


Abstract
Mammalian synthetic biology may provide novel therapeutic strategies, help decipher new paths for drug discovery and facilitate synthesis of valuable molecules. Yet, our capacity to genetically program cells is currently hampered by the lack of efficient approaches to streamline the design, construction and screening of synthetic gene networks. To address this problem, here we present a framework for modular and combinatorial assembly of functional (multi)gene expression vectors and their efficient and specific targeted integration into a well-defined chromosomal context in mammalian cells. We demonstrate the potential of this framework by assembling and integrating different functional mammalian regulatory networks including the largest gene circuit built and chromosomally integrated to date (6 transcription units, 27kb) encoding an inducible memory device. Using a library of 18 different circuits as a proof of concept, we also demonstrate that our method enables one-pot/single-flask chromosomal integration and screening of circuit libraries. This rapid and powerful prototyping platform is well suited for comparative studies of genetic regulatory elements, genes and multi-gene circuits as well as facile development of libraries of isogenic engineered cell lines.
INTRODUCTION
Programming mammalian cells with large synthetic gene networks is expected to play a central role in helping elucidate complex regulatory cellular mechanisms (1–4), implementing new useful biological functions (5–7) and accelerating the design of novel tailor-made therapeutic treatments (8–14). However, our limited ability to precisely engineer and predict the behavior of these genetic programs in mammalian cells remains a major challenge (8). Toward systematic and rational engineering of mammalian cells, new tools and methods are required that enable rapid prototyping and validation of genetic circuits in a standardized manner.
Stable chromosomal integration of genetic payloads can help achieve long-term expression of transgenes. Given the pleiotropic effect of the integration locus on transgene expression, it is critical to be able to study and compare the function of the integrated genetic components, genes or networks in the same genomic context (15). Gene transfer methods, such as retroviruses, lentiviruses and transposons, are therefore not well suited because they result in random integration and the copy number of the integrated payload is not controlled well. Moreover, such techniques often limit the size of the payload to a few kilobases and do not tolerate the presence of repetitive sequences, which is often essential for genetic circuits comprising multiple transcription units. Several approaches have been developed that focus on targeted integration of foreign DNA into a transcriptionally active locus. Recent engineering of meganucleases, zinc finger nucleases (ZFN), TALENs and CRISP/Cas9 systems enable efficient integration of small DNA fragments at the locus of choice in mammalian chromosomes (16–19). However, such strategies involve double-strand break repair by homologous recombination or non-homologous end joining, which can lead to frequent head-to-tail concatemer integrations (15), partial integration of the DNA fragments (Supplementary Figure S1) or sequence alteration close to the target site (20) and are therefore not well suited for single copy integration of large multi-gene payloads. Moreover, time-consuming clonal expansion and insert verification are almost always required due to the high frequency of off-target and multi-copy integrations (21). Alternatively, precise integration of intact constructs can be achieved using site-specific recombination technologies (22–25), although the use of these techniques for integration of genetic networks in mammalian cells has not been demonstrated yet.
To address these challenges, we developed a comprehensive framework for simple and efficient generation of engineered cell lines that stably express multi-component genetic networks in the same chromosomal context (Figure 1). Our strategy consists of three main components: (i) engineering of monoclonal chassis (landing pad) cell lines, (ii) fast and modular assembly of large synthetic circuits and (iii) targeted integration of the assembled circuits into the landing pad of the chassis cell lines with an efficient Bxb1 site-specific recombinase. Once the chassis cell line is generated, our method allows us to proceed from genetic parts (genes, promoters of choice) to functional assays of assembled and integrated circuits in mammalian cells in as little as 20 days. We demonstrate that the unique combination of very high integration efficiency, specificity and integrity (intact, functional payload) provided with our method enables rapid generation of nearly isogenic polyclonal cell populations characterized by highly homogenous and correlated transgene expression. We show scalability of the approach by construction, targeted chromosomal integration and functional validation of the largest mammalian genetic circuit that has been integrated to date (42 parts, 6 transcription units, 27 kb). Finally, we performed a one-flask integration of a multi-gene payload library to demonstrate the suitability of our method for high-throughput screening of circuit libraries. As a whole, our method paves the way for robust and fast prototyping of synthetic genetic networks and provides a modular platform to streamline the generation of engineered isogenic stable cell lines, useful for a broad research community.
Figure 1.
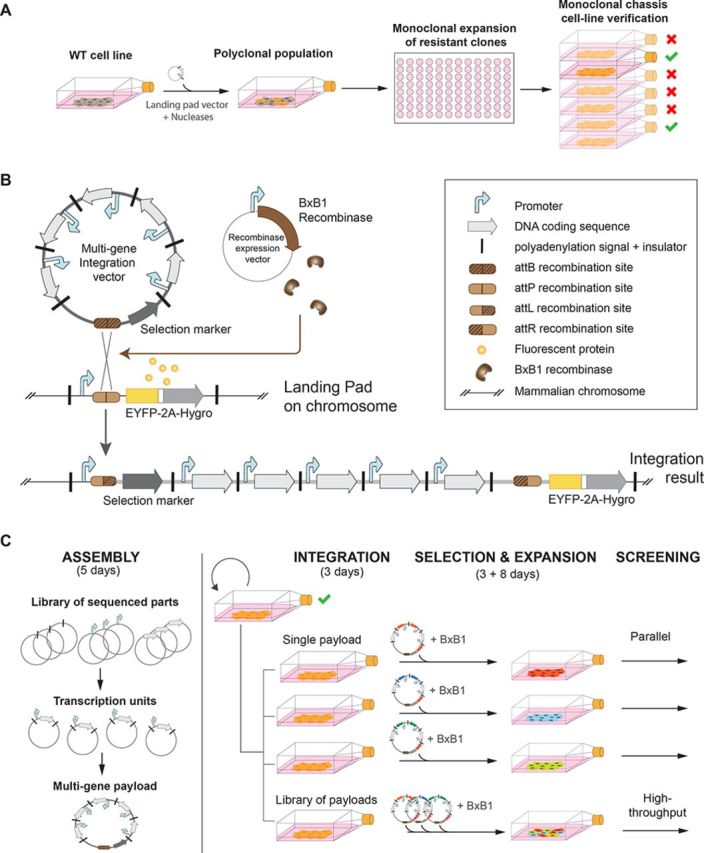
Overview of the circuit construction and integration framework. (A) First phase: chassis cell-line generation. A landing pad is integrated into the locus of a WT cell line of choice using a zinc-finger nuclease, TALEN or CRISPR-Cas system. Integration events are then selected with hygromycin and resistant clones are expanded. Finally, Southern blots and PCRs are performed on different clones to verify the insertion locus and integrity of the landing pad. (B) Multi-gene circuit integration details. Co-transfection of the integration vector and the recombinase expression plasmid results in site-specific recombination between the BxB1 attB site (from the integration vector) and the attP site (pre-integrated in the mammalian chromosome within the landing pad). As a result, expression of the promoterless selection marker from the integration plasmid is triggered since it is inserted after the constitutive promoter of the landing pad. (C) Second phase: Multi-gene circuit assembly and integration in the landing pad. The monoclonal chassis cell line can be used to integrate multi-gene circuits constructed with the mMoclo assembly method, which requires only two cloning steps from sequenced-verified basic genetic parts to complete multi-gene circuits. Depending on the application, either a single circuit or a library of circuits can be co-transfected with BxB1 recombinase expression plasmid. Cell populations are ready to be screened in less than 2 weeks after transfection.
MATERIALS AND METHODS
Golden gate reactions
For all Golden Gate assembly reactions we used: 0.4 μl of TypeIIS enzyme (either BsaI from NEB, or BpiI from Fermentas), 0.2 μl of T4 Ligase HC + 1 μl of T4 Ligase HC buffer (Promega), 1 μl of 10x bovine serum albumin (NEB), 40 fmol for all vectors used in the reaction, ddH20 up to a final total volume of 10 μl. The thermocycler program used for all assemblies included: 1 step of 15 min at 37°C; then 50 cycles of [2 min at 37°C followed by 5 min at 16°C]; 1 step of 15 min at 37°C, 1 step of 5 min at 50°C and 1 final step of 5 min at 80°C.
Bacterial cell cultures
Liquid cultures of Escherichia coli MG1655 were grown in LB Medium (Difco) at 37°C for plasmids up to 20 kb, and at 30°C for larger plasmids. When appropriate, antibiotics were added as follows: spectinomycin (100 μg/ml), ampicillin (100 μg/ml) and kanamycin (25 μg/ml). For blue/white screening, we used X-gal at a final concentration of 40 μg/ml.
Cell cultures, transfections and nucleofections
HEK293FT and HEK293 cell lines were purchased from Invitrogen. HeLa (CCL.2), CHO, COS, hESC cell lines were obtained from ATCC. HEK293FT, HEK293, HeLa and CHO cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Cellgro) supplemented with 10% Fetal Bovine Serum (FBS, PAA), 0.045 g/ml penicillin/streptomycin and non-essential amino acids (HyClone) at 37ºC, 100% humidity and 5% CO2. mESC were grown in DMEM supplemented with 15% Fetal Calf Serum (FCS, 10 ng/ml LIF/ESGRO (Millipore), 0.1 mM MEM non-essential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin and 100 μM Mercaptoethanol. COS cells were grown in DMEM supplemented with 10% FCS and 0.045 g/ml penicillin/streptomycin. hESC (CHB8, gift of George Daley, Harvard Medical School) were grown on Matrigel-coated plates in mTeSR-1 (Stem Cell Technologies, Vancouver, Canada). HEK293FT and HEK293 transfections were carried out in 24-well plates using Attractene reagent (Qiagen), 200 000 cells and 200–300 ng total DNA per well. Media were changed 24 h after transfection. mESC were transfected using Metafectene Pro (Biontex, Germany). Transfection was performed in 6-well plates using 800 000 cells and 2 μg of DNA. COS cells were transfected using Metafectene Pro. Transfection was performed in 6-well plates using 600 000 cells and 2 μg of DNA. For Hela, CHO, hESC we used the 4D Nucleofector (Lonza, Switzerland) to electroporate the vectors. For hESC we used 600 000 cells, 800 ng total of DNA and the nucleofection program CA-137 (Buffer P2). For CHO we used 600 000 cells, 600 ng total of DNA and the nucleofection program DT-133 (Buffer SF). For HeLa we used 800 000 cells, 800 ng total of DNA and the nucleofection program CN-114 (Buffer SE).
Landing pad integration using ZFN
To create the pLanding_Pad vector, an 800 bp sequence homologous to the AAVS1 sequence on the left of the ZFN cut site, was cloned into the p_TU1 position vector. Similarly, an 800 bp sequence homologous to the AAVS1 sequence on the right of the ZFN cut site, was cloned into the p_TU3 position vector. The following transcription unit was assembled into the p_TU2 position vector with a golden gate reaction: double cHS4 core insulator from a p_Insulator, CAG promoter from a p_Promoter, attP BxB1 from a p_5′UTR, EYFP-2A-Hygromycin from a p_Gene, inert 3′ UTR from a p_3′ UTR and rb glob polyAdenylation signal + double cHS4 core insulator from a p_polyA. The three verified position vectors were then assembled altogether into the Shuttle Vector, deleting the crt red operon cassette. To create the ZFN expressing vector, pLV_CAG_CN-2A-CN, the cDNA encoding the two ZFD for the AAVS1 locus, separated by a 2A tag was synthesized (GeneArt, Regensburg, Germany) and polymerase chain reaction (PCR)-amplified with Gateway attB1/attB2 tags. Upon gel extraction, the PCR product was recombined into a pENTR_L1_L2 vector using BP clonase (Life Technologies, Carlsbad, CA, USA), yielding the pENTR_L1_CN-2A-CN_L2 vector. In a next step, pENTR_L1_CN-2A-CN_L2 and pENTR_L4_CAG_R1 were recombined into pLV_R4R2_GTW using LR clonase II plus (Life Technologies), resulting in pLV_CAG_CN-2A-CN. To integrate the landing pad into the AAVS1 locus, we co-transfected cells with equimolar amount of the ZFN vector and the pLanding_Pad vector. Seventy-two hours post-transfection cell culture medium was supplemented with 200 μg/ml Hygromycin B (Invivogen) and the selection was maintained over a period of 2 weeks. Clonal cell lines were generated by serial dilutions of the surviving population.
Southern blots
Genomic DNA was extracted with the Quick-gDNA MidiPrep kit (ZYMO RESEARCH). Note that 4 μg of gDNA was digested over night with the EcoNI and XbaI (Landing Pad integration test) or with AseI (circuit integration test), separated on 0.8% agarose gel, transferred to a nylon membrane and probed with the indicated 32P-radiolabeled probe (Supplementary Text 1). Biomax MS film (KODAK) were stored 2 days in an exposition cassette with the membrane and revealed with a darkroom X-Ray processor (Velopex MD2000).
BxB1-mediated integration of circuits
To integrate circuits into the landing pad of HEK293FT chassis cell line, we co-transfected 150 ng of the appropriate multi-gene integration vector with 150 ng of BxB1 integrase expression vector using attractene (Qiagen) in 24-well format. Seventy-two hours post-transfection cells were transferred to 6-well plates and culture medium supplemented with 1 μg/ml puromycin (InvivoGen). Unless otherwise noted, selection was maintained for 5 days. Cells were typically assayed 14 days post-transfection. For induction of the TRE promoter, 2 ug/ml doxycycline was used.
Integration efficiency test
Cells were collected by trypsinisation from 24-well plates 24, 48 and 72 h after transfection. Cells were then pelleted (1600 g; 3 min) and resuspended in 300 μl of Phosphate Buffered Saline (PBS).
Lentiviral particle production and infection
We used Gateway (26) cloning to construct the integration vectors. Our lentiviral Gateway destination vectors contain pFUGW (27) (Addgene plasmid 14883) backbone and Gateway cassette (comprising chloramphenicol resistance and ccdB genes flanked by attR4 and attR2 recombination sites) followed by blasticidin or puromycin resistance markers expressed constitutively. LR reaction of the destination vectors with entry vectors carrying human elongation factor 1 alpha (hEF1a) promoter and either mKate2 or EBFP2 fluorescent proteins was used to create the following expression vectors: pLV-hEF1a-mKate2-P2A-Puromycin and pLV-hEF1a-EBFP2- P2A-Blasticidin. For production of lentiviral particles ∼2 × 106 HEK293FT cells (Invitrogen) in 3 ml of DMEM complete media were plated into gelatin-coated 60 mm dishes (Corning Incorporated). Three hours later the ∼80% confluent cells were co-transfected with 0.5 μg of the pLV-hEF1a-mKate2-P2A-Puromycin expression vector, 1.1 μg packaging plasmid pCMV-dR8.2 (Addgene plasmid 8455) and 0.55 μg envelope plasmid pCMV-VSV-G (Addgene plasmid 8454) (28) using Attractene reagent (Qiagen) and following manufacturer's protocol. Media containing viral particles produced from transfected HEK293FT cells were harvested ∼48 h post-transfection and filtered through a 0.45 μm syringe filter. Note that 1.5 ml of the filtrate was added to ∼20% confluent HEK293FT cells in 12-well plate seeded immediately before infection. After 48 h, media were changed and supplemented with 1 μg/ml puromycin (InvivoGen). Cells were maintained under selection for 5 days. After selection and expansion, cells were infected again with lentiviral particles produced using pLV-hEF1a-EBFP2-P2A-Blasticidin expression vector, following the same protocol. Blasticidin selection (10 μg/ml) (InvivoGen) was applied for 7 days. Cells were subsequently analyzed by flow cytometry.
Flow cytometry and data analysis
Cells were analyzed with LSRFortessa flow cytometer, equipped with 405, 488 and 561 nm lasers (BD Biosciences). We collected 30 000–50 000 events, using a forward scatter threshold of 5000. Fluorescence data were acquired with the following cytometer settings: 488 nm laser and 530/30 nm bandpass filter for EYFP, 561-nm laser and 610/20 nm filter for mKate2, 405 laser and 525/50 filter for AmCyan and 405 nm laser, 450/50 filter for Pacific Blue. Data analysis was performed with FACSDiva software (BD Biosciences) and FlowJo (http://www.flowjo.com/). For histogram analysis, flow cytometry data in .FCS format were exported into text format using FCS Extract 1.02 software (E.F. Glynn, Stowers Institute for Medical Research) and analyzed in Microsoft Excel. We used bi-exponential scales for visualizing fluorescence-activated cell sorting (FACS) data. For cell sorting, cells were collected directly into an 8-well micro-slide (Ibidi) by a FACSAria cell sorter.
Microscope measurements and image processing
Fluorescence microscopy images of live cells were taken in glass-bottom dishes or 12-well plates using Zeiss Axiovert 200 microscope and Plan-Neofluar 10x/0.30 Ph1 objective. The imaging settings for the fluorophores were S430/25x (excitation) and S470/30m (emission) filters for AmCyan, and S565/25x (excitation) and S650/70m (emission) for mKate2. Data collection and processing were performed using AxioVision software (Zeiss). For the circuit library experiment, we evaluated 500 cells from three different fields of views for each replicate transfection (total of 4500 cells examined). We manually marked all cells with a specific tag corresponding to its observed phenotype and used an Adobe Illustrator automated script to sum the number of cell instances for each cell type and each field of view.
RESULTS
Chassis cell lines generation
To generate the chassis cell lines (Figure 1a), we created a landing pad vector for integration into the chromosomal locus of choice using engineered ZFN. The landing pad contains a constitutive promoter driving co-expression of a fluorescent protein (EYFP) and a selection marker (Hygromycin) (Figure 1b). Between the promoter and the coding sequence we inserted an attP BxB1 phage attachment site (29) to enable site-specific recombination in the landing pad. Co-transfection in the chassis cell line of a vector carrying the corresponding attB BxB1 attachment site with a vector expressing the BxB1 recombinase results in attB/attP site-specific recombination and insertion of the complete vector in the landing pad. To limit interference with surrounding host genes, we placed insulator sequences on both sides of the transcription unit (30). We integrated the landing pad into the AAVS1 locus (21,31) of one simian (Cos) and four human model cell lines (HEK293FT, HEK293, Hela and human Embryonic Stem Cells). This locus was chosen because it promotes sustained expression of transgenes (32). Similarly, we targeted the Rosa26 locus of a mouse Embryonic Stem Cells line and a Chinese Hamster Ovary cell line, as it also supports robust gene expression (19). In this study we focus primarily on characterization and testing of the HEK293FT chassis cell line. After selection and expansion of clonal populations resistant to hygromycin, we isolated a HEK293FT-Landing Pad (LP) monoclonal cell line that showed correct mono-allelic integration of the landing pad, as confirmed with Southern blot and PCRs (Supplementary Figures S2 and S3). EYFP expression in the confirmed chassis cell lines was sustained at high levels for over 40 passages (maximal duration of culture) without further antibiotic selection. While time-consuming (5–6 weeks), monoclonal selection, expansion and verification of the landing pad cell line is required only once with our method, as the cell line can be stored and reused for all subsequent BxB1-mediated integrations (Figure 1c).
Mammalian modular cloning (mMoClo)
To create a framework for modular multi-gene circuits construction and integration into the landing pad of our chassis cell lines, we extended the previously developed Modular Cloning strategy (33) (Figure 2 and Supplementary Figure S4). This highly efficient assembly method (95–100% correctly assembled constructs) (34) relies on flanking the different circuit basic elements with Type IIs restriction enzyme sites that create unique 4 bp overhangs in order to obtain predetermined positioning of the parts within the overall circuit. The mMoclo we introduce here includes 6 parts positioning vectors, 9 transcription unit positioning vectors, 9 linker vectors and 1 destination vector (Figure 2a). The parts positioning vectors are used to create a sequence-verified library of all the basic components necessary to construct an insulated mammalian transcription unit: insulators (in pInsulator), promoters (in pPromoter), 5′ UTR sequences (in p5′ UTR), gene coding sequences (in pGene), 3′ UTR sequences (in p3′ UTR) and polyA signals (in ppolyA). These parts are used to assemble either (i) functional transcription units into the ‘transcription unit positioning vectors’ (Figure 2b) or (ii) a promoter trap cassette carrying attB BxB1 recombination site followed by a promoterless selection marker of choice into the destination vector, to create an integration vector (Figure 2c). A complex multi-unit circuit can then be assembled into this integration vector by combining transcription units with the corresponding linker vector chosen according to the number of transcription units of the circuit (Figure 2d). The vector backbone used for the assembly of large circuits carries a pBR322 origin of replication and can be used to amplify circuits up to 31 kb. While transformed bacteria were grown at 37°C for circuits up to 20 kb, we reduced the growth temperature to 30°C for circuits above this size in order to minimize deletions in the circuits.
Figure 2.

Overview of mammalian circuit assembly (mMoClo). (A) mMoClo is based on a library of empty backbones used to assemble parts (Parts Positioning Vectors), transcription units (Transcription Unit Positioning Vectors) and complex multi-unit circuits (Destination Vector) to be integrated into the landing pad. The Parts Library is made of sequence-verified basic genetic parts (Insulators, Promoters, 5′ UTR, Genes, 3′UTR and polyA sequences). (B) One-step creation of an integration vector carrying BxB1 attB site and promoterless selection marker. (C) One-step assembly of different functional transcription units in specific position vectors. (D) One-step assembly of a multi-unit genetic circuit by combining the desired transcription units, the corresponding linker vector and the previously built integration vector carrying the promoter-trap cassette. Details of the overhangs used in the assembly process are provided in Supplementary Figure S4.
Site-specific integration into the landing pad
To assess integration efficiency, we created an integration vector that contains mKate2 fluorescent reporter fused to a promoterless resistance marker such that integration events can be monitored easily with flow cytometry (Figure 3a). Constitutively expressed Cerulean fluorescent reporter gene was also included in the integration plasmid to determine the transfection efficiency. We co-transfected our HEK293FT-LP chassis cell line with the integration plasmid and a plasmid expressing BxB1 recombinase (wild-type or codon optimized versions), and monitored expression of the fluorescent reporters. Three days after transfection, 10% of the transfected cells expressed mKate2 (8% of the entire population) (Figure 3b). BxB1 codon optimization yielded a 3-fold improvement in integration efficiency over wild-type BxB1 coding sequence. No integration events were observed when the integration plasmid was transfected without the BxB1 expressing plasmid. Addition of a nuclear localization signal (NLS) to the recombinase did not increase the integration efficiency (Supplementary Figure S5). We also tested different ratios of the integration plasmid versus the BxB1 expression plasmid to determine whether vector titration would influence the integration efficiency (Supplementary Figure S6) and found that a 1:1 ratio resulted in the highest integration efficiency. After confirming the proper function of the platform in all our chassis cell lines (Supplementary Figure S7), we assessed the integration efficiency in two of them (CHO and HeLa). We found that a similar integration efficiency was obtained in these cell lines (Supplementary Figure S8). To assess the integration specificity of the promoter-trap strategy, we monitored expression of mKate2 and EYFP among the resistant cells after puromycin selection. More than 99% of the resistant cells carried a single targeted integration of the full vector into the landing pad (mKate2 positive, EYFP negative) while less than 1% of resistant cells expressed both mKate2 and EYFP (0.23% for HEK293FT, 0.43% for HeLa and 0.86% for CHO, Supplementary Figure S9), accounting for rare integration events into cryptic acceptor sites placed after an endogenous constitutive promoter. To validate these flow cytometry statistics and further demonstrate the platform's ability to generate nearly isogenic engineered cell populations, we randomly isolated and expanded 42 resistant clones derived from the HEK293FT-LP cell line and analyzed their genomic DNA with Southern blots. Correct integration in the landing pad was confirmed for all 42 clones (Supplementary Figure S10). We also sequenced the flanking sequences of the insert for these 42 isolated clones and confirmed that the integration process did not trigger any small insertions or deletions close to the recombination site (Supplementary Figure S11). To assess the ability to transfer the platform to other cell lines, we co-transfected both the integration plasmid and the BxB1 expressing plasmid in all our chassis cell lines.
Figure 3.
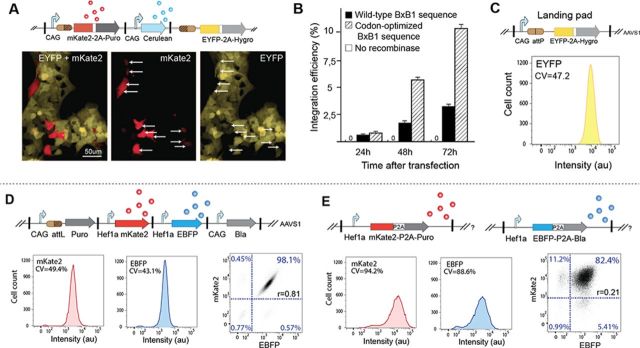
Integration efficiency and homogeneity of payload expression. (A) Fluorescence microscopy images of HEK293FT–LP chassis cell line 3 days after co-transfection of the integration vector and the recombinase expression plasmid. Site-specific integration events can be monitored by expression of mKate2 and concomitant decrease in EYFP levels (integrated circuits shown on top). Cells that undergo the yellow to red switch, indicating successful integration, are denoted with arrows. (B) Integration efficiency assayed by flow cytometry defined as the percentage of cells expressing mKate2 among the population of transfected cells, without any selection. Error bars represent standard deviation from three biological replicates. (C) Homogeneity of transgene (EYFP) expression in HEK293FT-LP cell line. (D) Schematic representation of integrated multi-gene circuits and representative FACS analysis histograms of cells selected with puromycin after BxB1-mediated integration or (E) lentivirus-mediated random integration. Targeted integration results in high homogeneity of transgene expression as compared to lentiviral integrations. The representative fluorescence 2D density plots show high correlation between expression of the fluorescent reporters after single-copy integration in the landing pad as compared to lentiviral integrations.
Expression homogeneity of multi-genic integrated constructs in a polyclonal cell population
The high integration efficiency and specificity of the recombinase-based method, combined with its ability to preserve intact payload, should result in homogenous expression of transgenes from a multi-genic construct even in polyclonal cell populations. To validate this hypothesis, we created a more complex integration vector containing a promoterless puromycin resistance marker followed by three transcription units constitutively expressing mKate2, EBFP2 and Blasticidin each using a separate promoter. To monitor fluorescence levels, we co-transfected this circuit together with the BxB1 expression vector, initiated a 5-day puromycin selection 3 days after the transfection and grew the surviving cells for 6 additional days without any selection before FACS analysis. As desired, we obtained highly homogenous expression of the two fluorescent proteins. We observed that the variability of transgene expression in the polyclonal population after integration measured by the coefficient of variation (CV) (mKate2: = 49.4%; EBFP: CV = 43.1%) is similar to that of the monoclonal chassis cell line population (EYFP: CV = 47.2%) (Figure 3c and d). We also observed that expression of transgenes placed on the same payload is highly correlated (coefficient of correlation r = 0.81, Figure 3d). Similar results were confirmed with other chassis cell lines (Supplementary Figure S12). In comparison, polyclonal populations generated by widely used viral-based integration strategies exhibited a stronger signal due to a high multiplicity of infection but larger variances and a markedly lower correlation in transgene expression (mKate2: CV = 94.2%; EBFP: CV = 88.6%; r = 0.21) (Figure 3e). To generate these polyclonal populations, we had to infect the cells with two separate lentiviruses (given the limited packaging capabilities of the lentivirus particles), one encoding the Hef1a_EBFP-P2A-Blasticidin transcription unit and the other encoding the Hef1a_mKate2-P2A-Puromycin. To select for integrations, we first applied puromycin for 5 days followed by blasticidin for 7 days before FACS analysis.
Integration of a 7-gene payload encoding an inducible memory device
To test the scalability of our method with respect to payload size and number of transgenes, we constructed and integrated a large gene circuit (42 basic parts, 6 transcription units, 27 kb, Supplementary Figure S13a) encoding an inducible memory device (Figure 4). At the initial state, prior to doxycycline induction, the circuit constitutively expresses the Cerulean fluorescent protein. Upon induction, expression of both EYFP and B3 site-specific recombinase (orthogonal to BxB1) are triggered, which results in precise genomic rearrangement within the circuit. Specifically, B3 recombinase recognizes the B3 recombination sites flanking the Cerulean coding sequence, which is then excised. The promoterless downstream mKate2 coding sequence is thus placed in frame with the constitutive promoter previously activating the Cerulean gene expression. The circuit is therefore locked into a final state in which the output has been switched from constitutive Cerulean expression to constitutive mkate2 expression. Following transfection, integration and selection with puromycin for 5 days, we monitored expression levels of the different fluorescent reporters before, during and after induction of genomic rearrangement with doxycycline (Figure 4b). At the end of the selection, after circuit transfection but before doxycycline induction, about 40% of the selected cells had already switched from Cerulean to mKate2 expression (Supplementary Figure S13b). As this ratio was stable over time and essentially no cells had an intermediate level of either Cerulean or mKate2, we hypothesized the B3-mediated switch had occurred right after transfection since leaky TRE promoter expression from the transfected circuits in the presence of high levels of constitutively expressed rtTA3 from the same plasmids was sufficient to trigger expression of B3 integrase at low levels. This would then result in moderate excision of the Cerulean expression cassette in the pool of plasmids, whether or not they had been already integrated in the landing pad.
Figure 4.

Transcriptional regulation and genomic rearrangement for a 7-gene circuit integrated into the landing pad of HEK293FT chassis cell line. (A) Schematic representation of the integrated circuit. In its initial state, the circuit expresses puromycin resistance gene, Cerulean, rtTA3 and Blasticidin resistance gene. After dox induction, rtTA3 activates TREt promoter and triggers expression of EYFP and B3 recombinase (Step 1). This leads to the intermediate state, during which B3 integrase mediates excision of the Cerulean cassette by recombination of the two attB3 sites. Subsequently, mKate2 gene is expressed. When dox is removed, EYFP is no longer expressed. In the final state, mKate2 is the only reporter that is expressed. (B) Fluorescent microscopy images and flow cytometry results confirmed the correct behavior of the circuit and show homogeneity of transgene expression in the polyclonal cell population. (Intermediate state: 3 days after dox induction; Final state: 7 days after dox removal.)
To gain improved understanding, we performed a doxycycline induction experiment (Figure 4b) with cells harboring a non-rearranged payload only (mKate2-negative sorted cells). We monitored expression levels of the different fluorescent reporters before, during and after induction of genomic rearrangement (Figure 4b). Before induction with doxycycline, the circuit remained in its stable initial state with a highly homogenous expression of Cerulean in the population. Efficient transition between the device's states could be successfully monitored 3 days after induction of the circuit with doxycycline at the same time through the strong activation of EYFP and mkate2 expression, and the slow reduction in Cerulean levels. The B3-induced genetic rearrangement therefore locks the circuit into a new state with constitutive expression of mKate2. After withdrawal of doxycycline, mKate2 is still constitutively expressed while EYFP expression diminishes, as the induced B3-mediated genetic rearrangement is irreversible. Flow cytometry analysis of the polyclonal population showed homogenous expression in the population for all reporters at the different time points (Figure 4b). These results suggest that our framework preserves the genetic integrity of such large payloads and provides not only homogenous expression of all transgenes without monoclonal expansion but also reliable dynamic behavior.
Integration of a library of 18 circuits
Finally we examined the suitability of our platform for high-throughput screening of multi-genic payload libraries. As a proof of concept, we performed three replicate co-transfections of the HEK293FT-LP chassis cell line with an equimolar mix of 18 different plasmids (each containing one distinct circuit) together with the BxB1 expression plasmid (Figure 5). Each circuit encoded a fluorescent reporter fused to a NLS and a different fluorescent reporter with variable cellular localization tags such that each circuit generates a distinct fluorescence phenotype (Figure 5b and Supplementary Figure S14). Fourteen days after transfection, we evaluated the phenotype of 1500 cells from each of the three polyclonal populations (total of 4500 cells examined) and observed that all 18 phenotypes were represented well (Figure 5e).
Figure 5.

One pot integration of payload library. (A) Architecture of the payload library. The first fluorescent reporter (EYFP, mKate2 or Cerulean) is fused to a nuclear localization tag. The second fluorescent reporter, different from the first one, is tagged either with an utrophin localization signal, a mitochondrial localization signal or with no localization signal. All possible combinations result in a library of 18 different payloads. (B) Fluorescent microscopy images of microcolonies 7 days after selection with puromycin (10 days after transfection of equimolar amount of the 18 plasmids and BxB1 expression plasmid in HEK293FTLP chassis cell line) and no cell passaging. All 18 phenotypes were observed and are shown. (C and D) Fluorescence microscopy images of polyclonal population 14 days after transfection, with 10x (C) and 40x (D) magnification, after cell passaging. (E) Average number of each cell phenotype appearance in 500 classified cells. Error bars represent the standard deviation from these classifications performed over three different fields of views from three independently repeated experiments (total of 4500 classified cells).
DISCUSSION
In this study, we introduce and validate a comprehensive framework for rapid prototyping of mammalian multi-gene synthetic networks. The framework combines a modular and rapid DNA assembly method to create the circuits together with a platform for stable integration of large synthetic gene networks in a predefined location in a mammalian chromosome. We demonstrate that our method provides an efficient and reliable strategy for correct and sustained execution of complex genetic programs in mammalian cells.
Engineering of cellular behavior remains a challenge and is still strongly dependent on the ability to screen many variants of a designed genetic program in a manner that allows direct comparison of the results. To accelerate the development of such libraries, we developed mMoClo, an extension of the MoClo assembly method, specifically tailored for construction of mammalian genetic circuits. mMoClo enables modular and combinatorial assembly of sequence-verified basic genetic parts into mammalian transcription units and then multi-unit gene circuits in only two cloning steps. Our extension confers an important advantage compared to other assembly methods previously developed for mammalian systems (35–37). In particular, the design of the library reflects the structure of a functional mammalian transcription unit and allows combinatorial generation of a vast diversity of transcription units and circuits from basic sequence-verified genetic parts, from short 3′ and 5′ UTR sequences to large insulator sequences. We believe such a method could provide a new and broadly used standard for assembly of mammalian transcription units and complex circuits. To disseminate our platform, we deposited in Addgene a library of more than 120 parts and 20 assembly vectors (2 different insulators, 8 promoters, 20 5′ UTRs, 90 coding sequences, 8 3′ UTRs and 6 polyadenylation signals). We assembled circuits up to 31 kb, although larger plasmids exhibited deletions or rearrangements during the bacterial cloning phase, likely due to the limited stability conferred by the pBR322 origin of replication for large plasmids. As an alternative more suitable for larger circuits, one could potentially replace the origin of replication of our destination vector with a BAC origin of replication, often used for the amplification of large circular DNA fragments (38).
To study and compare the different circuits we assembled with the mMoClo method, we created a stable integration platform, the landing pad, which can be inserted in specific loci of different cell types using engineered nucleases. While we used ZFN in this study, other efficient systems, such as CRISPR/Cas or TALEN, could also be chosen for chromosomal insertion of the landing pad. Even though variation in DNA integration efficiency will differ according to both the nuclease used and the cell line, it is important to note that only a very limited number of candidate clones that survive selection are necessary in order to isolate and validate one functional chassis cell line. In this case, any of the available nucleases are therefore good candidates to generate chassis cell lines. The landing pad platform we created carries a BxB1 recombination site located downstream of a constitutive promoter and therefore acts as a synthetic promoter trap to allow for quick selection of targeted integration events. Such a strategy significantly reduces the survival likelihood of cells with circuit integration in cryptic pseudo-att sites. Once a monoclonal landing pad chassis cell line is created, it can then be repeatedly used for single copy targeted integration of synthetic circuits mediated by our efficient codon-optimized BxB1 site-specific recombinase. While this is not the first study to use recombination sites for the creation of engineered cell lines, we demonstrate here a comprehensive platform based on the targeted insertion of a tailor-made landing pad in a wide range of cell lines, which can then be used to efficiently generate nearly isogenic cell populations carrying single and targeted copies of modularly assembled complex genetic circuits. This combination of mMoClo and our landing pad platform therefore becomes a convenient framework to significantly reduce the time from transfection of an integration vector to assay (from 2 months to 2 weeks) and to avoid the monoclonal expansion typically required when engineering cells with (multi)gene circuits.
We validated the approach by assembling the largest known functional mammalian synthetic circuit to date, integrating it into the landing pad of a HEK293FT monoclonal chassis cell line, and assessing its behavior within the polyclonal population of selected cells 2 weeks after transfection. Due to the unique combination of high integration efficiency, little to no off-target effect and robust preservation of payload integrity, we obtained sustained and homogeneous expression of the genetic program. The desired genomic rearrangement was triggered with the same dynamics in the selected polyclonal cell population after induction of specific transgenes with doxycycline. For various applications, it would be important to integrate vectors without any bacterial sequences, as the presence of CpG nucleotides in these sequences has been shown to potentially lead to post-integrative silencing of the transgene (39). One possibility is to use the Minicircle (40) strategy to excise such sequences before transfection of the vectors, or use efficient excisionases, such as Cre or Dre (41), after integration of the circuit.
To validate the suitability of our framework for the combinatorial assembly and screening of large circuit libraries, we also created a proof-of-concept library of 18 different circuits. We co-transfected all of them at the same time together with the BxB1 recombinase expression vector in our landing pad chassis cell line. Since there were no significant differences between the occurrences of the 18 different phenotypes within the polyclonal selected cell population, we expect that our framework could readily support the screening of a significantly higher number of circuits. In comparison to transient transfections and random stable transfections which lead to heterogeneous and often inconsistent gene expression, our approach enables the evaluation of libraries of genetic constructs in the same genomic context, and therefore the side-by-side comparison of circuit behavior over an extended period of time. At the part level, our platform could be useful to test the activity of libraries of synthetic DNA promoters or enhancers in order to better understand the architecture of gene regulatory regions since gene activity is modulated by a complex interplay between these cis- and trans- acting DNA elements. At the circuit level, creating and stably integrating libraries of circuits in which the position and orientation of specific parts or transcription units are shuffled could be used to evaluate promoter interference. Preliminary results indeed indicate that part positioning within a circuit can influence transgene expression levels and overall dynamics. Libraries of therapeutically relevant circuits, such as prosthetic networks (14) or T cell proliferation controllers (42), could be rapidly evaluated in parallel to tune their parameters and optimize their sensitivity. In summary, our framework could accelerate the development of a well-characterized and standardized parts libraries for genetic engineering and also help create rules for the design of synthetic networks with better prediction accuracy, useful for a broad range of applications from systems and synthetic biology to biotechnology and medicine.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr. G. Felsenfeld (National Institute of Diabetes and Digestive and Kidney Diseases, USA) for providing the chicken 2xHS4 insulator sequences. We thank Dr. Pascal Hersen for his continued support and input during the study.
Author Contributions: X.D. conceived and designed the experiments, assembled the landing pad and the circuits with help from J.R. to create the mammalian parts library, and L.W. in experimental design. X.D. and L.W. performed integration of the circuits and analyzed the data, P.G. created the zinc-finger expression vector and generated landing pad chassis cell lines, Y.L. performed the ZFN mediated integration of the three color circuit and prototyping of testing circuits, J.E. performed Southern blots, G.B and R.W. supervised the research, X.D., L.W., G.B and R.W. wrote the manuscript.
FUNDING
DARPA CCM [HR0011–12-C-0067]; DARPA Synbio BBN [DARPA-BAA-11–23]; NIGMS of the NIH [P50GM098792]; Syne2arti [ANR-10-COSINUS-007]; Iceberg [ANR-IABI-3096] from the French National Research Agency. Funding for open access charge: DARPA Synbio BBN [DARPA-BAA-11-23].
Conflict of interest statement. None declared.
REFERENCES
- 1.Bleris L., Xie Z., Glass D., Adadey A., Sontag E., Benenson Y. Synthetic incoherent feedforward circuits show adaptation to the amount of their genetic template. Mol. Syst. Biol. 2011;7:519. doi: 10.1038/msb.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukherji S., Ebert M.S., Zheng G.X.Y., Tsang J.S., Sharp P.A., van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011;43:854–859. doi: 10.1038/ng.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kramer B.P., Viretta A.U., Baba M.D.-E., Aubel D., Weber W., Fussenegger M. An engineered epigenetic transgene switch in mammalian cells. Nat. Biotechnol. 2004;22:867–870. doi: 10.1038/nbt980. [DOI] [PubMed] [Google Scholar]
- 4.Tigges M., Marquez-Lago T.T., Stelling J., Fussenegger M. A tunable synthetic mammalian oscillator. Nature. 2009;457:309–312. doi: 10.1038/nature07616. [DOI] [PubMed] [Google Scholar]
- 5.Siciliano V., Menolascina F., Marucci L., Fracassi C., Garzilli I., Moretti M.N., di Bernardo D. Construction and modelling of an inducible positive feedback loop stably integrated in a mammalian cell-line. PLoS Comp. Biol. 2011;7:e1002074. doi: 10.1371/journal.pcbi.1002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ausländer S., Ausländer D., Müller M., Wieland M., Fussenegger M. Programmable single-cell mammalian biocomputers. Nature. 2012;487:123–127. doi: 10.1038/nature11149. [DOI] [PubMed] [Google Scholar]
- 7.Lanitis E., Poussin M., Klattenhoff A.W., Song D., Sandaltzopoulos R., June C.H., Powell D.J. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013;1:43–53. doi: 10.1158/2326-6066.CIR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grushkin D. The new drug circuit. Nat. Med. 2012;18:1452–1454. doi: 10.1038/nm1012-1452. [DOI] [PubMed] [Google Scholar]
- 9.Ruder W.C., Lu T., Collins J.J. Synthetic biology moving into the clinic. Science. 2011;333:1248–1252. doi: 10.1126/science.1206843. [DOI] [PubMed] [Google Scholar]
- 10.Folcher M., Fussenegger M. Synthetic biology advancing clinical applications. Curr. Opin. Chem. Biol. 2012;16:345–354. doi: 10.1016/j.cbpa.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Bacchus W., Aubel D., Fussenegger M. Biomedically relevant circuit-design strategies in mammalian synthetic biology. Mol. Syst. Biol. 2013;9:691. doi: 10.1038/msb.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye H., Aubel D., Fussenegger M. Synthetic mammalian gene circuits for biomedical applications. Curr. Opin. Chem. Biol. 2013;17:910–917. doi: 10.1016/j.cbpa.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Ye H., Baba M.D.E., Peng R.W., Fussenegger M. A synthetic optogenetic transcription device enhances blood-glucose homeostasis in mice. Science. 2011;332:1565–1568. doi: 10.1126/science.1203535. [DOI] [PubMed] [Google Scholar]
- 14.Kemmer C., Gitzinger M., Daoud-El Baba M., Djonov V., Stelling J., Fussenegger M. Self-sufficient control of urate homeostasis in mice by a synthetic circuit. Mol. Ther. 2010;28:355–360. doi: 10.1038/nbt.1617. [DOI] [PubMed] [Google Scholar]
- 15.Eyquem J., Poirot L., Galetto R., Scharenberg A.M., Smith J. Characterization of three loci for homologous gene targeting and transgene expression. Biotechnol. Bioeng. 2013;110:2225–2235. doi: 10.1002/bit.24892. [DOI] [PubMed] [Google Scholar]
- 16.Moehle E.A., Rock J.M., Lee Y.L., Jouvenot Y., Dekelver R.C., Gregory P.D., Urnov F.D., Holmes M.C. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson D.F., Fahrenkrug S.C., Hackett P.B. Targeting DNA with fingers and TALENs. Mol. Ther. Nucleic Acids. 2012;1:e3. doi: 10.1038/mtna.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reyon D., Tsai S.Q., Khayter C., Foden J.A., Sander J.D., Joung J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Pinera P., Ousterout D.G., Brown M.T., Gersbach C.A. Gene targeting to the ROSA26 locus directed by engineered zinc finger nucleases. Nucleic Acids Res. 2012;40:3741–3752. doi: 10.1093/nar/gkr1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendel A., Kildebeck E.J., Fine E.J., Clark J.T., Punjya N., Sebastiano V., Bao G., Porteus M.H. Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep. 2014;7:293–305. doi: 10.1016/j.celrep.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lombardo A., Cesana D., Genovese P., Di Stefano B., Provasi E., Colombo D.F., Neri M., Magnani Z., Cantore A., Lo Riso P., et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Meth. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- 22.Zhu F., Gamboa M., Farruggio A.P., Hippenmeyer S., Tasic B., Schüle B., Chen-Tsai Y., Calos M.P. DICE, an efficient system for iterative genomic editing in human pluripotent stem cells. Nucleic Acids Res. 2014;42:e34. doi: 10.1093/nar/gkt1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olivares E.C., Hollis R.P., Calos M.P. Phage R4 integrase mediates site-specific integration in human cells. Gene. 2001;278:167–176. doi: 10.1016/s0378-1119(01)00711-9. [DOI] [PubMed] [Google Scholar]
- 24.Zhou H., Liu Z.-G., Sun Z.-W., Huang Y., Yu W.-Y. Generation of stable cell lines by site-specific integration of transgenes into engineered Chinese hamster ovary strains using an FLP-FRT system. J. Biotechnol. 2010;147:122–129. doi: 10.1016/j.jbiotec.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 25.Sakurai K., Shimoji M., Tahimic C.G.T., Aiba K., Kawase E., Hasegawa K., Amagai Y., Suemori H., Nakatsuji N. Efficient integration of transgenes into a defined locus in human embryonic stem cells. Nucleic Acids Res. 2010;38:e96. doi: 10.1093/nar/gkp1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartley J.L., Temple G.F., Brasch M.A. DNA cloning using in vitro site-specific recombination. Genome Res. 2000;10:1788–1795. doi: 10.1101/gr.143000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lois C., Hong E.J., Pease S., Brown E.J., Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 28.Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S.Y., Hahn W.C., Sharp P.A., et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai H., Sun M., Ghosh P., Hatfull G.F., Grindley N.D.F., Marko J.F. Single-molecule analysis reveals the molecular bearing mechanism of DNA strand exchange by a serine recombinase. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7419–7424. doi: 10.1073/pnas.1018436108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma N., Hollensen A.K., Bak R.O., Staunstrup N.H., Schrøder L.D., Mikkelsen J.G. The impact of cHS4 insulators on DNA transposon vector mobilization and silencing in retinal pigment epithelium cells. PLoS ONE. 2012;7:e48421. doi: 10.1371/journal.pone.0048421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B., et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sadelain M., Papapetrou E.P., Bushman F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2012;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 33.Weber E., Engler C., Gruetzner R., Werner S., Marillonnet S. A modular cloning system for standardized assembly of multigene constructs. PLoS ONE. 2011;6:e16765. doi: 10.1371/journal.pone.0016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engler C., Gruetzner R., Kandzia R., Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torella J.P., Boehm C.R., Lienert F., Chen J.-H., Way J.C., Silver P.A. Rapid construction of insulated genetic circuits via synthetic sequence-guided isothermal assembly. Nucleic Acids Res. 2014;42:681–689. doi: 10.1093/nar/gkt860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guye P., Li Y., Wroblewska L., Duportet X., Weiss R. Rapid, modular and reliable construction of complex mammalian gene circuits. Nucleic Acids Res. 2013;41:e156. doi: 10.1093/nar/gkt605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid-Burgk J.L., Xie Z., Frank S., Virreira Winter S., Mitschka S., Kolanus W., Murray A., Benenson Y. Rapid hierarchical assembly of medium-size DNA cassettes. Nucleic Acids Res. 2012;40:e92. doi: 10.1093/nar/gks236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shizuya H., Birren B., Kim U.J., Mancino V., Slepak T., Tachiiri Y., Simon M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. U.S.A. 1992;89:8794–8797. doi: 10.1073/pnas.89.18.8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chevalier-Mariette C., Henry I., Montfort L., Capgras S., Forlani S., Muschler J., Nicolas J.F. CpG content affects gene silencing in mice: evidence from novel transgenes. Genome Biol. 2003;4:R53. doi: 10.1186/gb-2003-4-9-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z.-Y., He C.-Y., Kay M.A. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum. Gene Ther. 2005;16:126–131. doi: 10.1089/hum.2005.16.126. [DOI] [PubMed] [Google Scholar]
- 41.Anastassiadis K., Fu J., Patsch C., Hu S., Weidlich S., Duerschke K., Buchholz F., Edenhofer F., Stewart A.F. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis. Models Mech. 2009;2:508–515. doi: 10.1242/dmm.003087. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y.Y., Jensen M.C., Smolke C.D. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc. Natl. Acad. Sci. U.S.A. 2010;107:8531–8536. doi: 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.