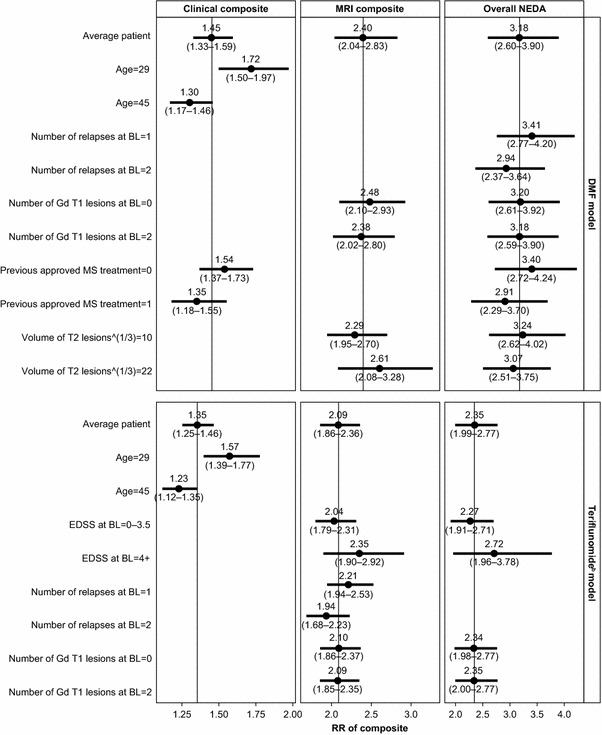
Impact of baseline characteristics on predicted RRs for fingolimod versus placeboa (final model). An RR above 1.0 indicates an improved outcome for treatment relative to placebo. aFor non-categorical covariates, the model predicts the treatment effect for setting that variable at the 1st and 3rd quartile of the distribution while holding all other covariates constant. bVolume of T2 lesions at baseline was not included in the initial model for the teriflunomide analysis, and EDSS-defined progression was reported differently (0–3.5 instead of 0–1.5 in the DMF analysis). BL baseline, DMF dimethyl fumarate, EDSS expanded disability status scale, Gd gadolinium, MRI magnetic resonance imaging, MS multiple sclerosis, NEDA no evidence of disease activity, RR relative risk
Final Model Predictions
The covariates included in the final models can predict the efficacy of fingolimod versus placebo in an alternative trial population. When the covariates that best predicted the clinical composite measure, age and previous DMT use, were included in the final models for the DMF comparisons, the RR of showing NEDA for the clinical composite for fingolimod versus placebo was increased in younger or treatment-naïve patients (or decreased in older and previously treated patients). As individuals in the pooled DEFINE and CONFIRM population were, on average, younger and more likely to be treatment naïve than in the pooled FREEDOMS population, the model predicted a marginally increased RR of achieving NEDA status for the clinical composite in an average patient from the pooled DEFINE and CONFIRM population (RR: 1.58) than from the pooled FREEDOMS population (RR: 1.54). For the final teriflunomide model, which only included age as a covariate because there was only a small difference in age between the trial populations, fingolimod was predicted to not perform differently in the TEMSO population compared with the pooled FREEDOMS population.
Comparisons After Adjustment for Baseline Characteristics
Estimated RRs for fingolimod versus placebo in the FREEDOMS trial populations were similar to those predicted in the final models for fingolimod versus placebo in a pooled FREEDOMS population for all three composite measures in both sets of analyses (data not shown). This demonstrates the predictive ability of the model. Furthermore, the goodness-of-fit assessed by Hosmer–Lemeshow grouping showed that the predicted probabilities of achieving NEDA status were consistent with the reported data in the FREEDOMS trials (see Supplementary Materials S3 and S5). The final models were used to predict the efficacy of fingolimod versus placebo in an average patient from the trial populations of DEFINE and CONFIRM, and TEMSO. The predicted RRs for fingolimod versus placebo in an average individual from each of these trial populations were marginally increased or similar to estimated RRs for fingolimod in the pooled FREEDOMS population for the three composite measures of NEDA (Fig. 2, rows 1 and 3 for DMF and rows 4 and 7 for teriflunomide). The only exception was for the overall NEDA for the teriflunomide comparison in that the predicted RRs were slightly lower than estimates observed for fingolimod (2.57 [2.16–3.06] for teriflunomide versus 2.78 [2.22–3.49] for fingolimod). The predicted RRs for fingolimod versus placebo in the DEFINE and CONFIRM population, and TEMSO population were greater than those calculated for DMF and teriflunomide, respectively, for all three composite measures of NEDA (Fig. 2, rows 2 and 3 for DMF and rows 5, 6 and 7 for teriflunomide).
Indirect Comparison of Oral DMTs for Measures of NEDA
In indirect comparisons, RRs were greater than 1 for fingolimod versus DMF or teriflunomide in the DEFINE and CONFIRM, or TEMSO populations, respectively. Similar results were seen for the three composite measures of NEDA with both estimated (Fig. 4, rows 1, 3 and 5) and predicted values for fingolimod (Fig. 4, rows 2, 4 and 6). Indirect comparisons using predicted RRs for fingolimod in an alternative trial population were significantly greater than 1 for all analyses versus DMF in a pooled DEFINE and CONFIRM population and for analyses versus teriflunomide 7 and 14 mg in a TEMSO population (with the exception of the clinical composite for the teriflunomide 14 mg comparison in which only a positive trend was observed). For the overall NEDA, the indirect comparison RRs for fingolimod in the trial populations of DMF and teriflunomide were: 1.67 (1.08–2.57) versus DMF and 2.01 (1.38–2.93) and 1.61 (1.12–2.31) versus teriflunomide 7 and 14 mg, respectively (Fig. 4, rows 2, 4 and 6). Results for the individual composite measures for the DMF comparisons are presented in Supplementary Material S6; data at this level are not available for teriflunomide.
Fig. 4.
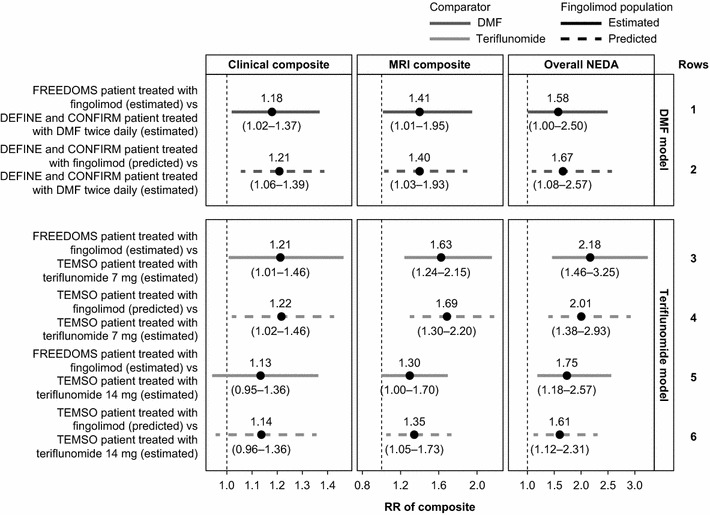
Indirect comparison of RRs of achieving NEDA status for fingolimod versus DMF or teriflunomide. An RR above 1.0 indicates an improved outcome for fingolimod relative to comparator. Indirect comparisons were performed using estimated RR for fingolimod in a pooled FREEDOMS and FREEDOMS II population (solid lines, estimated) or using predicted RRs for fingolimod in a pooled DEFINE and CONFIRM or TEMSO population (dashed line, predicted). CONFIRM comparator and an oral fumarate in relapsing–remitting multiple sclerosis, DEFINE determination of the efficacy and safety of oral fumarate in relapsing–remitting multiple sclerosis, DMF dimethyl fumarate, FREEDOMS FTY720 research evaluating effects of daily oral therapy in multiple sclerosis, MRI magnetic resonance imaging, NEDA no evidence of disease activity, RR relative risk, TEMSO teriflunomide multiple sclerosis oral
Discussion
It is often useful for neurologists, health policy makers and patients to compare the efficacy of therapies for MS, and with the recent introduction of these oral therapies, there is much interest in their comparative effectiveness. This study was a comparison of the efficacy of oral DMTs using a statistical modeling approach to account for differences between the individual, placebo-controlled, phase 3 trials conducted in patients with RRMS. The approach estimated what the RRs of achieving NEDA status would be between two treatments using a comparison in the form A is to B and C is to B to infer the comparison of A to C. The results estimated that in comparisons without covariate adjustment, the RR of achieving NEDA status was higher for fingolimod versus placebo than for DMF and teriflunomide versus placebo, for the three composite measures of NEDA. These results remained similar when models adjusted for differences between the phase 3 trial patient populations. In addition, the indirect comparisons of oral DMTs estimated that fingolimod was more efficacious than both DMF and teriflunomide (i.e., RRs >1) in their respective trial populations for all three composite measures of NEDA, and in most cases these results were statistically significant.
Randomized head-to-head trials are the best method for evaluating the efficacy of different treatments. There is, however, a lack of head-to-head clinical trials, so indirect comparisons provide a means to assess the treatments. The method proposed by Bucher et al. [33], in which an indirect comparison of two therapies is adjusted according to the results of their direct comparisons with placebo, is valid only if differences in the patient populations do not affect the treatment effect and endpoints are equally defined. Given that the FREEDOMS trials were not sufficiently similar to the DEFINE, CONFIRM and TEMSO trials, use of the Bucher methodology without any adaptation may not have provided a valid comparison. In addition, we sought to use individual patient-level data, which were available for the FREEDOMS trials but not for the DEFINE, CONFIRM and TEMSO trials. We therefore developed a modeling approach for indirect comparisons, which was built upon the Bucher method that adjusted for differences in patient characteristics and methodologies across the trials and allowed for the combination of individual- and population-level data to be used. The model was created by expressing key outcomes from the pooled FREEDOMS trials as a function of baseline characteristics, and then applying this model to an average patient in the pooled DEFINE and CONFIRM trials, as well as to an average patient in TEMSO, to predict the efficacy of fingolimod versus placebo on three composite measures of NEDA.
While alternate modeling approaches are possible (see Table 2), these methods are less suitable because they do not allow for all of the following to be appropriately achieved: (1) controlling for differences in patient populations; (2) accounting for differences in endpoint definitions; (3) accounting for the way in which non-completers are dealt with; and (4) using individual patient data where they are available. For example, a Bayesian mixed treatment comparison has been used to compare the efficacy of teriflunomide with other approved DMTs in the treatment of MS [24]. Mixed treatment comparisons using Poisson, mixed-log binomial, time-to-event and continuous models have been used to compare the efficacy and safety of DMF with other approved DMTs including fingolimod. However, these analyses could not adjust for differences in trial methodology or endpoint definitions across trials [26], and although this could be achieved by performing sub-analyses, these methods require data to be available from several studies to enable reasonable estimation of the random effects. Meta-analysis methods are also available to synthesize individual patient and aggregate data, and enable adjustment for patient baseline characteristics [37]. Such methods would also allow differences in treatment effect due to differences in patient population to be accounted for, using a treatment–covariate interaction, but again these methods would be hindered by not having enough studies in the network to enable reasonable estimation of the random effects. The small number of studies and the need to account for endpoint definitions by performing additional sub-analyses (which would reduce the number of studies even further) made this method inappropriate in our case. An alternate method that could have been applied is the propensity score method of Signorovitch et al. [31]. This method adjusts for a predefined set of patient baseline characteristics, whereas our approach selects from such a set that best predicts the treatment effect. In the case of MS, in which studies have largely deduced potential treatment modifiers, our approach avoids over-parameterization of the model and enables selection of a parsimonious model.
Table 2.
Modeling methods for indirect treatment comparisons
| Model | Why it was not suitable for our analysis |
|---|---|
| Mixed treatment comparison using summary level data | Does not take into account differences in patient population, endpoint definitions and ways of dealing with non-completers between trials and does not make use of individual patient-level data |
| Differences in patient populations could be accounted for using meta-regression by including study-level treatment–covariate interactions [45], but adjustments at the study level can be susceptible to the ecological fallacy, where the relationship between outcome and covariate may not be the same at the study and individual level | |
| Differences in trial methodology could be accounted for using sub-analyses but this requires a larger number of studies than is available in the present case to enable estimation of the random effects assuming that there is heterogeneity in treatment effect between studies [46] | |
| Mixed treatment comparison using individual and summary level patient data [47] | Enables the use of individual patient data and adjustment for patient populations, but it does not take into account differences in endpoint definitions or the different ways of dealing with non-completers |
| This methodology can also be susceptible to ecological fallacy, require a random effects model and a separate analysis to adjust for endpoint definitions or the different ways of dealing with non-completers | |
| Bucher pair-wise indirect comparison [33] | Enables endpoint definitions or the different ways of dealing with non-completers to be adjusted for, but does not make use of individual patient data and adjust for patient populations |
| This methodology can be built on to adjust for patient characteristics and use individual patient data as demonstrated in our study | |
| Matching-adjusted indirect comparison [31] | Enables the use of individual patient data, adjustment for patient populations and trial methodology. This methodology uses individual patient data from trials of one treatment to match baseline summary statistics reported from trials of another treatment |
| This method adjusts for a predefined set of patient baseline characteristics and may over-fit the prediction model. This approach may not have sufficient power for all treatments being assessed |
In this analysis, our modeling approach suggests that differences in average patient characteristics between the populations of the clinical trials of the oral therapies have a marginal impact on indirect comparisons of NEDA outcomes, because model outputs before adjustment for baseline covariates are similar to the outputs after adjustment. Taking previous DMT use as an example, the pooled FREEDOMS population had a higher rate of previous DMT use than the other trial populations. A smaller effect on achieving NEDA status might therefore be expected in this population than in one with less previous DMT use, and this was observed. Thus, adjusting for previous DMT use is likely to improve the comparative effectiveness of fingolimod relative to other therapies studied in a population with lower rates of DMT use. However, other differences in trial populations might lead to a greater effect on achieving NEDA status and the effects of different variables may eventually cancel each other out. Our methodology is indeed designed to improve on simply comparing raw event rates across studies. Our modeling approach showed that differences in trial methodologies had a greater impact on NEDA outcomes than differences in patient characteristics, thus highlighting the importance of adjusting for these methodological differences. The impact of these differences was exemplified by the RR predicted when using the DEFINE and CONFIRM approach of dealing with non-completers compared with using the TEMSO method.
This study assessed treatment efficacy using three composite measures of NEDA that were based on the absence of relapses, disability progression, Gd-enhancing T1 lesions, and new or newly enlarged T2 lesions. These individual component measures are well-established indicators of disease activity and are commonly assessed in clinical trials [17–22]. As the effectiveness of treatments for MS increases, the composite endpoint of NEDA is becoming an important measure for clinicians and patients [16]. The use of these composite endpoints, however, does have some limitations because they do not take into account other potentially important indicators of disease activity, such as brain volume loss or cognitive function. In addition, some analytical adjustment to account for the dominance of one component measure may potentially be required. For example, one analysis has shown that the overall composite endpoint is driven to a large extent by MRI outcomes, with minimal contribution from clinical measures [32]. Finally, the number and timing of MRI scans were identical for the FREEDOMS trials and DEFINE and CONFIRM, but different for TEMSO. Imbalances in the timing or scheduling of scans could have an impact on MRI outcomes and the extent to which these outcomes contribute to the overall NEDA. Further research is needed to define the best combination of criteria that represents NEDA in MS and the best population in which to adjust the results, but this study provides a valuable exploration into the concepts.
Endpoint definitions also impact the results. In an analysis of the CombiRx trial, which evaluated interferon beta-1a and glatiramer acetate in patients with RRMS, using a 1.0-point increase in EDSS score as definition of progression, 15% of individuals whose screening EDSS score was greater than baseline “progressed” by month 3; that is, many went back to their screening value leading to false positive progressions and diminishing the treatment effect. When a 1.5-point definition of progression was used instead, the false positive progressions were reduced, enhancing the treatment effect [38]. A similar impact on treatment effect was observed in the FREEDOMS trials, where the treatment effect with respect to 3- and 6-month confirmed disability progression was numerically greater when requiring a 1.5-point change [28, 39]. Thus, in our study, treatment effect may be lower in the teriflunomide comparisons using the FREEDOMS and TEMSO definition of disability progression (1.0-point increase in patients with a baseline EDSS score of 0), compared with the DMF comparisons, which used the DEFINE and CONFIRM definition (1.5-point increase in patients with a baseline EDSS score of 0).
As with all statistical modeling, limitations exist based on assumptions that are necessary to make the modeling feasible. Firstly, indirect treatment comparisons are a type of observation research, owing to the non-randomized selection of studies for inclusion in these analyses, and are subject to confounding. Our modeling approach, in contrast to several alternative methodologies, reduces this confounding by controlling for differences in patient populations. In addition, our approach is based on the Bucher method and is therefore subject to the same assumptions as this methodology, for example, the transitivity of the treatment effects assumes we can learn about the effect of A versus C via B [40]. Furthermore, it was assumed that the outcomes of the trials were influenced by a specific set of covariates, but it is possible and indeed likely that results are affected by additional variables not included in the models, such as the treatment environment at the time these studies were conducted and/or the countries or practices involved. We adjusted for known baseline variables, but we could not account for subtle unmeasured selection criteria as sources of influence or bias. Controlled trials in MS have demonstrated the relevance of such hidden selection biases because identical selection criteria have resulted in similar baseline characteristics, but widely different responses to placebo across studies [41]. In addition, we had to make several assumptions about the methodology used in the TEMSO trial, because this information was not available at the time of planning the analysis. We assumed that the TEMSO trial used the same method of dealing with non-completers as the FREEDOMS trials, but it is possible that an alternative method was used that should have been controlled for in the models. There may also have been additional differences in study methodologies that could affect the results, which we did not account for, such as differences between trials in the use of unscheduled visits for assessing suspected relapses or disability progression. For example, if unscheduled visits (in contrast to scheduled visits) were used to confirm disability progression, an impact on the overall disability progression rate could occur. There is also uncertainty regarding the standard population chosen in which to adjust the results. Statistical analyses usually assume that all patients are at a similar risk of disease activity, but if the adjusted covariate is a key variable, the results could differ considerably in different populations. We also assumed that non-completers had the same likelihood of being disease free as those who completed a trial. This might have led the efficacy results of two therapies to be more similar than in reality if the less effective DMT was associated with higher dropout rates but the number of completers was similar to completers taking the more effective therapy. Lastly, we assumed that the probability of achieving NEDA status could be reasonably predicted using a linear model. The goodness-of-fit assessment demonstrated that the predicted probability of achieving NEDA status was similar to the observed probability of achieving NEDA status, suggesting that this was an appropriate choice of model. Our conclusions must be interpreted with caution because of the assumptions inherent in any indirect comparison.
Conclusions
Our modeling approach, which controlled for known or suspected treatment modifiers and differences in patient characteristics between the trials, predicted that those treated with fingolimod in some comparisons have a significantly higher probability of achieving NEDA status compared with those treated with DMF and teriflunomide for the three composite measures in both unadjusted and adjusted indirect comparisons. The statistical modeling suggests that differences in patient characteristics between the trials have a marginal impact on indirect comparisons of these treatments. In the absence of direct, head-to-head comparisons, our modeling approach can be used to make informed conclusions about the comparative efficacy of oral DMTs in patients with MS. These findings should, however, be interpreted with caution, owing to the assumptions inherent in any modeling approach.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Novartis Pharma AG provided funding for this project and the article processing charges. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. Richard Nixon and Niklas Bergvall conceived and designed the study, analyzed the data, interpreted the results, and critically reviewed and approved the manuscript. Davorka Tomic, Nikolaos Sfikas, Gary Cutter and Gavin Giovannoni designed the study, interpreted the results, and critically reviewed and approved the manuscript. All named authors meet the ICMJE criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. The authors take full responsibility for the content of the paper. They thank Dr Gemma Carter and Hilary Phelps (Oxford PharmaGenesis Ltd, Oxford, UK) for medical writing support, editorial assistance, and collation and incorporation of comments from all authors. Support for this assistance was funded by Novartis Pharma AG.
Conflict of interest
Richard Nixon is a paid employee of Novartis Pharma AG. Niklas Bergvall is a paid employee of Novartis Pharma AG. Davorka Tomic is a paid employee of Novartis Pharma AG. Nikolaos Sfikas is a paid employee of Novartis Pharma AG. Gary Cutter has received personal compensation for participation in Data and Safety Monitoring Committees for Sanofi-Aventis, Cleveland Clinic, Daiichi Sankyo, GlaxoSmithKline Pharmaceuticals, Genmab, Eli Lilly, Medivation, Modigenetech, Ono Pharmaceutical, PTC Therapeutics, Teva Pharmaceuticals, Vivus, University of Pennsylvania, National Heart, Lung, and Blood Institute, National Institute of Neurological Disorders and Stroke, and National Multiple Sclerosis Society. He has also received consulting and speaking fees, and served on advisory boards for Alexion Pharmaceuticals, Bayhill Therapeutics, Bayer Pharmaceuticals, Celgene, Novartis, Consortium of Multiple Sclerosis Centers (grant), Genzyme, Klein Buendel Inc., Nuron Biotech, Peptimmune, Somnus Therapeutics, Sandoz, Teva Pharmaceuticals, University of Texas Southwestern, and Visioneering Technologies Inc. He is President of Pythagoras Inc. and has received Consortium of Multiple Sclerosis Centers task orders that involve research for various pharmaceutical organizations. Gavin Giovannoni has received honoraria from Bayer HealthCare, Biogen Idec, Canbex, Genzyme, GlaxoSmithKline, Merck Serono, Novartis, Protein Discovery Laboratories, Roche, Synthon, Teva Neuroscience, and UCB; research support from Biogen Idec, Ironwood, Merck Serono, Merz, and Novartis; and compensation from Elsevier as co-chief editor of MS and Related Disorders.
Compliance with ethics guidelines
The analysis in this article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
- 1.World Health Organization. Atlas: multiple sclerosis resources in the world. 2008. Available from: http://whqlibdoc.who.int/publications/2008/9789241563758_eng.pdf?ua=1. Accessed November 13, 2014
- 2.Derwenskus J. Current disease-modifying treatment of multiple sclerosis. Mt Sinai J Med. 2011;78:161–175. doi: 10.1002/msj.20239. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds MW, Stephen R, Seaman C, Rajagopalan K. Healthcare resource utilization following switch or discontinuation in multiple sclerosis patients on disease modifying drugs. J Med Econ. 2010;13:90–98. doi: 10.3111/13696990903579501. [DOI] [PubMed] [Google Scholar]
- 4.European Medicines Agency. European public assessment report: Gilenya. 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002202/WC500104530.pdf. Accessed October 16, 2014.
- 5.US Food and Drug Administration. Teriflunomide (marketed as Aubagio) prescribing and labeling information. Latest label approved on September 12, 2012. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202992s000lbl.pdf. Accessed September 9, 2013.
- 6.European Medicines Agency. Aubagio summary of product characteristics. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002514/WC500148682.pdf. Accessed September 9, 2013.
- 7.US Food and Drug Administration. Dimethyl fumarate (marketed as Tecfidera) prescribing and labeling information. Latest label approved on March 27, 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204063lbl.pdf. Accessed September 9, 2013.
- 8.European Medicines Agency. European public assessment report: Tecfidera. 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002601/WC500162072.pdf. Accessed March 7, 2014.
- 9.Calabresi PA, Goodin D, Jeffery D, et al. Efficacy and safety of fingolimod versus placebo: primary outcomes from the phase 3 FREEDOMS II study in patients with relapsing–remitting multiple sclerosis. Mult Scler. 2012;18(4 Suppl):P491. [Google Scholar]
- 10.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 11.Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 12.Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 13.O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 14.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 15.Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler. 2013;14:14. doi: 10.1177/1352458513507821. [DOI] [PubMed] [Google Scholar]
- 16.Havrdova E, Galetta S, Stefoski D, Comi G. Freedom from disease activity in multiple sclerosis. Neurology. 2010;74:S3–S7. doi: 10.1212/WNL.0b013e3181dbb51c. [DOI] [PubMed] [Google Scholar]
- 17.Giovannoni G, Arnold DL, Cohen JA, et al. Disease activity free status in CARE-MS I phase 3 study. 2012. Available from: http://www.slideshare.net/gavingiovannoni/disease-activity-free-status-on-alemtuzumab-vs-ifnbeta1a-carems-1-study. Accessed August 13, 2012.
- 18.Giovannoni G, Cook S, Rammohan K, et al. Sustained disease-activity-free status in patients with relapsing–remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post hoc and subgroup analysis. Lancet Neurol. 2011;10:329–337. doi: 10.1016/S1474-4422(11)70023-0. [DOI] [PubMed] [Google Scholar]
- 19.Havrdova E, Galetta S, Hutchinson M, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the natalizumab safety and efficacy in relapsing–remitting multiple sclerosis (AFFIRM) study. Lancet Neurol. 2009;8:254–260. doi: 10.1016/S1474-4422(09)70021-3. [DOI] [PubMed] [Google Scholar]
- 20.Havrdova E, Gold R, Fox RJ, et al. BG-12 (dimethyl fumarate) treatment for relapsing–remitting multiple sclerosis (RRMS) increases the proportion of patients free of measured clinical and neuroradiologic disease activity in the phase 3 studies. Neurology. 2013;80(Meeting Abstracts 1):P07.106.
- 21.Kappos L, Radue E, O’Connor P, et al. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis: results from a phase 3, placebo-controlled study (FREEDOMS) Neurology. 2011;76(Suppl 4):A563. [Google Scholar]
- 22.Khatri B, Barkhof F, Comi G, Jin J, Francis G, Cohen J. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis compared to IFN-b1a: results from a phase 3, active-controlled study (TRANSFORMS). Neurology. 2012;78(Meeting Abstracts 1):PD5.006.
- 23.Freedman M, O’Connor P, Wolinsky J, et al. Teriflunomide increases the proportion of patients free from disease activity in the TEMSO phase III study. Neurology. 2012;78(Meeting Abstracts 1):PD5.007.
- 24.Cutter G, Fahrbach K, Huelin R, et al. Relative efficacy of teriflunomide 14 mg among the approved disease-modifying therapies in relapsing forms of multiple sclerosis: a mixed treatment comparison. 2013. Available from: https://cmscactrims.confex.com/cmscactrims/2013/webprogram/Paper1826.html. Accessed March 5, 2014.
- 25.Hadjigeorgiou GM, Doxani C, Miligkos M, et al. A network meta-analysis of randomized controlled trials for comparing the effectiveness and safety profile of treatments with marketing authorization for relapsing multiple sclerosis. J Clin Pharm Ther. 2013;38:433–439. doi: 10.1111/jcpt.12090. [DOI] [PubMed] [Google Scholar]
- 26.Hutchinson M, Fox RJ, Havrdova E, et al. Efficacy and safety of BG-12 (dimethyl fumarate) and other disease-modifying therapies for the treatment of relapsing–remitting multiple sclerosis: a systematic review and mixed treatment comparison. Curr Med Res Opin. 2014;30:613–627. doi: 10.1185/03007995.2013.863755. [DOI] [PubMed] [Google Scholar]
- 27.Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 28.Bergvall N, Sfikas N, Alsop J, Chin P, von Rosensteil P, Kappos L. Consequences of different definitions of confirmed disability progression across randomised trials of MS therapies. 2012. Available from: http://novartis.medicalcongressposters.com/Default.aspx?doc=9ce32. Accessed March 5, 2014.
- 29.Nixon R, Eckert B, Cutter G, Mercier F, Francis G, Kappos L. Indirect comparisons of oral fingolimod vs natalizumab and cladribine for the treatment of relapsing–remitting multiple sclerosis based on data from FREEDOMS, AFFIRM and CLARITY. J Neurol. 2012;259:P344. [Google Scholar]
- 30.Caro JJ, Ishak KJ. No head-to-head trial? Simulate the missing arms. Pharmacoeconomics. 2010;28:957–967. doi: 10.2165/11537420-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 31.Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28:935–945. doi: 10.2165/11538370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 32.Nixon R, Bergvall N, Sfikas N, et al. Indirect comparisons of oral fingolimod versus natalizumab on measures of disease freedom in patients with multiple sclerosis, based on results from FREEDOMS and AFFIRM. Neurology. 2013;80(Meeting Abstracts 1):P01.209.
- 33.Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol. 1997;50:683–691. doi: 10.1016/S0895-4356(97)00049-8. [DOI] [PubMed] [Google Scholar]
- 34.Lumley T. Network meta-analysis for indirect treatment comparisons. Stat Med. 2002;21:2313–2324. doi: 10.1002/sim.1201. [DOI] [PubMed] [Google Scholar]
- 35.Devonshire V, Havrdova E, Radue EW, et al. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double-blind, randomised, placebo-controlled FREEDOMS study. Lancet Neurol. 2012;11:420–428. doi: 10.1016/S1474-4422(12)70056-X. [DOI] [PubMed] [Google Scholar]
- 36.Hutchinson M, Fox RJ, Miller DH, et al. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing–remitting multiple sclerosis: subgroup analyses of the CONFIRM study. J Neurol. 2013;260:2286–2296. doi: 10.1007/s00415-013-6968-1. [DOI] [PubMed] [Google Scholar]
- 37.Riley RD, Steyerberg EW. Meta-analysis of a binary outcome using individual participant data and aggregate data. Res Synth Methods. 2010;1:2–19. doi: 10.1002/jrsm.4. [DOI] [PubMed] [Google Scholar]
- 38.Cofield SS, Cutter GR, Lublin FD, Wolinsky JS, Conwit R. Variability in the EDSS: what does it mean for progression? 2008. Available from: http://www.combirx.org/Results/PosterPres/AAN_2008_EDSS_poster.pdf. Accessed March 5, 2014.
- 39.Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing–remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13:545–556. doi: 10.1016/S1474-4422(14)70049-3. [DOI] [PubMed] [Google Scholar]
- 40.Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res Synth Methods. 2012;3:80–97. doi: 10.1002/jrsm.1037. [DOI] [PubMed] [Google Scholar]
- 41.Kappos L, Weinshenker B, Pozzilli C, et al. Interferon beta-1b in secondary progressive MS: a combined analysis of the two trials. Neurology. 2004;63:1779–1787. doi: 10.1212/01.WNL.0000145561.08973.4F. [DOI] [PubMed] [Google Scholar]
- 42.Havrdova E, Gold R, Fox RJ, et al. Relapses requiring intravenous steroid use and MS-related hospitalizations: findings from the phase 3 DEFINE and CONFIRM studies. 2012. Available from: http://www.biogenidecconferences.com/ECTRIMS2012/BG-12_Havrdova_DEFINE-CONFIRMIVSteroid.html. Accessed March 5, 2014.
- 43.Miller AE, O’Connor P, Wolinsky JS, et al. Pre-specified subgroup analyses of a placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis. Mult Scler. 2012;18:1625–1632. doi: 10.1177/1352458512450354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller DH, Gold R, Fox RJ, et al. Effects of BG-12 on magnetic resonance imaging outcomes in relapsing–remitting multiple sclerosis: an integrated analysis of the phase 3 DEFINE and CONFIRM studies. 2012. Available from: http://www.biogenidecconferences.com/ECTRIMS2012/BG-12_Miller_PooledMRI.html. Accessed March 5, 2014.
- 45.Nixon RM, Bansback N, Brennan A. Using mixed treatment comparisons and meta-regression to perform indirect comparisons to estimate the efficacy of biologic treatments in rheumatoid arthritis. Stat Med. 2007;26:1237–1254. doi: 10.1002/sim.2624. [DOI] [PubMed] [Google Scholar]
- 46.Thompson SG, Higgins JP. How should meta-regression analyses be undertaken and interpreted? Stat Med. 2002;21:1559–1573. doi: 10.1002/sim.1187. [DOI] [PubMed] [Google Scholar]
- 47.Riley RD, Lambert PC, Staessen JA, et al. Meta-analysis of continuous outcomes combining individual patient data and aggregate data. Stat Med. 2008;27:1870–1893. doi: 10.1002/sim.3165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.