

Abstract
In “evolve-and-resequence” (E&R) experiments, whole-genome sequence data from laboratory-evolved populations can potentially uncover mechanisms of adaptive change. E&R experiments with initially isogenic, asexually reproducing microbes have repeatedly shown that beneficial de novo mutations drive adaptation, and these mutations are not shared among independently evolving replicate populations. Recent E&R experiments with higher eukaryotes that maintain genetic variation via sexual reproduction implicate largely different mechanisms; adaptation may act primarily on pre-existing genetic variation and occur in parallel among independent populations. But this is currently a debated topic, and generalizing these conclusions is problematic because E&R experiments with sexual species are difficult to implement and important elements of experimental design suffer for practical reasons. We circumvent potentially confounding limitations with a yeast model capable of shuffling genotypes via sexual recombination. Our starting population consisted of a highly intercrossed diploid Saccharomyces cerevisiae initiated from four wild haplotypes. We imposed a laboratory domestication treatment on 12 independent replicate populations for 18 weeks, where each week included 2 days as diploids in liquid culture and a forced recombination/mating event. We then sequenced pooled population samples at weeks 0, 6, 12, and 18. We show that adaptation is highly parallel among replicate populations, and can be localized to a modest number of genomic regions. We also demonstrate that despite hundreds of generations of evolution and large effective population sizes, de novo beneficial mutations do not play a large role in this adaptation. Further, we have high power to detect the signal of change in these populations but show how this power is dramatically reduced when fewer timepoints are sampled, or fewer replicate populations are analyzed. As ours is the most highly replicated and sampled E&R study in a sexual species to date, this evokes important considerations for past and future experiments.
Keywords: experimental evolution, population genomics, quantitative trait loci
Introduction
Evolutionary biology has long taken advantage of laboratory selection experiments to test fundamental questions about how populations change over time. Investigators can impose selection on a particular trait of interest with the goal of dissecting the genetic and physiological components of the trait. Disregarding the phenotype of interest, selection experiments can also be used to describe the properties and dynamics of adaptation. In recent years, the ubiquity of whole-genome sequencing has added a new dimension to selection experiments often termed “evolve-and-resequence” (E&R). Genomic data from evolved populations can be probed to associate particular sequence changes to adaptive phenotypes, as well as describe what happens to the genome under selection.
E&R experiments have been performed in many biological systems, including bacteria (Barrick et al. 2009; Woods et al. 2011; Tenaillon et al. 2012), yeast (Kao and Sherlock 2008, Lang et al. 2013), phage (Miller et al. 2011), Drosophila (Burke et al. 2010; Zhou et al. 2011; Turner et al. 2011, Turner and Miller, 2012), and mouse (Chan et al. 2012). E&R experiments with microbes have generally been more successful than those with sexually reproducing organisms in terms of both identifying the genes responsible for adaptive phenotypes and the depth of understanding of adaptive dynamics in the experiment. This is true for a number of reasons, the simplest being the sheer ease of handling. Microbes such as bacteria and yeast can be easily maintained at huge population sizes, in parallel with a large number of replicate populations, and can achieve hundreds of generations in a matter of weeks. Their small genome sizes make the cost of sequencing entire populations to high coverage reasonable, even for multiple populations at multiple timepoints over the course of an experiment. These features allow for improved experimental design and provide a wealth of information that simply cannot be obtained from selection experiments in sexual models.
On the other hand, microbial models lack important features of higher eukaryotes, chiefly the ability to sexually reproduce and shuffle genotypes via recombination, restricting generalizations across taxa. Asexual E&R experiments are typically initiated from isogenic base populations and hence adapt to novel environments via the accumulation of de novo beneficial mutations; except for the period during which selection acts upon a handful of competing mutations, populations remain devoid of genome-wide variation (see Burke 2012 for review). Although an asexual E&R experiment could be initiated from a genetically variable base population, theory predicts that one founder clone will eventually fix and all the initial variation lost. This situation is quite different from outbred sexual populations harboring standing variation that is present at the start of, and maintained throughout, a selection experiment. Recombination decouples flanking variants from sites under selection allowing different regions of the genome to evolve independently. The degree to which adaptation in sexual species is driven by standing genetic variation is currently a topic of debate (Burke et al. 2010; Hernandez et al. 2011; Sattath et al. 2011). An E&R experiment with Drosophila illustrates this tension; the authors concluded the vast majority of the evolutionary response was due to selection on standing variation (Burke et al. 2010), suggesting that the combination of standing variation and sex renders adaptation in sexual higher eukaryotes fundamentally different than what occurs with asexual microbes. However, comparing this experiment directly with microbial E&R experiments presents a “straw man” problem, because so many features of experimental design differ. For one, while effective population sizes were large with respect to other Drosophila work (Ne∼103), they were several orders of magnitude smaller than what is typical with microbes. This is notable because the likelihood of a de novo beneficial mutation arising in an evolving population will be higher in a larger population. In addition, the Drosophila experiment implicated several dozen large regions of the genome, and hundreds of genes, as being important for evolutionary change. An important application of the E&R technique is to identify regions of the genome associated with quantitative traits, there is a clear need to improve the ability to localize candidate genes. We have therefore designed an experiment using yeast that incorporates the ability to maintain genetic variation via sexual recombination. With this design, we present a model system with features that help us draw general conclusions about the nature of adaptation in sexual populations as well as resolve the signature of adaptive change in evolved populations to small regions of the genome.
The budding yeast Saccharomyces cerevisiae has a facultatively sexual cycle; it can reproduce through asexual cell division or sexual recombination under permitting conditions. Here, we take advantage of a panel of S. cerevisiae natural isolates that have been made stable heterothallic haploid strains (i.e. mating type switching does not occur) and can be easily outcrossed (Cubillos et al. 2009). This panel constitutes a substantial resource for classical and molecular genetic methods and samples natural variation from a variety of sources and locations worldwide (Liti et al. 2009, Bergström et al. 2014). Using these strains, Cubillos et al. (2013) devised a general strategy to generate pools of outbred diploid individuals from multiple rounds of recombination between four founder haplotypes. We used this outbred population (the SGRP-4X; Cubillos et al. 2013), as the common ancestor for a selection experiment (fig. 1, supplementary fig S1, Supplementary Material online). We then continue to impose outcrossing regularly over the course of the experiment, shuffling genomes and effectively establishing a sexual yeast model. In this study, we established 12 replicate populations that adapt to a novel environment—forced outcrossing—for 18 weeks. We obtained whole-genome sequence data from these populations at four different timepoints over the course of the experiment: initially, then after 6, 12, and 18 weeks. These weekly timepoints correspond to approximately 180, 360, and 540 mitotic generations (see Materials and Methods for discussion). While we impose a complex life cycle involving sex as well as vegetative growth, there is a precedent for this type of long-term culture regime in the literature (e.g. to test hypotheses regarding advantages of sex; cf. Goddard et al. 2005; Gray and Goddard 2012).
Fig. 1.
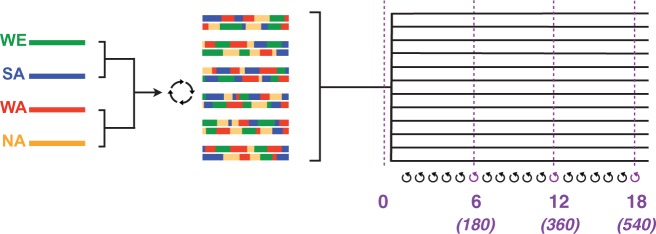
Schematic illustrating the experimental strategy. A four-way cross of diploid strains from different geographic origins (DBVPG6765, wine/European or WE in green; Y12, sake/Asian or SA in blue; DBVPG6044, W. African or WA in red; YPS128, N. American or NA in gold) generated a “synthetic population” that we used as the ancestral source for experimental populations (the SGRP-4X; Cubillos et al. 2013). Twelve replicate populations evolved in parallel and were sampled for sequencing at four timepoints: initially, and after 6, 12, and 18 rounds of forced outcrossing. Numbers in parentheses indicate the approximate number of mitotic divisions that have elapsed by each week.
We used this novel sexual outbred yeast system to address two evolutionary questions of fundamental importance. First, how replicable is evolutionary change at the molecular level? Highly replicated evolution experiments with Escherichia coli (Tenaillon et al. 2012) and yeast (Lang et al. 2013) suggest that adaptation is rarely repeatable at the level of individual mutations. As asexual microbial populations evolve via the accumulation of beneficial mutations, it is unclear the extent to which this lack of repeatability is a consequence of the first appropriate mutation becoming fixed, rather than the most appropriate mutation. Perhaps when selection can act on standing variation, evolution experiments are more reproducible. Here, we observe strong evidence that adaptation in these populations is fundamentally driven by standing variation, not new mutations; furthermore, results from the genomic regions showing the most change in this study suggest that the variants involved in adaptation are largely the same in independently evolving populations.
Second, how does the repeatability of evolution impact our ability to uncover mechanisms of adaptive change? The nascent E&R field has yet to converge on gold standards of experimental design, and these design features affect the power to detect the quantity and effect size of causative variants. E&R experiments in sexual systems must balance practical limitations against the goals of maintaining large population sizes, extensive replication, and repeated sampling over the course of a long period of time. Recent simulation studies (Baldwin-Brown et al. 2014; Kofler and Schlötterer 2014) suggest that high-powered E&R experiments should employ large population sizes, more generations of selection, and many more independent replicates than are currently employed in published Drosophila studies (Burke et al. 2010; Turner et al. 2011; Zhou et al. 2011; Orozco-ter Wengel et al. 2012; Turner and Miller 2012). Our work demonstrates that maximizing at least two of these parameters—the number of independent replicate populations and the number of sampled timepoints—is absolutely essential to an investigator’s ability to identify candidate genomic regions. We surveyed our complete data set for alleles that changed most significantly over time and identified a small number of highly localized regions genome-wide. However, applying the same analysis to smaller “downsampled” subsets of our data set suggests a much weaker signal involving a larger portion of the genome; this has important implications for the field, as E&R experiments with Drosophila and other sexual organisms historically have not implemented extensive replication schemes. Ours is the first empirical demonstration of the idea that the degree to which an E&R experiment is replicated determines the nature and scope of what we can learn about adaptive evolution.
Results
SNP/Indel Identification
We identified 75,410 high-quality biallelic single nucleotide polymorphisms (SNPs) in our data set that met the following conditions: 1) a mean coverage of between 50 and 150 averaged over all 36 evolved populations (12 independent replicate populations × 3 timepoints); 2) an observation of greater than 10 × coverage in each of the four haploid mat α founder strains; and 3) fixed in each of the founder strains (since the founders are isogenic haploid strains, polymorphic SNPs in a founder are artifacts). At SNPs, the average coverage depth among all 36 evolved populations was 115 × and the ancestral population, which received an additional two lanes of sequencing, had an average coverage of 175 × . We also identified 3,456 positions at which Genome Analysis Toolkit (GATK) called small insertion and deletion polymorphisms, and otherwise met the same criteria as SNPs in terms of coverage and founder genotypes. We created custom tracks for the UCSC genome browser such that the properties of these SNPs and indels can be easily visualized across the genome (see Materials and Methods).
We examined our 36 evolved population/timepoints for potential de novo mutations responding to selection. We find 15 such SNPs with a frequency ≥0.15 in at least one replicate population for at least one timepoint. The largest allele frequency over all 15 such SNPs and populations is 0.2, and there is a clear tendency to observe candidate de novo alleles at higher frequencies at more advanced generations of experimental evolution. Although the majority of these SNPs are intergenic, 3 of them are predicted to result in a nonsynonymous amino acid change; a leucine to phenylalanine polymorphism in YBR184W, a histidine to arginine polymorphism in YNL318C, and a glycine to serine polymorphism in UFE1 (supplementary table S1, Supplementary Material online). Despite these SNPs having allele frequencies of zero in the founder and ancestral populations, the derived alleles were always present in more than a single replicate population at some low frequency (supplementary table S1, Supplementary Material online). Thus it is possible that these mutations were present in the ancestral population at some very rare frequency or that they represent some sort of complex sequencing or alignment error, implying that they are in fact standing variants rather than mutations that arose over the course of the experiment.
SNP Frequency Change over Time
To identify sites that changed the most over time consistently in all 36 experimental populations, we fit a regression model to transformed allele frequencies as a function of time. We found the results from a regression on time to be essentially the same as those from more complex models, for example treating time as a factor or including a quadratic term in time (Materials and Methods, supplementary fig. S2, Supplementary Material online), so we focus on the simple model throughout the manuscript. We also did not find evidence of allele frequency change that was unique to individual replicate populations relative to the others (Materials and Methods, supplementary fig. S3, Supplementary Material online). Based on permutations, we identify two localized regions, on chromosomes 9 and 11, that exceed a genome-wide false positive rate of 5% (fig. 2a). If we consider a more liberal threshold that holds the genome wide false positive rate at 50%, we identify three additional significant regions on chromosomes 7, 13, and 16, suggesting a false discovery rate at this more liberal threshold of 10%. The five regions harboring significant SNPs are labeled peaks A-E in fig. 2a; significant SNPs in each region are generally contained in a 10 - to 20-kb interval. Supplementary fig. S4a–e, Supplementary Material online, provide closeup views of these regions including LOD confidence intervals and predicted effects of significant SNPs based on the snpEFF tool (Cingolani et al. 2012). We observe a total of 39 SNPs and 3 indels that exceed the false positive rate of 50% (supplementary tables S2 and S3, Supplementary Material online). Of these 42 polymorphisms, 29 occur in protein-coding regions of genes (11 are predicted to result in nonsynonymous substitutions, while 18 are predicted to result in synonymous substitution), while the remaining 13 are intergenic. The majority of these synonymous SNPs (13/18) are in phase, in terms of the four founder alleles, with a nonsynonymous SNP in the same gene; this is consistent with an explanation involving hitchhiking in response to selection acting on nonsynonymous SNPs. Significant SNPs that are intergenic tend to occur in “footprints” of the genome that are sensitive to cleavage by DNase I, implying candidate transcription factor binding sites (Hesselberth et al. 2009; Materials and Methods). For example, the three most significant SNPs in the data set occur in very close proximity to one another (peak C, chr11:614-615 kb) and the average measure of DNAse 1 hypersensitivity at these sites is 99; by contrast, the average measure of DNase 1 hypersensitivity among all significant sites is only 54 (supplementary table S2, Supplementary Material online).
Fig. 2.

Evidence of allele frequency change across the genome. (a) Sites that have changed the most over time in all replicate populations. Results of a genome scan for SNPs with significant variation in allele frequency between sampled timepoints (via an ANOVA on square root arcsin transformed allele frequencies treating “generation” as a continuous variable). y-Axis values indicate transformed p-values from this ANOVA, such that higher values indicate higher levels of significance; the blue and red horizontal lines represent our empirically determined genome-wide alpha of 0.5 and 0.05, respectively. We find five peak regions that exceed our lower threshold, and label these A–E. (b) Haplotype change across the genome, represented as the average difference between founder haplotype frequency at week 18 from the ancestral founder haplotype frequency. We find three additional peaks and label these F–H. Note that absolute frequency differences are plotted for easier visualization; relative frequency differences are plotted in supplementary fig. S6 for comparison.
Haplotype Frequency Change over Time
To quantify change in founder haplotype frequencies over time, we first estimated the frequency of each of the four founder alleles at every SNP for each experimental population at each timepoint (Materials and Methods). Figure 2b shows the difference between the evolved populations at the final timepoint (generation 540) relative to the ancestral frequency, for each of the four founder alleles, averaged across all replicate populations. The regions of high haplotype differentiation are localized and appear to implicate a greater number of candidate regions than the SNP frequencies alone (see Materials and Methods). This being said, the three regions of highest SNP frequency change in fig. 2a are all supported by high haplotype frequency differences. The additional regions where we observe evidence of high haplotype divergence but not high SNP frequency change are labeled peaks F–H (fig. 2b; supplementary fig. S4, Supplementary Material online).
Regions of high haplotype divergence are largely driven by a single founder haplotype. Peaks A, E, F, and G are all driven by founder Y12 (Asian sake strain), while peaks B and C are driven by founder DBVPG6044 (West African strain) and peak D is driven by founder DBVPG6765 (European wine strain). Peak H involves more complicated haplotype dynamics and shows evidence of change in two founder haplotypes (DBVPG6765 and Y12). Peak G occurs at a region where SNP representation is poor (supplementary fig. S5, Supplementary Material online), and while this does not appear to affect the haplotype signal, it is an unusual feature of the data set that deserves mention. There is a gap in the number of SNPs on chromosome 7 from 701.357 to 712.818 kb, and within this region a long terminal repeat retrotransposon (YGRWTy3-1) occurs from 707.196 to 712.545 kb. Short reads cannot be used to reliably call SNPs in repeats such as this Ty3 element. Directly upstream of this element, SNPs were filtered out of the data set because coverage at these sites was zero in at least some of the evolved replicate populations, and we suspect the proximity to YGRWTy3-1 made the alignment of short reads difficult in this interval. Interestingly, Sanger sequencing reads from this region (Liti et al. 2009) indicate the presence of a Ty3 element in the Y12 strain, but not the other three founder strains; this is notable as Y12 is the founder haplotype driving change at this peak, suggesting a causative effect of the retrotransposon insertion.
Dynamics of Allele Frequency Change
Among the most significant markers in the data set, average allele frequencies across all 12 replicate populations tend to change in a linear fashion over the course of the experiment, and as these frequencies approach fixation their rate of change slows (fig. 3). This same pattern is observed in the most significant markers in regions with high haplotype divergence. Although allele frequency trajectories in the individual 12 replicate populations at these sites do exhibit varying degrees of heterogeneity (supplementary fig. S7a–h, Supplementary Material online), for the most part this heterogeneity is subtle. In addition to this repeatability at the level of individual SNPs, we also observe a striking level of repeatability at the level of founder haplotype frequencies. Haplotype frequencies from each of the four founders are also consistent across replicate populations (supplementary fig. S8a–h, Supplementary Material online, show founder frequency change in peak regions over the course of the experiment). Because most significant regions are driven by change in a single founder haplotype, this means that the “driver” haplotype frequency changes in the same direction in all 12 replicate populations, while the other three “nondriver” haplotype frequencies change in the opposite direction in all 12 replicate populations (supplementary fig. S7a–h, Supplementary Material online).
Fig. 3.
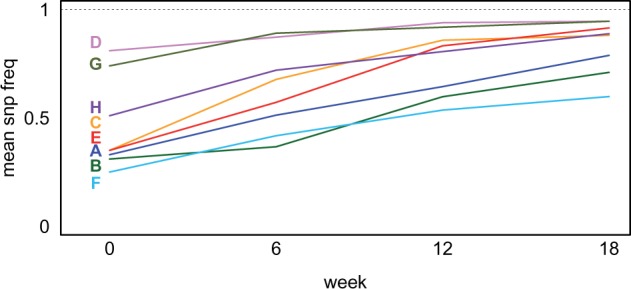
Allele frequency trajectories at the most significant SNPs in the data set. Allele frequencies at each timepoint, averaged over all replicate populations for peaks A-H, The allele being modeled is the most significant SNP in each peak region.
We also addressed the question of whether different linear models best describe different regions of the genome, as this observation implies that an allele frequency trajectory might depend on the individual locus under selection (e.g. alleles at some loci might plateau at intermediate frequencies, while others might increase until they become fixed). We fit multiple models to the data, including a linear model incorporating a quadratic term, and a linear model including time as a factor (Materials and Methods, supplementary fig. S2, Supplementary Material online). As all three of these models predicted the same regions as having the most significant allele frequency change over time, we conclude that a general linear response is the path that adaptive alleles take, across all sites in the genome. This linear response does “flatten” at later timepoints of the experiment, but only when frequencies approach zero or one (e.g. peaks C, D, E, G, H). For sites where allele frequencies are not extreme at later timepoints (e.g. peaks A, B, F), the trajectory of the selected allele will presumably continue to change in a linear fashion in future generations of the experiment.
Effects of Replication and Sampling
Evolution experiments with sexually reproducing organisms have historically employed low levels of replication (N ≤5) and few sampled timepoints (t ≤ 3) for practical reasons. We evaluated the signal of allele frequency change that would be detectable from the present data set, had we sampled fewer replicate populations or fewer timepoints. Figure 4 presents the results of such analysis. Applying our models to data from only five of the replicate populations results in much weaker signals of change across the genome; this identifies only a single peak (peak C, the most significant peak in the data set) and this peak only exceeds our more liberal 50% significance cutoff. Only considering two of our four timepoints has a similar effect when the model is applied to data from all 12 replicate populations sampled at generation 0 and generation 360, only peak C emerges. It is difficult to make a quantitative statement about the power to detect peaks when there are only two timepoints, given that this data structure is not amenable to permutation test we use to establish a null distribution. However, the qualitative signal appears dampened compared with the signal observed from four timepoints, insofar as far fewer localized peaks emerge, and any small peaks that do emerge do not necessarily match peaks A–H. Downsampling the data to simulate an experiment with only five replicate populations and only two timepoints leads to a noisy signal and essentially no ability to identify peaks A–H.
Fig. 4.

Effects of replication and sampling on signal. ANOVA analysis used to generate fig. 2 was carried out on (a) the entire data set consisting of all 12 replicate populations sampled at four timepoints (0, 180, 360, and 540 generations); (b) a data set consisting of only five replicate populations sampled at the same four timepoints; (c) a data set consisting of all twelve replicate populations sampled at two timepoints (zero and 360 generations) and (d) a data setconsisting of five replicate populations sampled at these two timepoints. Increasing both replication and sampling results in stronger, more localized signals of change.
Discussion
Selection Acts on Standing Genetic Variation, not New Mutations
We find that adaptation is overwhelmingly due to standing genetic variation in this experiment. The observation that the same candidate de novo SNPs are present in multiple replicate populations, as well as the observation of so few candidate de novo SNPs in the data set, provide strong evidence that in these large sexual populations with standing genetic variation, de novo mutations play a small role in the first several hundred generations of evolution. This result has been observed before, in E&R experiments with smaller effective population sizes (Burke et al. 2010; Johansson et al. 2010; Chan et al. 2012), but with the caveat that experimental populations are not large enough for beneficial de novo mutations to be likely in a single generation. Yeast populations in this study were bottlenecked to a minimum of 106 cells per generation, and the spontaneous mutation rate in S. cerevisiae is estimated at 3.3 ± 0.1 × 10−3 per diploid genome per generation (Zhu et al. 2014). It is pertinent to consider that the genetic background on which a de novo mutation arises is likely to play a key role in whether that mutation has a phenotypic effect; thus, the probability that a de novo mutation arises and has beneficial consequences effectively decreases Ne in this study. Although we cannot quantify this probability directly, we expect the emergence of potentially beneficial de novo mutations to be orders of magnitude higher in this experiment relative to experiments with other sexual species; however, these mutations did not contribute to adaptation. This begs the question of what conditions need to exist for new mutations to become an important source of variation for adaptive evolutionary change. The populations in this experiment have achieved over 500 generations, which is appreciable on an ecological timescale. Our results suggest the interplay between selection coefficient, population size, and duration of sustained selection in determining the role of new mutations versus standing variation in a population adapting to a novel environment; this is ultimately an empirical question, and we are optimistic that future E&R data sets will help resolve this ambiguity.
Founder Haplotype Data Improves Inference
The “synthetic population mapping” approach to the study of quantitative traits involves crossing a small number of inbred founder strains to generate a recombinant mapping population in which phenotype–genotype associations can be resolved to relatively small genomic regions (e.g. King et al. 2012). Associating haplotype frequency change, as well as individual marker frequency change, allows for better detection of candidate regions in synthetic mapping studies (e.g. Burke et al. 2014). We have found this general principle to apply to the current study. Measuring differences in founder haplotype frequencies from the ancestral state, per genomic position, uncovers an additional three localized regions that have dramatically differentiated over the course of the experiment (peaks F–H), compared to the five that were uncovered by screening for SNP frequency change alone (peaks A–E). Thus moving forward, applying the methods of synthetic population mapping to E&R studies will optimize opportunities to pinpoint regions of the genome important for adaptation in evolved laboratory populations.
Identifying Putatively Causative Sites
In E&R experiments, the chance that a causative SNP occurs within a 2-LOD support interval of the most significant marker in a region is high (>80%) when long-term Ne is greater than 103 and there are four or more founder haplotypes in the ancestral population (Baldwin-Brown et al. 2014, cf. fig. 4). The distance between the most significant marker in region and the actual causative site is also predicted to diminish as the number of replicate populations increases (r > 10), and the number of generations is high (n > 500; Baldwin-Brown et al. 2014, cf. fig. 3). Our experiment achieves these aspects of design, and implicates a small number of genes in a few narrow regions of the genome. We identified 2-LOD support intervals for the most significant marker of each peak; among peaks A–E, the smallest 2-LOD support interval is 8.8 kb (peak C), and the largest spans 20.5 kb (peak A). These intervals overlap a median of ten genes each. Therefore, we are able identify a much more modest number of genes/variants as potentially causative than what has generally been reported in E&R experiments with Drosophila (e.g. Burke et al. 2010; Zhou et al. 2010; Turner et al. 2011; Turner and Miller 2012; Orozco-ter Wengel et al. 2012). We attribute this to our improved ability to maximize experimental design parameters. The most significant SNP in peaks A, B, and D, are each predicted to be nonsynonymous (in genes KEL2, ATG32, and RSF1, respectively). The most significant marker in peak E is a synonymous polymorphism in LGE1. In peak C, the most significant marker does not occur in a coding region, but is associated with high values of DNAse 1 hypersensitivity, providing evidence for a regulatory consequence.
The intercrossed base population ancestral to our evolved populations was developed as a resource for quantitative-genetic experiments, and was generated by crossing the four founder strains 12 times. This intercrossing primarily imposed selection for increased sporulation efficiency, and the loci associated with this period of selection have been described (Cubillos et al. 2013). It is thus relevant to ask how these regions of change compare to those we identify in this study. If the regions of change overlap, this would suggest similar selection pressures in the 12 intercross rounds and the 18 weeks of forced outcrossing; if the regions do not overlap, this would provide evidence that the forced outcrossing regime of this study involves wholly different selective pressures. Our results support the latter scenario, as the regions of high allele frequency change reported by Cubillos et al. (2013) generally do not coincide with our peaks A–H (supplementary fig. S6, Supplementary Material online; compare with supplementary table S1, Supplementary Material online). Peak H is an exception to this pattern; the most significant SNP in this peak occurs 436,416 bases into chromosome 12, which is located 8 kb away from a position that changed in frequency during the intercross. After the 12 intercross rounds, the Asian and European founder haplotypes increased while the west African and north American founder haplotypes decreased. Over the 18 weeks of the current experiment, the Asian founder haplotype increased further while the European founder haplotype decreased. Thus at the region corresponding to peak H, we do observe evidence of change in the Asian founder haplotype during both the intercross and our longer-term experiment. However, the absence of such correspondence at the other peaks suggests that the selective pressures are different enough between these two studies that they can be considered independent regimes. This is not surprising as the sporulation conditions used during the intercross were quite different from those in the forced outcrossing treatments, including a much longer incubation time in sporulation media. In this context, it is potentially relevant that the most significant markers in our peaks D and E occur in genes annotated as influencing sporulation efficiency; both LGE1 and RSF1 null mutants reportedly have decreased sporulation efficiencies (Deutschbauer et al. 2002).
It is outside the scope of this article to validate potentially functional consequences of these allelic substitutions (via gene replacement experiments, for example). We find it notable that there are clear candidate alleles to point to in each peak, though it would be difficult to connect these to particular phenotypes in our experimental evolution regime, which likely imposes selection on several traits simultaneously. Our protocol involves several potentially competitive phases including mating, sporulation, exposure to reagents important for spore isolation, and growth in multiple types of media. In particular, growth in rich media in culture plates may incur stresses related to gas exchange or extended periods in stationary phase. That our selection regime requires both sex and asexual growth creates an additionally complex environment; it is probable that selection favors different alleles in these different phases, perhaps in an antagonistic manner. Clear tradeoffs have been demonstrated between the choice to outcross versus grow vegetatively (Zeyl et al. 2005), and each strategy has adaptive advantages in particular environments (Grimberg and Zeyl 2005; Tannenbaum 2008). Ultimately, we present a case of populations adapting to a novel environment, and are not preoccupied with the particular traits under direct selection. Our results show clear evidence that allelic change is the result of adaptation in these populations, rather than a null expectation of drift. Our system is thus more useful for addressing general questions about adaptation than questions about physiological mechanisms of adaptation, so we will limit speculation into the biological relevance of the candidate genes we have uncovered.
Selection on Standing Genetic Variants Is Repeatable and Elicits an Initial Linear Response
At loci where we observe evidence of significant allele frequency change over time, we also observe a general convergence among independent replicate populations (supplementary fig. S6a–h, Supplementary Material online). Generally, the selected allele changes in the same direction over time in all 12 replicate populations, though there are some rare instances of a single replicate population behaving differently from the others, at least for a part of the experiment (perhaps most notably in peaks A and E, supplementary fig. S7a and e, Supplementary Material online). These exceptions could conceivably represent examples of higher-order epistatic interactions, or an example of a modifier allele interfering with balancing selection. It bears noting that the analysis we have employed here was designed to detect sites in the genome that behaved similarly among all replicate populations. In an attempt to identify regions of the genome with more heterogeneous patterns of allele frequency change among replicates, we applied similar analyses to each population independently, but failed to identify convincing evidence of relevant sites (supplementary fig. S3, Supplementary Material online). Thus we feel that our original analysis, which resulted in an observed signal of highly localized significant P values, is strong evidence that convergence dominates the dynamics of this experiment. It is not clear whether additional replicate populations would improve our ability to detect a signal of heterogeneous allele frequency change, and this ambiguity represents an unresolved challenge in the field of experimental evolution.
Recent E&R work with asexual isogenic microbes suggests that mutations driving adaptive change are rarely shared among independently evolving replicate populations (Tenaillon et al. 2012; Lang et al. 2013). We might expect adaptation to proceed differently in populations of sexual, outbreeding eukaryotes. If adaptation is driven by standing variation, present in the multiple identical starting replicate populations, we would expect the same response in the direction of maximum additive genetic variance in all of them. Surveys of some wild populations are consistent with this expectation; for example, an ancestral haplotype for armor plating in three-spine sticklebacks has driven parallel evolution in multiple isolated populations of this species worldwide (Colosimo et al. 2005). We observe both evidence of adaptation on standing variation and evidence of highly parallel allele frequency trajectories among replicate populations. These observations contribute to a growing body of empirical work suggesting that initially isogenic asexual populations adapt to novel environments via different mechanisms than do sexual populations harboring a large amount of genetic variation.
In sexual E&R studies to date, it has not been possible to establish a general pattern of long-term allele frequency change. Experiments may be long, but as a consequence of history have not archived multiple timepoints (e.g. Burke et al. 2010); or experiments may have been sampled over time but with a relatively small number of generations elapsed (e.g. Orozco-ter Wengel et al. 2012). Our outbred sexual yeast system clearly shows the potential for characterizing the dynamics of alleles under selection, as 18 weeks of real time still represents a significant passage of evolutionary time (540 generations). As a point of reference, the longest running E&R study in a sexual species to date are Drosophila populations that have achieved over 600 generations but have been continually evolving in the lab since 1991 (Chippindale et al. 1997, Burke et al. 2010). Our yeast system therefore provides an opportunity to address the question of whether allele frequencies change in a consistent linear fashion over time, or plateau at an intermediate frequency at some late stage.
We conclude that our data are more consistent with a model of long-term allele frequency plateaus than a standard model of linear allele frequency change. Most of our significant alleles have a slowed rate of change at later stages of the experiment (fig. 3), and while this is expected of an allele with a positive selection coefficient in an infinite population, the initial rate of allele frequency increase is much greater, and the onset of its slowing is much earlier, than what the standard theory would predict (Falconer and Mackay 1996, p. 28). One scenario that could result in allele frequency plateaus has been formally modeled and invokes a phenotypic optimum being achieved before the allele being modeled reaches fixation, perhaps due to the concerted action of multiple loci (Chevin and Hospital 2008). If the response to selection is polygenic, as we expect in outbred sexual higher eukaryotes, selection coefficients at specific loci may not be constant over time and depend on responses at other loci; this dynamic view of selection coefficients provides a possible explanation for empirical observations of incomplete fixation. Another scenario consistent with allele frequency plateaus posits that adaptation in populations of diploids should generally lead to heterozygote advantage (Sellis et al. 2011). In this model, adaptation systematically generates genetic variation by promoting balanced polymorphisms that are expected to segregate at high frequencies. As such, adaptation proceeds through a succession of balanced states that should leave a signature of incomplete selective sweeps. It is important to note that both of these scenarios have been explicitly modeled for newly-arising mutations, and not standing variants; thus, our results foretell a need for these types of models that incorporate standing genetic variation to best interpret E&R data sets from outbred sexual populations.
Replication and Sampling in E&R Experiments
This E&R experiment employs a much larger number of replicate populations, much larger number of sampled timepoints, and many more generations than is typical for E&R experiments in sexual outbred populations. We previously published an E&R study with Drosophila populations from an exceptional resource in the field; with 5-fold replication, sampling at two timepoints, and over 600 generations of sustained evolution (Burke et al. 2010), this resource may represent the limit of what is feasible with a Drosophila model. To investigate the degree to which the number of replicate populations assayed impacts the detection of candidate regions, we repeated analysis of replicable SNP frequency change over time (i.e. the analysis of fig. 2a) on downsampled data sets including 1) a random set of five of our replicate populations; 2) the data from all of the 12 replicate populations at only generations 0 and 360; and 3) the data from only five replicate populations at these two timepoints. We find that increasing the number of replicate populations is crucial for our ability to detect significant sites; surveying the data from five replicates would have only identified a single peak exceeding our 50% false-positive threshold, and this peak would have been regarded as having 50% chance of being a false positive (fig. 4b). Thus, genomic data from a small number of replicate populations may lead investigators to poorly resolve those regions that are responding to selection. In addition, sampling genomic data from populations at the start of the experiment and again at only one other point has a very similar effect. The signal at all peak regions but one is dramatically decreased, and several non-peak regions (i.e., likely false positive) emerge. When the analysis is run on the data sampling only two timepoints and only five replicate populations, it becomes essentially impossible to distinguish peak regions.
We infer that surveying the genomes of populations from an E&R experiment with a small number of replicate populations and sampled timepoints (e.g., the design of Burke et al. 2010; Zhou et al. 2011; Turner et al. 2011, Turner and Miller, 2012), might lead investigators to conclude that a large proportion of the genome is responding to evolution, but that no individual candidate region is a major determinant of change. By contrast, the same analysis applied to a 12-fold replicated experiment that sample four timepoints implies that only a few (5–8) highly significant regions are responsible for adaptation genome-wide. We therefore conclude that replication and generational sampling are of paramount importance to experimental design in E&R experiments.
While experimental evolution is a widely used method in evolutionary biology research (Garland and Rose 2009), the incorporation of whole-genome sequencing is new and optimal design standards for E&R projects have yet to be established. Recent theoretical work has shown that the idealized parameter space for an E&R experiment, in terms of the power to detect candidate causal sites, includes a design with more than 25 independent populations, effective population sizes of >103, and a total number of generations >500 (Baldwin-Brown et al. 2014). To the best of our knowledge, ours is the first empirical validation of the idea that within the framework of a single E&R experiment, the number of replicate populations and generations sampled dramatically impacts the inferences that can be made about how adaptive evolution proceeds.
Materials and Methods
Forced Outcrossing Scheme
Yeast biology limits the ability to exclusively force cells to undergo meiosis rather than mitotic budding, so we imposed forced outcrossing once per week (supplementary fig. S1, Supplementary Material online). Diploids from a four-way cross of strains from different geographic origins (DBVPG6765, wine/European or WE; DBVPG6044, W. African or WA; YPS128, N. American or NA; Y12, sake/Asian or SA) were used as the ancestral source for experimental populations. This ancestor was developed via 12 rounds of outcrossing as detailed in previous work (Parts et al. 2011; Cubillos et al. 2013). This base population is extremely efficient at sporulation, such that random spore analysis techniques (adapted from Bahalul et al. 2010) are fairly easy to implement.
Essentially, populations are transferred to liquid potassium acetate sporulation media on Fridays, and sporulate for ∼60 h (supplementary fig. S1, Supplementary Material online). Random spores are recovered on Mondays via a combination of exposure to Y-PER protein extraction reagent (Thermo Scientific) to kill unsporulated cells and mechanical agitation with 400 µm silica beads (OPS Diagnostics) to disrupt asci. Haploid cells then mate and dropout plates (ura- lys-) were used to recover a/α diploids. In addition to undiluted “culture” plates, multiple “titer” plates were prepared using dilute culture from each population to provide accurate estimates of the number of diploids that resulted from successful mating. After ∼48 h of growth, colonies were counted on titer plates to estimate the number of colony forming units present in the “culture” plate. We then scraped a proportion of each culture plate into 1 ml of yeast peptone dextrose (YPD) media, aiming to impose an effective population size on each replicate as close as possible to 106 (for example, if we estimated there to be 10 million colonies growing on the culture plate, only 1/10 of the surface of this plate was sampled for the next phase of culture). One microliter of this population sample was then transferred to 1 ml of liquid YPD in every other well of 24-well culture plates (Phenix); alternate wells contained YPD only to monitor for contamination events. 6 mm glass beads were added to wells for improved aeration, and culture plates were sealed with gas-permeable adhesive film (ABgene). Culture plates were shaken in humid chambers at 30 °C for 24 h, after which 1 µl (an estimated 106 cells) of overnight culture was transferred to a fresh culture plate containing 1 ml of YPD media. The remainder of each population was archived in 15% glycerol and frozen at −80 °C. To avoid potential contamination, seals were never removed from plates and instead were punctured by pipette tips. After a second period of overnight growth, cells were washed and again transferred to 10 ml sporulation media in individual flasks. Multiple times throughout the experiment, we assayed individual populations for both the number of cells that initiated growth in liquid YPD, and the density of cells in those populations at stationary phase 24 h later. Each assay of population growth indicated that the input of cells into liquid YPD was consistently between 1 and 5 million cells among populations, and that cell density at stationary phase was between 0.8 and 4 billion cells/ml. Based on these samples of cell density, we estimate that approximately 10 cell doublings occurred during each phase of growth in liquid media, for a total of 20 “generations” per week. Some noncompetitive growth occurs immediately following mating on dropout plates, and we were also able to estimate that this period of growth involves 10 cell doublings. We therefore use 30 as our measure of the number of generations that occur between forced outcrossing events, although it is important to note that this involves a complicated life cycle including 10 generations of noncompetitive growth as well as 20 generations of competitive growth in liquid media (during which we would expect selection pressure for allele frequency change to be higher).
DNA Isolation and Sequencing
DNA was isolated from 1 ml of overnight culture in YPD (∼109 diploid cells) from all experimental populations at four timepoints: week 0 (the ancestor), week 6, week 12, and week 18. The standard protocol for the Gentra Puregene Yeast/Bacteria Kit (Qiagen, USA) was used to collect DNA from the entire mixed population at once. DNA was also collected from the four isogenic founder strains in the same fashion. Sequencing libraries were constructed using the Nextera Library Preparation Kit (Illumina), and the 36 experimental populations, four founder strains, the ancestor were each given unique barcodes, normalized, and pooled together. Libraries were run on SR75 lanes of a HiSeq2500 instrument at the UCI Genomics High Throughput Facility (additional details of library prep and sequencing are available upon request).
We have developed a processing pipeline for estimating allele frequencies in each population directly from our pooled sequence data. We created bam files from the raw reads using Stampy (Lunter and Goodson 2011), which we empirically found performed better than BWA when there is the potential for highly diverged regions of the genome. We then merged the bam files and called SNPs and INDELs using the GATK pipeline (McKenna et al. 2010). We have custom PERL scripts for creating SNP frequency tables from the “.vcf” files (which contain the reference and non-reference counts) suitable for downstream analysis in R (www.R-project.org, last accessed September 9, 2014).
We downloaded the database of (DNAse I) hypersensitivity footprints described in (Hesselberth et al. 2009; http://noble.gs.washington.edu/proj/footprinting/, last accessed September 9, 2014), aligned it to the reference genome using bwa, and combined the resulting aligned reads to make a coverage “.bed” file (genomeCoverageBed -bg -trackline -ibam PMC2668528.bam -g ref/S288c.fasta.fai > coverage.bed). We similarly made bed files from the locations of indel polymorphisms in the data set. Files and information for visualizing these tracks in the UCSC Genome Browser are available as supplementary data, and we also host them separately so that the following commands can be pasted directly into “custom tracks”:
track type=vcfTabix name="yeast SNPs” bigDataUrl=http://wfitch.bio.uci.edu/∼tdlong/SantaCruzTracks/only-PASS-Q30-SNPs.vcf.gz
track type=vcfTabix name="yeast INDEL” bigDataUrl=http://wfitch.bio.uci.edu/∼tdlong/SantaCruzTracks/only-PASS-Q30-INDEL.vcf.gz
http://wfitch.bio.uci.edu/∼tdlong/SantaCruzTracks/coverage.bed.gz
SNP Frequency Change over Time
To identify genetic polymorphisms evolving according to different evolutionary forces we took two approaches: 1) a genome scan for SNPs exhibiting dramatic variation in frequency among timepoints and 2) a genome scan for de novo mutations that appeared in our population(s) and subsequently respond to selection. First, we carried out an ANOVA on square root arcsin transformed allele frequencies, for three different linear models: A) treating “generation” as a continuous regressor, B) treating “generation” as a factor, and C) treating “generation” as a continuous regressor but including a quadratic term in the model. These models detect SNPs with large frequency changes from one sampled generation to another, as well as SNPs where frequency change is dramatic but nonlinear (e.g., a plateau in frequency at later generations), assuming a homogeneous response among replicate populations. All of these linear models were weighted by the square root of the population allele count, or coverage, per position.
The –log10 transformed P values from these analyses are very similar to logarithm of the odds (LOD) scores (Broman and Sen 2009). To generate null distributions for these scores (i.e., distributions of these scores associated with a null expectation of genetic drift rather than selection), we shuffled the population identifiers of replicate population number and generation for the entire data set, such that frequencies were still associated with their “real” position. We chose not to shuffle the first timepoint (generation 0) as it was shared among all populations. Thousand of these dummy data sets were created, and the previously described linear models were fit to allele frequencies in each. For each of the models we took the most significant marker from the entire genome scan from each permutation and used the quantile function in R to define thresholds that hold the genome wide false positive rate at 5 or 50%. We applied the same permutation test to the downsampled data set consisting of all four timepoints but only five replicate populations. We could not apply this method to the down-sampled data sets with two replicate timepoints, due to all timepoints sharing the same ancestor, there are only 13 or 6 possible permutations of these data.
We also attempted to detect significant heterogeneity among populations, that is to say, positions at which alleles changed significantly in only one or a few populations, with an analysis of covariance approach to test for interactions between “replicate population” and “generation”. We fit the data to a linear model regressing square root arcsin transformed allele frequencies on both generation and replicate population, and we also fit the data to a more complex model including an interaction term (generation:replicate population). An ANOVA comparing these two models gave us P-values for significance of the interaction term, which would be indicative of heterogeneous allele frequency change across replicate populations. We also fit these models to null data sets as described above to establish significance thresholds. Supplementary fig. S3, Supplementary Material online, shows the results of this ANCOVA across the genome. While three points exceed our 5% genome-wide false positive rate, these do not show the characteristic localization of our major results (compare to fig. 2 and supplementary fig. S2, Supplementary Material online). Thus it is not likely that these are examples of true heterogeneity in this context; visualizing allele frequency trajectories at these candidate sites also does not reveal a pattern of heterogeneity.
Finally, to identify candidate de novo beneficial mutations, we scanned the genomes of the 12 evolved replicate populations for alleles that satisfied three criteria: 1) those not present in the ancestral base population (i.e. the base population after the initial intercross rounds); 2) those not present in any of the four founder populations; and 3) those that had achieved a frequency ≥0.15 in at least one evolved population, at least one timepoint. Thus, we conditioned candidate de novo beneficial mutations on those that would have arisen after the start of our experiment. It is possible that some of these mutations could have been present in the ancestor at a very rare frequency, as our initial SNP ascertainment was conditioned on a mean coverage of between 50 and 150 averaged over all populations.
Estimating Founder Haplotype Frequencies
As genotypes of the four founder strains are completely known, for any small region of the of the genome we can estimate the most likely set of founder haplotype frequencies that explains the vector of SNP frequency estimates for that region. Using a 10-kb sliding window with a 2 kb step size, at a given genomic position, we used the set of SNPs within 5 kb on either side of that position to determine the most likely set of founder haplotype frequencies that would produce the observed set of SNP frequencies. We considered the set of founder haplotype frequencies that minimized the following quantity to be the most likely set of founder haplotype frequencies at the focal position:
where MAFi is the minor allele frequency at the ith SNP, n = number of SNPs within 5 kb of either side of the ith SNP, fi,j is the allelic state (1 = minor allele, 0 = major allele) of the jth founder at the ith SNP, and hj is the haplotype frequency of the jth founder for the window under consideration. The set of four haplotype frequencies (the hj’s) for the window are the quantities being optimized. Optimization was achieved using the optim function in R. Individual haplotype frequencies were bounded by 0.001 on the lower end. Individual haplotype frequencies were bounded by 0.001 rather than zero to avoid convergence to a local minima.
We used a quantile–quantile plot approach to identify critical values for haplotype frequency change, since we could not devise a permutation test we could apply to estimated haplotype frequencies. We calculated the mean absolute difference in square root arcsin transformed allele frequency between each haplotype’s frequency in the ancestral population and it’s frequency at the final timepoint over all 12 replicate populations and 4 haplotypes. This statistic, , as the mean of 48 independent identically distributed random variables (of unknown distribution) should converge to a normally distributed random variable of unknown mean and variance. We sorted all the values of from the genome-wide scan and estimated the mean and variance of the normal distribution most consistent with the data from the smallest 4,000 such values (by minimizing sums of squares between expected normal quantiles and observed quantiles). We fit only the first 4,000 quantiles, since visually there does not appear to be haplotype divergence over most of the genome, if there is in fact divergence, our test is conservative. Given estimates of the mean and variance we can plot observed values of
against expected values, a Q-Q plot (supplementary fig. S9a, Supplementary Material online). From the Q-Q plot it is visually apparent that values of bigger than 0.2 are very unlikely under the null hypothesis of no divergence. We then plotted against genome position, and observe that the regions corresponding to values greater than 0.2 correspond to regions of high haplotype and allele frequency divergence (supplementary fig. S9b, Supplementary Material online, fig. 2).
Supplementary Material
Supplementary figs. S1–S9 and tables S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank E. Morimoto for assistance with the yeast population culture. M.K.B. was awarded a scholarship to attend the 2011 Yeast Genetics and Genomics Course, where valuable discussion of the project took place. This work was supported by NIH R01 RR024862, NIH R01 GM085251, and a Borchard Foundation Scholar-in-Residency grant to A.D.L.
References
- Bahalul M, Kaneti G, Kashi Y. Ether-zymolyase ascospore isolation procedure: an efficient protocol for ascospores isolation in Saccharomyces cerevisiae yeast. Yeast. 2010;27:999–1003. doi: 10.1002/yea.1808. [DOI] [PubMed] [Google Scholar]
- Baldwin-Brown JG, Long AD, Thornton KR. The power to detect quantitative trait loci using resequenced, experimentally evolved populations of diploid, sexual organisms. Mol Biol Evol. 2014;31(4):1040–1055. doi: 10.1093/molbev/msu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1274. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Bergström A, Simpson JT, Salinas F, Barré B, Parts L, Zia A, Nguyen Ba A, Moses AM, Louis EJ, Mustonen V, et al. A high-definition view of functional genetic variation from natural yeast genomes. Mol Biol Evol. 2014;31(4):872–888. doi: 10.1093/molbev/msu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Sen S. A guide to QTL mapping with R-qtl. New York: Springer; 2009. [Google Scholar]
- Burke MK. How does adaptation sweep through the genome? Insights from long-term selection experiments. Proc Roy Soc B. 2012;279:5029–5038. doi: 10.1098/rspb.2012.0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke MK, Dunham JP, Shahrestani P, Thornton KR, Rose MR, Long AD. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467:587–590. doi: 10.1038/nature09352. [DOI] [PubMed] [Google Scholar]
- Burke MK, King EG, Shahrestani P, Rose MR, Long AD. Genome-wide association study of extreme longevity in Drosophila melanogaster. Genome Biol Evol. 2014;6(1):1–11. doi: 10.1093/gbe/evt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YF, Jones FC, McConnell E, Bryk J, Buenger L, Tautz D. Parallel selection mapping using artificially selected mice reveals body weight control loci. Curr Biol. 2012;22:794–800. doi: 10.1016/j.cub.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Chevin LM, Hospital F. Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics. 2008;180:1645–1660. doi: 10.1534/genetics.108.093351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chippindale AK, Alipaz JA, Chen HW, Rose MR. Experimental evolution of accelerated development in Drosophila. 1. Developmental speed and larval survival. Evolution. 1997;51:1536–1551. doi: 10.1111/j.1558-5646.1997.tb01477.x. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Jr., Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM, et al. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science. 2005;307:1928–1933. doi: 10.1126/science.1107239. [DOI] [PubMed] [Google Scholar]
- Cubillos FA, Louis EJ, Liti G. Generation of a large set of genetically tractable haploid and diploid Saccharomyces strains. FEMS Yeast Res. 2009;9:1217–1225. doi: 10.1111/j.1567-1364.2009.00583.x. [DOI] [PubMed] [Google Scholar]
- Cubillos FA, Parts L, Salinas F, Bergström A, Scovacricchi E, Zia A, Illingworth CJR, Mustonen V, Ibstedt S, Warringer J, et al. High-resolution mapping of complex traits with a four-parent advanced intercross yeast population. Genetics. 2013;195:1141–1155. doi: 10.1534/genetics.113.155515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, Williams RM, Chu AM, Davis RW. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99(24):15530–15535. doi: 10.1073/pnas.202604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. 1996 Introduction to quantitative genetics. 4th ed. Harlow: Pearson/Prentice Hall. [Google Scholar]
- Garland T, Rose MR. Experimental evolution. Berkeley: University of California Press; 2009. [Google Scholar]
- Goddard MR, Godfray CJ, Burt A. Sex increases the efficacy of natural selection in experimental yeast populations. Nature. 2005;434:636–640. doi: 10.1038/nature03405. [DOI] [PubMed] [Google Scholar]
- Gray JC, Goddard MR. Sex enhances adaptation by unlinking beneficial from detrimental mutations in experimental yeast populations. BMC Evol Biol. 2012;12:43. doi: 10.1186/1471-2148-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimberg B, Zeyl C. The effects of sex and mutation rate on adaptation in test tubes and to mouse hosts by Saccharomyces cerevisiae. Evolution. 2005;59(2):431–438. [PubMed] [Google Scholar]
- Hernandez RD, Kelley JL, Elyashiv E, Melton SC, Auton A, McVean G, 1000 Genomes Project. Sella G, Przeworski M. Classic selective sweeps were rare in recent human evolution. Science. 2011;331:920–924. doi: 10.1126/science.1198878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson AM, Pettersson ME, Siegel PB, Carlborg Ö. Genome-wide effects of long-term divergent selection. PLoS Genet. 2010;6:e1001188. doi: 10.1371/journal.pgen.1001188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40:1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EG, Merkes CM, McNeil CL, Hoofer SR, Sen S, Broman KW, Long AD, Macdonald SJ. Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 2012;22:1558–1566. doi: 10.1101/gr.134031.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Schlötterer C. A guide for the design of evolve and resequencing studies. Mol Biol Evol. 2014;31:474–483. doi: 10.1093/molbev/mst221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500:571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, et al. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–341. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CR, Joyce P, Wichman HA. Mutational effects and population dynamics during viral adaptation challenge current models. Genetics. 2011;187:185–202. doi: 10.1534/genetics.110.121400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco-ter Wengel P, Kapun M, Nolte V, Kofler R, Flatt T, Schlötterer C. Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles. Mol Ecol. 2012;21:4931–4941. doi: 10.1111/j.1365-294X.2012.05673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Cubillos FA, Warringer J, Jain K, Salinas F, Bumpstead SJ, Molin M, Zia A, Simpson JT, Quail MA, et al. Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res. 2011;21:1131–1138. doi: 10.1101/gr.116731.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattath S, Elyashiv E, Kolodny O, Rinott Y, Sella G. Pervasive adaptive protein evolution apparent in diversity patterns around amino acid substitutions in Drosophila simulans. PLoS Genet. 2011;7:e1001302. doi: 10.1371/journal.pgen.1001302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellis D, Callahan BJ, Petrov DA, Messer PW. Heterozygote advantage as a natural consequence of adaptation in diploids. Proc Natl Acad Sci U S A. 2011;108(51):20666–20671. doi: 10.1073/pnas.1114573108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannenbaum E. A comparison of sexual and asexual replication strategies in a simplified model based on the yeast life cycle. Theory Biosci. 2008;127:323–333. doi: 10.1007/s12064-008-0049-5. [DOI] [PubMed] [Google Scholar]
- Tenaillon O, Rodriguez-Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS. The molecular diversity of adaptive convergence. Science. 2012;335:457–461. doi: 10.1126/science.1212986. [DOI] [PubMed] [Google Scholar]
- Turner TL, Miller PM. Investigating natural variation in Drosophila courtship song by the evolve and resequence approach. Genetics. 2012;191:633–642. doi: 10.1534/genetics.112.139337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TL, Stewart AD, Fields AT, Rice WR, Tarone AM. Population-based resequencing of experimentally evolved populations reveals the genetic basis of body size variation in Drosophila melanogaster. PLoS Genet. 2011;7:e1001336. doi: 10.1371/journal.pgen.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, Lenski RE. Second-order selection for evolvability in a large Escherichia coli population. Science. 2011;331:1433–1436. doi: 10.1126/science.1198914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyl C, Curtin C, Karnap K, Beauchamp E. Antagonism between sexual and natural selection in experimental populations of Saccharomyces cerevisiae. Evolution. 2005;59(10):2109–2115. [PubMed] [Google Scholar]
- Zhou D, Udpa N, Gersten M, Visk DW, Bashir A, Xue J, Frazer KA, Posakony JW, Subramaniam S, Bafna V, et al. Experimental selection of hypoxia-tolerant Drosophila melanogaster. Proc Natl Acad Sci U S A. 2011;108:2349–2354. doi: 10.1073/pnas.1010643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 2014;111(22):E2310–E2318. doi: 10.1073/pnas.1323011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.