

Abstract
We previously demonstrated that activated microglia release excessive glutamate through gap junction hemichannels and identified a novel gap junction hemichannel blocker, INI-0602, that was proven to penetrate the blood–brain barrier and be an effective treatment in mouse models of amyotrophic lateral sclerosis and Alzheimer disease. Spinal cord injury causes tissue damage in two successive waves. The initial injury is mechanical and directly causes primary tissue damage, which induces subsequent ischemia, inflammation, and neurotoxic factor release resulting in the secondary tissue damage. These lead to activation of glial cells. Activated glial cells such as microglia and astrocytes are common pathological observations in the damaged lesion. Activated microglia release glutamate, the major neurotoxic factor released into the extracellular space after neural injury, which causes neuronal death at high concentration. In the present study, we demonstrate that reduction of glutamate-mediated exitotoxicity via intraperitoneal administration of INI-0602 in the microenvironment of the injured spinal cord elicited neurobehavioral recovery and extensive suppression of glial scar formation by reducing secondary tissue damage. Further, this intervention stimulated anti-inflammatory cytokines, and subsequently elevated brain-derived neurotrophic factor. Thus, preventing microglial activation by a gap junction hemichannel blocker, INI-0602, may be a promising therapeutic strategy in spinal cord injury.
Key words: : functional recovery, gap junction hemichannel inhibitor, microglia, glutamate, spinal cord injury
Introduction
Spinal cord injury (SCI) causes tissue damage in two successive mechanisms. The initial injury is mechanical and directly causes primary tissue damage, which induces subsequent ischemia, inflammation, and neurotoxic factor release resulting in the secondary tissue damage. Accumulation of activated glial cells such as microglia and astrocytes is a common pathological observation in the damaged lesion.1 Activated microglia release a variety of neurotoxic factors that induce neurodegeneration at the injury site. The major neurotoxic factor released into the extracellular space after neural injury is glutamate,2 which causes neuronal death at high concentration.3 Microglial glutamate induces excitotoxicity via N-methyl-D-aspartate (NMDA) receptor signaling and increase in Ca2+ influx.4–6 As a result, calmodulin-dependent protein kinase is activated, which increases nitric oxide (NO) production and inhibits the mitochondrial respiratory chain complex IV activity followed by intracellular energy loss, leading to eventual neuronal death.7
Axon regeneration in the adult central nervous system (CNS) during the chronic phase is limited by axon-growth inhibitors. Axon-growth inhibitors include myelin-associated proteins expressed by oligodendrocytes and chondroitin sulfate proteoglycans (CSPGs) expressed by astrocytes.8 Inhibition of secondary tissue damage can reduce these chronic phase problems. Thus, preventing microglial activation has been considered as a candidate of promising therapeutic strategy. Microglia, however, also play a neuroprotective role by mediating neurotrophic factor release and sequestering neurotoxic substances.4,9–13 Therefore, the required therapy is specific inhibition of neurotoxicity by activated microglia.
Recently, we found that activated microglia release excessive glutamate through gap junction hemichannels (connexine 32 hemichannels).4 Our previous study revealed that lipopolysaccharide-induced glutamate release only from microglia among the CNS cells in vitro and INI-0602 significantly ameliorated the elevated glutamate levels.14 Both in vitro and in vivo, such gap junction blockers as glycyrrhetinic acid, carbenoxolone (CBX), and connexin mimetic peptides decreased the amount of glutamate released by activated microglia and prevented neuronal death in a dose-dependent manner.15 Nevertheless, these compounds are unlikely therapeutic strategies because they cannot penetrate an intact blood–brain barrier (BBB) and can induce pseudoaldosteronism. These problems were solved with the invention of the novel gap junction hemichannel blocker, INI-0602.14 As a gap junction hemichannel blocker and a BBB permeable molecule, INI-0602 is a proven and effective treatment in mice models of amyotrophic lateral sclerosis (ALS) and Alzheimer disease.
Previous report described a number of activated microglia cells in the injured spinal cord.16 In fact, activated microglia can be more prevalent in SCI than brain injury.16 Nevertheless, whether the role microglia plays in SCI is neurotoxic or neuroprotective, or both, remains uncertain.17,18 Considering that activated microglia induce neurotoxicity by releasing excessive glutamate, we hypothesize that blocking microglial glutamate release can protect against secondary tissue damage after SCI. Blocking microglial glutamate release could therefore provide a promising therapeutic strategy in SCI.
Methods
Reagents
The gap junction hemichannel blocker INI-0602 was developed as described previously.14 The INI-0602 used in this study was chemically synthesized and purified by the Nard Institute (Osaka, Japan). The purity of the compound was greater than 99%.
Surgical procedures
We used 7–9-week-old, adult, female C57BL/6 mice (30 mice in total; weight, 16–21 g; SLC, Shizuoka, Japan). All experiments were performed in accordance with the ethical guidelines of the Nagoya University Institutional Animal Care and Use Committee. The mice were provided free access to water and food.
After anesthesia was induced with 1.5% halothane in an oxygen/nitrous oxide gas mixture, we performed a laminectomy at vertebral level T9–T10. The dura was opened, and the dorsal half of the spinal cord was transected to a 1-mm depth using a pair of extra-fine microscissors. The muscle layers and skin were then closed with suture. After the surgery, the mice recovered for 1 h on a warming blanket.
Administration of drug
Just before surgery, the mice were randomly divided into two groups that were injected (intraperitoneally) with either phosphate buffered saline (PBS, group A) or INI-0602 (100 mg/kg, group B). After surgery, administrations were continued with 100 mg/kg of INI-0602 (group B) or an equal volume of PBS (group A) every other day for 4 weeks until day 28 (Fig. 1).
FIG. 1.
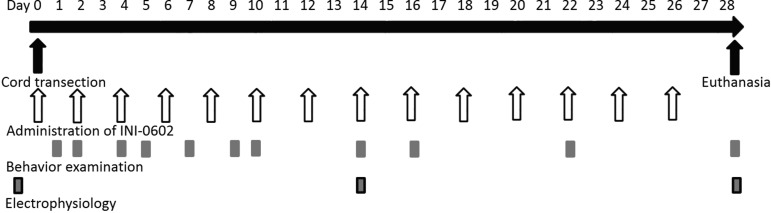
Diagram of the experimental protocol. Time schedules of each procedure are shown including INI-0602 administration, spinal cord transection, behavioral testing (Basso mouse scale, inclined plane test), electrophysiology, and the day of euthanasia.
Immunohistochemistry
Ten mice were deeply anesthetized with a barbiturate overdose and intracardially perfused with PBS, followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer. The lesioned spinal cord was removed and postfixed overnight in the same fixative. The fixed tissues were embedded in paraffin, and 3-μm sections were prepared for hematoxylin and eosin (H&E) stain and immunohistochemistry.
Immunohistochemical staining was performed according to the manufacturer's instructions. Briefly, sections were pre-blocked with the blocking reagent (REAL Peroxidase-Blocking Solution, DAKO, Glostrup, Denmark), and antigen retrieval was performed. After the sections reacted with rabbit anti-glial fibrillary acidic protein (GFAP; DAKO), anti-neurofilament (NF; Nichirei, Tokyo, Japan), and anti-CD68 antibodies (DAKO), we used diaminobenzidine to develop a subsequent polymer reagent (Chemomate Envision kit/HRP, DAKO). The corresponding tissue sections were then routinely stained for morphological evaluation using H&E stain.
Accumulation data for these stains were determined automatically using Image J software (NIH). Image mean intensity and stain area were analyzed statistically, as described below. These data were obtained from four animals in each group.
RNA extraction and quantitative reverse transcription-polymerase chain reaction (RT-PCR)
Previously frozen injured spinal cords were removed at the length of 1 mm at 2 weeks after injury. Total RNA was isolated from these samples using an RNeasy extraction kit (Qiagen, Hilden, Germany) with DNase treatment. cDNA was prepared using SuperScript III RNase H-Reverse Transcriptase (Invitrogen) and 0.2 ng random hexamer primer (6-mer, Fermentas). cDNA was pooled from three animals from each group treatment group (PBS or INI-0602).
Quantitative RT-PCR (qRT-PCR) was performed on a Light Cycler 480 II real-time PCR system (Roche) using STAR oligo designed primers with THUNDERBIRD SYBR qPCR Mix (Toyobo, Japan) under the following thermocycling conditions: initial denaturation at 95°C for 1 min followed by 45 cycles at 95°C for 10 sec, 55°C for 10 sec, and 72°C for 1 min, melting at 95°C for 5 sec and 55°C for 1 min, and cooling at 50°C for 30 sec. All samples were run in triplicate. The primer sequences are listed in Table 1. The resulting values were normalized to the endogenous control (glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) that was consistent among all samples. Results were compared using the fits point method. GADPH was indicated as 1 standard value; other values were shown as the ratio.
Table 1.
Primer Sequences
| GAPDH | F: CACGAACGAGTCCCTAGAGC |
| R: TCACATCACCACGTCCTTGT | |
| TNF-α | F: AGCCCCCAGTCTGTATCCTT |
| R: CTCCCTTTGCAGAACTCAGG | |
| IL-1 | F: GCCCATCCTCTGTGACTCAT |
| R: AGGCCACAGGTATTTTGTCG | |
| IL-6 | F: AGTTGCCTTCTTGGGACTGA |
| R: TCCACGATTTCCCAGAGAAC | |
| BDNF | F: GCGGCAGATAAAAAGACTGC |
| R: CTTATGAATCGCCAGCCAAT |
Basso Mouse Scale (BMS) for locomotion
We measured hindlimb motor function recovery in 15 mice from each treatment group using the BMS for locomotion.19 We videotaped each animal's behavior for 5 min, and two investigators scored (on a scale of 0–9) both hindlimbs at 1, 2, 4, 5, 7, 9, 14, 21, and 28 days after SCI. The scores for both hindlimbs were averaged, which provided a single value per animal at each time point.
Inclined plane test
Inclined plane test was performed to test each animal's ability to maintain its position on the incline at 1, 2, 4, 5, 7, 9, 14, 16, 22, and 28 days after injury. The mice were placed on a flat plane in the horizontal position, and the board was slowly inclined to the vertical position. The maximum inclination at which a mouse could maintain itself for 5 sec was recorded. Three trials were performed for each testing session. In practice, the angle was either increased or decreased by 5-degree intervals.
Electrophysiology
Transcranial electrical motor nerve evoked-action potentials (MEPs) measured signal conduction in motor pathways at 2 and 4 weeks after SCI. When MEPs was recorded, the mice were anesthetized with 1.5% halothane and maintained in 1.0% halothane in oxygen. All recordings were performed using standard clinical electromyographical analysis with a 3000-Hz hi-cut filter and a 30-Hz low-cut filter (Nicolet, Endeavor, Madison, WI). Electrical stimulation (1.1 Hz, 20 mA) was delivered over the cranium at an area 1 mm lateral to bregma. Evoked responses were recorded from the contralateral femoral muscle.
The data measured on day 0 (before SCI) was indicated as 1 for standard value; other values were shown as the ratio each mouse. The data were obtained from five animals in each group.
Statistics
Averages for the two experimental groups are expressed as mean±standard error of the mean, which were compared statistically using the Student t test. Statistical significance was determined at the p<0.05 level.
Results
Intraperitoneal administration of INI-0602 protects the spinal cord against injury
SCI resulted in severe neural tissue damage (Fig. 2A) and activated reactive astrocytes, which subsequently induced glial scarring (Fig. 2B). Axons were destroyed at the injury site and collapsed at the lateral funiculus (Fig. 2C). In addition, CD68, a marker for activated microglia, indicated robust accumulation of activated microglia (Fig. 2D). Finally, H&E staining revealed destruction of the dorsal half of the spinal cord, above the central canal, with neutrophil (segmented nuclei) and lymphocyte (small nuclei) accumulation (Fig. 2-I).
FIG. 2.

Intraperitoneal administration of INI-0602 protects the spinal cord against injury. (A) Hematoxylin and eosin (H&E) stained sections of spinal cord injury sites, showing severe neural tissue damage of the dorsal half of the spinal cord. (B) Glial fibrillary acidic protein (GFAP) immunohistochemical staining, showing activated reactive astrocytes, which subsequently induced glial scarring hypertrophy of GFAP-positive reactive astrocytes. (C) Neurofilament (NF) immunohistochemical staining. NF-stained transverse sections demonstrate significant loss of neural fibers. (D) CD68, a marker for activated microglia, indicated robust accumulation of activated microglia. (E) The neural tissue damage was decreased in H&E stained sections. (F) GFAP-positive reactive astrocytes were restricted in the dorsal half of the spinal cord. (G) NF-stained tissue were relatively preserved in the ventral half of the spinal cord. (H) CD68 positive cells were scarcely detectable. (I) Magnification image of H&E stained sections revealed destruction of the dorsal half of the spinal cord, above the central canal, with neutrophil (segmented nuclei) and lymphocyte (small nuclei) accumulation. (J) The immunopositive staining intensities of GFAP, NF, and CD68 were calculated using image J software. We observed that INI-0602 treatment significantly reduced the intensities of GFAP and CD68 staining whereas it increased that of NF staining.*p<0.05. All error bars represent standard error of the mean.
To investigate the effect of INI-0602 on SCI, we examined the expression of GFAP, NF, and CD68 at the injury site 2 weeks post-injury. The immunopositive staining intensities of GFAP, NF, and CD68 were calculated using image J software.20 We observed that INI-0602 treatment significantly reduced the intensities of GFAP and CD68 staining, whereas it increased that of NF staining (Fig. 2E–H and J). These results indicate that INI-0602 treatment reduced glial scar formation and microglia activation, and thereby prevented subsequent neurofilament destruction.
INI-0602 decreased pro-inflammatory cytokine expression and increased brain-derived neurotrophic factor (BDNF) expression
In ALS models, active microglia release pro-inflammatory mediators that cause neuroinflammation and neural tissue damage.21 To quantify the extent to which INI-0602 affected the microenvironment of the injured spinal cord, we used qRT-PCR to examine both pro-inflammatory cytokine and neurotrophic factor expression (Fig. 3). We observed that INI-0602 treatment decreased tumor necrosis factor (TNF)-α and interleukin (IL)-1 at 1 and 2 weeks, but increased BDNF levels mostly at 1 week. Of interest, the Il-6 level increased at 1 week and decreased at 2 weeks.
FIG. 3.

INI-0602 decreased pro-inflammatory cytokine expression and increased brain-derived neurotrophic factor (BDNF) expression. INI-0602 treatment decreased tumor necrosis factor (TNF)-α and interleukin (IL)-1 at 1 and 2 weeks, but increased IL-6 and BDNF levels mostly at 1 week. *p<0.05. The values represent relative expression compared with non-treatment controls at each time point. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
INI-0602 improved behavior scores
The recovery of hindlimb locomotor function was quantified with the BMS and the inclined plane test. Mice were evaluated for locomotor recovery for 4 weeks after SCI. On day 1 post-injury, all mice exhibited an average BMS score for 2 hindlimbs <1.5, and there was no significant difference between the two groups (1.03±0.39 vs. 0.83±0.44). The BMS of the mice treated with INI-0602 showed immediate improvement beginning with day 2 post-injury, and then improved gradually over the assessment period (from 1.03±0.39 on day 1 to 7.5±1.69 on day 28). Those treated with PBS also gradually improved (from 0.83±0.44 on day 1 to 3.7±1.60 on day 28) but at a much lesser rate than those treated with INI-0602 (Fig. 4A). The inclined plane test also revealed improvement of motor function (Fig. 4B).
FIG. 4.

Behavioral studies. We assessed functional recovery by using two independent behavioral tasks: the Basso Mouse Scale (BMS) score (A) and the inclined plane test (B). All injured mice exhibited an average BMS score for 2 hindlimbs <1.5 on the first day post-spinal cord injury (SCI). The BMS of the mice treated with INI-0602 showed immediate improvement beginning with day 2 post-injury, and then improved gradually over the assessment period; on the other hand, those treated with phosphate buffered saline also gradually improved but at a much lesser rate than those treated with INI-0602. *p<0.05 (A). The inclined plane test also revealed improvement of motor function. *p<0.05 (B). All error bars represent standard error of the mean.
INI-0602 improved electrophysiological scores
Measurement of the MEP revealed a marked decrease in the action potential amplitude and prolonged latency after SCI (Fig. 5). The amplitude showed a 76% decrease (Fig. 5A), and latency was prolonged 1.08 times at 4 weeks after SCI in the control group (Fig. 5B). Animals in the INI-0602 group showed significantly better recovery at 2 and 4 weeks after SCI (p<0.05).
FIG. 5.

Electrophysiology. Transcranial electrical motor nerve evoked-action potentials (MEPs). Amplitude (A) and latency (B) were measured at days 14 and 28 after injury. INI-0602 treatment significantly increased the amplitude whereas it also significantly decreased the latency.*p<0.05. All error bars represent standard error of the mean.
Discussion
In the present study, we provide the first evidence that the gap junction hemichannel inhibitor, INI-0602, can treat SCI. INI-0602 reduced glutamate release and optimized the host microenvironment by disrupting glial scar formation and increasing growth factor expression that is normally deficient in SCI. INI-0602 also successfully promoted motor function repair in mice with SCIs, as well as ischemic brain injury, ALS, and Alzheimer disease.14,15
Our previous studies demonstrate that glutamate is released by microglia through connexin 32 hemichannels.4 On the other hand, there are reports that connexin 43 is up-regulated after SCI and that blocking of connexin 43 may become a potential therapeutic target.22,23 Recent studies, however, showed that mimetic peptides specific for connexin 32 (32gap 24 and 32gap 27), connexin 43 (43gap 27), or Panx1 (10panx1) non-specifically block both connexins and pannexins.24 Moreover, INI-0602 also nonspecifically blocks these hemichannels. Therefore, our data have clarified that hemichannels play a pivotal role in the lesion development within SCI; however, further investigation is needed to determine which hemichannel is critical for this process, although many challenges remain because of technical limitations.
One issue is the heterogeneity of gap junctions and hemichannels. The different connexin isoforms structurally interact in various ways. Homomeric hemichannels consist of a single connexin isoform. Heteromeric hemichannels contain two or more different connexin isoforms. Homotypic gap junction channels are composed of two identical hemichannels, whereas heterotypic gap junction channels are formed by two different hemichannels. Thus, compositional patterns of gap junctions can be categorized into four types: homomeric and homotypic; heteromeric and homotypic; homomeric and heterotypic; and heteromeric and heterotypic. As has been observed in connexin-knockout studies, the potential for various connexins to compensate for the loss of other isoforms complicates the analysis of this system.25
Another problem is the dysfunction of fluorescently tagged and/or overexpressed connexins. Tagging and/or overexpression of connexins in cultured cells often produces abnormally large gap junction plaques.26–28 Tagging the amino termini of connexins also results in nonfunctional channels, whereas tagging the carboxyl termini alters the properties of the channels.29,30 Unfortunately, few good quality antibodies against each connexin are available for immunostaining/immunoblotting. Moreover, as mentioned, existing inhibitors of specific connexins often non-specifically block connexins and pannexins. Therefore, future studies should detail spatiotemporal expression profiles of connexin isoforms under pathological conditions in the CNS, which will necessitate specific blockers and tracers for each connexin isoform, hemichannel, and gap junction.
Although neurons also release glutamate, microglia release larger quantities of excitatory amino acid than neurons.31 We previously observed that extracellular glutamate concentration was reduced by blockade of either gap junction hemichannel or NMDA receptor in a dose-dependent manner in ischemic neural tissue injury.15 Hemichannels in activated microglia may be more exposed to the extracellular space and provide a unique surface for molecular drug target than those in unactivated microglia and other CNS cells. Thus, an inhibitor of hemichannel may preferentially suppress glutamate release from microglia, although it also may act on neuronal and astrocytic gap junctions.14 Further, microglia appear in the spinal cord at levels higher than those observed in the brain after injury.16 Thus, INI-0602 shows considerable potential for treating SCI. By reducing glutamate release, INI-0602 reduced detrimental neuron-glia interactions that subsequently reduced secondary tissue damage in both subacute and chronic phases.
The INI-0602 dose in a previous study using an ALS model mice was 20 mg/kg. In the present study, however, we used the dose of 100 mg/kg. One reason for increasing the dose is that neural tissue damage in SCI is immediate and more severe than what is seen in degenerative diseases such as ALS. The intraperitoneal administration in this study did limit the quantity of this drug that could be dispensed because of the small volume of mice. Microglia proliferation increases after SCI, and peaks from 48 h to 72 h after the injury.32 Therefore, INI-0602 should be administered immediately after the injury, or at least in the early phase of SCI.
Microglial activation is considered the upstream of a vicious feed-forward cycle of neuronal damage and glial activation, which is likely fueled by microglia-derived glutamate. In this study, INI-0602 treatment suppressed both neuronal damage and glial activation while INI-0602 itself did not alter microglial activation. A previous study has demonstrated that blocking connexin 43 ameliorated SCI and was accompanied by a reduction in glial activation, suggesting that connexin blockade effectively inhibits the propagation and amplification of neuroinflammation initiated by the injury.22,23
Injured cells contain toxic molecules at high concentrations, such as Ca2+, K+, NO, and reactive oxygen species (ROS). These toxic molecules are propagated from injured cells to healthier cells through gap junctions. Pathological conditions also induce uncoupled hemichannels to open, leading to a paracrine transfer of toxic molecules.33,34 These waves of death signaling activate astrocytes and microglia, causing the release of more toxic molecules including glutamate, ROS, NO, and pro-inflammatory cytokines and chemokines. This vicious amplification spiral of signaling could worsen neuroinflammation by recruiting leukocytes and thereby increase the lesion area.35,36
INI-0602 is also suggested to halt this feed-forward cycle and reduced glial activation indirectly by inhibiting microglial glutamate release. As a result, surviving neurons and neuroprotective glial cells (astrocytes and microglia) is suggested to enhance BDNF production. Increased BDNF release helps to reduce detrimental neuron-glia interactions and subsequently increase neural tissue survival. Moreover, blocking gap junctions may confer neuroprotection because both neural and astrocytic gap junctions contribute to ischemic neuronal damage.34,37 Taken together, it is likely the beneficial effect produced by INI-0602 improved the microenvironment at the SCI site in the present study.
Glial scars disrupt axonal growth after SCI. CSPGs in post-injury glial scars are undesirable for axonal regeneration, especially in chronic SCI. In a previous study, we found that suppressing reactive astrocytes reduced glial scar formation and improved motor function in mice with SCI. This was accomplished using a liposome-mediated interferon (IFN)-β gene delivery method, or by IFN-β delivery with intravenous injection of genetically engineered neural stem cells.38,39
Inhibition of glial scar formation is a potential strategy for treating SCI. Whether reactive astrocytes are beneficial is still controversial. Some studies report that reactive astrocyte migration is beneficial and limits inflammatory cell infiltration in the subacute injury phase.40 Regardless of whether the reactive astrocytes are beneficial, glial scar formation is unfavorable for motor function recovery, because it disrupts axonal regeneration.41 INI-0602 reduced the number of reactive astrocytes, and likely inhibited glial scar formation without directly inhibiting these astrocytes. Consistent with our previous results, this contributed to improved recovery as quantified by the scores in our behavioral measures.
Conclusion
Mice were administered INI-0602 after SCI, which reduced glutamate excitotoxicity by inhibiting microglial activation. Reducing glutamate excitotoxicity subsequently promoted self-regeneration within the injured spinal cord microenvironment and suppressed glial scar formation. By blocking neutrophil and pro-inflammatory cytokine infiltration, anti-inflammatory cytokines levels were increased and contributed to elevated BDNF levels. As a result, neurobehavioral recovery was attained, as indicated by the inclined plane test and the BMS for locomotion. INI-0602 has been used clinically for other purposes that further indicate INI-0602 decreases neurodegeneration mediated by microglia and protects neural tissue after SCI.
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science's Grant-in-Aid for Research Activity Start-up Grant Numbers 24890086 (DU), Japan Ministry of Education, Culture, Sports, Science and Technology's Grant-in-Aid for Scientific Research on Innovative Areas (AN, HT), the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (HT), a Grant-in Aid from the Ministry of Health, Labour and Welfare of Japan (HT) and Japan Society for the Promotion of Science's Grant-in-Aid for Strategic Young Researcher Overseas Visit Program for Accelerating Brain Circulation (TW).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Takeuchi H. (2010). Neurotoxicity by microglia: Mechanisms and potential therapeutic strategy. Clin. Exp. Neuroimmunol. 1, 12–21 [Google Scholar]
- 2.Benveniste H., Jorgensen M.B., Sandberg M., Christensen T., Hagberg H., and Diemer N.H. (1989). Ischemic damage in hippocampal CA1 is dependent on glutamate release and intact innervation from CA3. J. Cereb Blood Flow Metab. 9, 629–639 [DOI] [PubMed] [Google Scholar]
- 3.Choi D.W. (1985). Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 58, 293–297 [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi H., Jin S., Wang J., Zhang G., Kawanokuchi J., Kuno R., Sonobe Y., Mizuno T., and Suzumura A. (2006). Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 281, 21362–21368 [DOI] [PubMed] [Google Scholar]
- 5.Barger S.W., and Basile A.S. (2001). Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J. Neurochem. 76, 846–854 [DOI] [PubMed] [Google Scholar]
- 6.Piani D., Spranger M., Frei K., Schaffner A., and Fontana A. (1992). Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur. J. Immunol. 22, 2429–2436 [DOI] [PubMed] [Google Scholar]
- 7.Mungrue I.N., and Bredt D.S. (2004). nNOS at a glance: implications for brain and brawn. J. Cell Sci. 117, 2627–2629 [DOI] [PubMed] [Google Scholar]
- 8.Yiu G., and He Z. (2006). Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 7, 617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kempermann G., and Neumann H. (2003). Neuroscience. Microglia: the enemy within? Science 302, 1689–1690 [DOI] [PubMed] [Google Scholar]
- 10.Kipnis J., Avidan H., Caspi R.R., and Schwartz M. (2004). Dual effect of CD4+CD25+regulatory T cells in neurodegeneration: a dialogue with microglia. Proc. Natl. Acad. Sci. U. S. A. 101, Suppl 2, 14663–14669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koenigsknecht J., and Landreth G. (2004). Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J. Neurosci. 24, 9838–9846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwab J.M., and Schluesener H.J. (2004). Microglia rules: insights into microglial-neuronal signaling. Cell Death Differ. 11, 1245–1246 [DOI] [PubMed] [Google Scholar]
- 13.Zietlow R., Dunnett S.B., and Fawcett J.W. (1999). The effect of microglia on embryonic dopaminergic neuronal survival in vitro: diffusible signals from neurons and glia change microglia from neurotoxic to neuroprotective. Eur. J. Neurosci. 11, 1657–1667 [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi H., Mizoguchi H., Doi Y., Jin S., Noda M., Liang J., Li H., Zhou Y., Mori R., Yasuoka S., Li E., Parajuli B., Kawanokuchi J., Sonobe Y., Sato J., Yamanaka K., Sobue G., Mizuno T., and Suzumura A. (2011). Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer's disease. PloS one 6, e21108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeuchi H., Jin S., Suzuki H., Doi Y., Liang J., Kawanokuchi J., Mizuno T., Sawada M., and Suzumura A. (2008). Blockade of microglial glutamate release protects against ischemic brain injury. Exp. Neurol. 214, 144–146 [DOI] [PubMed] [Google Scholar]
- 16.Olson J.K. (2010). Immune response by microglia in the spinal cord. Ann. N. Y. Acad. Sci. 1198, 271–278 [DOI] [PubMed] [Google Scholar]
- 17.Popovich P.G., Guan Z., Wei P., Huitinga I., van Rooijen N., and Stokes B.T. (1999). Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp. Neurol. 158, 351–365 [DOI] [PubMed] [Google Scholar]
- 18.Horn K.P., Busch S.A., Hawthorne A.L., van Rooijen N., and Silver J. (2008). Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J. Neurosci. 28, 9330–9341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basso D.M., Fisher L.C., Anderson A.J., Jakeman L.B., McTigue D.M., and Popovich P.G. (2006). Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma 23, 635–659 [DOI] [PubMed] [Google Scholar]
- 20.Suzuki H., Fukuyama R., Hasegawa Y., Tamaki T., Nishio M., Nakashima T., and Tatematsu M. (2009). Tumor thickness, depth of invasion, and Bcl-2 expression are correlated with FDG-uptake in oral squamous cell carcinomas. Oral Oncol. 45, 891–897 [DOI] [PubMed] [Google Scholar]
- 21.Philips T., and Robberecht W. (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 10, 253–263 [DOI] [PubMed] [Google Scholar]
- 22.O'Carroll S.J., Gorrie C.A., Velamoor S., Green C.R., and Nicholson L.F. (2013). Connexin43 mimetic peptide is neuroprotective and improves function following spinal cord injury. Neurosci. Res. 75, 256–267 [DOI] [PubMed] [Google Scholar]
- 23.Cronin M., Anderson P.N., Cook J.E., Green C.R.,. J. and Becker D.L. (2008). Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Mol. Cell. Neurosci. 39, 152–160 [DOI] [PubMed] [Google Scholar]
- 24.Wang J., Ma M., Locovei S., Keane R.W., and Dahl G. (2007). Modulation of membrane channel currents by gap junction protein mimetic peptides: size matters. Am. J. Physiology Cell Physiol. 293, C1112–C1119 [DOI] [PubMed] [Google Scholar]
- 25.Bedner P., Steinhauser C. and Theis M. (2012). Functional redundancy and compensation among members of gap junction protein families? Biochim. Biophys. Acta 1818, 1971–1984 [DOI] [PubMed] [Google Scholar]
- 26.Gaietta G., Deerinck T.J., Adams S.R., Bouwer J., Tour O., Laird D.W., Sosinsky G.E., Tsien R.Y., and Ellisman M.H. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503–507 [DOI] [PubMed] [Google Scholar]
- 27.Hunter A.W., Jourdan J., and Gourdie R.G. (2003). Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun. Adhes. 10, 211–214 [DOI] [PubMed] [Google Scholar]
- 28.Lopez P., Balicki D., Buehler L.K., Falk M.M., and Chen S.C. (2001). Distribution and dynamics of gap junction channels revealed in living cells. Cell Commun. Adhes. 8, 237–242 [DOI] [PubMed] [Google Scholar]
- 29.Contreras J.E., Saez J.C., Bukauskas F.F., and Bennett M.V. (2003). Gating and regulation of connexin 43 (Cx43) hemichannels. Proc. Natl. Acad Sci. U. S. A. 100, 11388–11393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bukauskas F.F., Jordan K., Bukauskiene A., Bennett M.V., Lampe P.D., Laird D.W., and Verselis V.K. (2000). Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Aci. U. S. A. 97, 2556-–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogata T., Nakamura Y., Shibata T., and Kataoka K. (1992). Release of excitatory amino acids from cultured hippocampal astrocytes induced by a hypoxic-hypoglycemic stimulation. J. Neurochem. 58, 1957–1959 [DOI] [PubMed] [Google Scholar]
- 32.Morino T., Ogata T., Horiuchi H., Takeba J., Okumura H., Miyazaki T., and Yamamoto H. (2003). Delayed neuronal damage related to microglia proliferation after mild spinal cord compression injury. Neurosci. Res. 46, 309–318 [DOI] [PubMed] [Google Scholar]
- 33.De Vuyst E., Decrock E., De Bock M., Yamasaki H., Naus C.C., Evans W.H., and Leybaert L. (2007). Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. . Biol. Cell 18, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson R.J., Zhou N., and MacVicar B.A. (2006). Ischemia opens neuronal gap junction hemichannels. Science 312, 924–927 [DOI] [PubMed] [Google Scholar]
- 35.Orellana J.A., Saez P.J., Shoji K.F., Schalper K.A., Palacios-Prado N., Velarde V., Giaume C., Bennett M.V., and Saez J.C. (2009). Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antiox. Redox. Signal. 11, 369–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeuchi H. (2013). Roles of glial cells in neuroinflammation and neurodegeneration. Clin. Exp. Neuroimmunol. 4, 2–16 [Google Scholar]
- 37.Lin J.H., Weigel H., Cotrina M.L., Liu S., Bueno E., Hansen A.J., Hansen T.W., Goldman S., and Nedergaard M. (1998). Gap-junction-mediated propagation and amplification of cell injury. Nat. Neurosci. 1, 494–500 [DOI] [PubMed] [Google Scholar]
- 38.Ito M., Natsume A., Takeuchi H., Shimato S., Ohno M., Wakabayashi T., and Yoshida J. (2009). Type I interferon inhibits astrocytic gliosis and promotes functional recovery after spinal cord injury by deactivation of the MEK/ERK pathway. J. Neurotrauma 26, 41–53 [DOI] [PubMed] [Google Scholar]
- 39.Nishimura Y., Natsume A., Ito M., Hara M., Motomura K., Fukuyama R., Sumiyoshi N., Aoki I., Saga T., Lee H.J., Wakabayashi T., and Kim S.U. (2013) Interferon-beta delivery via human neural stem cell abates glial scar formation in spinal cord injury. Cell Transplant. 22,2187–2201 [DOI] [PubMed] [Google Scholar]
- 40.Okada S., Nakamura M., Katoh H., Miyao T., Shimazaki T., Ishii K., Yamane J., Yoshimura A., Iwamoto Y., Toyama Y., and Okano H. (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 12, 829–834 [DOI] [PubMed] [Google Scholar]
- 41.Rolls A., Shechter R., and Schwartz M. (2009). The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 10, 235–241 [DOI] [PubMed] [Google Scholar]