


Abstract
The occurrence of a G-triplex folding intermediate of thrombin binding aptamer (TBA) has been recently predicted by metadynamics calculations, and experimentally supported by Nuclear Magnetic Resonance (NMR), Circular Dichroism (CD) and Differential Scanning Calorimetry (DSC) data collected on a 3′ end TBA-truncated 11-mer oligonucleotide (11-mer-3′-t-TBA). Here we present the solution structure of 11-mer-3′-t-TBA in the presence of potassium ions. This structure is the first experimental example of a G-triplex folding, where a network of Hoogsteen-like hydrogen bonds stabilizes six guanines to form two G:G:G triad planes. The G-triplex folding of 11-mer-3′-t-TBA is stabilized by the potassium ion and destabilized by increasing the temperature. The superimposition of the experimental structure with that predicted by metadynamics shows a great similarity, with only significant differences involving two loops. These new structural data show that 11-mer-3′-t-TBA assumes a G-triplex DNA conformation as its stable form, reinforcing the idea that G-triplex folding intermediates may occur in vivo in human guanine-rich sequences. NMR and CD screening of eight different constructs obtained by removing from one to four bases at either the 3′ and the 5′ ends show that only the 11-mer-3′-t-TBA yields a relatively stable G-triplex.
INTRODUCTION
The structural polymorphism of DNA interplays with nuclear proteins to regulate many cellular activities (1,2). Guanine-rich sequences at the end of human telomeres have been reported to fold into a variety of topologies, including single or multi-stranded G-quadruplex structures (3–6), depending on lengths, number of G tracts, cation identity and loop sequences. Also the bases sequence at 5′ and/or 3′ end influence the folding topology by forming base-stacking interactions with the core of the quadruplex structure. G-quadruplex structures have been characterized in vitro since the 1990s but only recently have been observed in human cells (7). The role of G-quadruplexes in cell replication makes G-quadruplex structures attractive drug-target candidates for cancer therapy (8,9). The growing importance of G-quadruplex structures has prompted the analysis of their folding mechanism and the search of new polymorphisms involving the guanine-rich sequences. G-quadruplex structures can be formed by a single oligonucleotide or by properly assembled two to four different oligonucleotides stabilized by Hoogsteen-like hydrogen bonds and base stacking (10–12). Several spectroscopic studies show that some guanine-rich sequences fold into compact intramolecular quadruplex structures by following a multistep pathway that proceeds through stable triplex intermediates (13–15). However, none of such triplex structures has been solved by nuclear magnetic resonance (NMR) or X-ray crystallography so far, because they are elusive and difficult to isolate.
Thrombin binding aptamer (TBA) is a DNA 15-mer oligonucleotide, namely, 5′-GGTTGGTGTGGTTGG-3′, that folds in a monomeric G-quadruplex structure and binds α-thrombin (10), a trypsin-like serine protease involved in hemostasis. In the chair-like structure of the G-quadruplex, two G-tetrads, which are connected by two TT loops and by a single TGT loop, coordinate a monovalent metal ion (10,16). Metadynamics simulations of the TBA unfolding pathway suggested the presence of three main energy minima (17). The deepest one corresponds to the well-characterized G-quadruplex structure of TBA (18), which evolves toward a second minimum where TBA shows a partial opening of the 3′ end with residue G15 oriented into the solvent (17). Following the proposed unfolding pathway, a third minimum is encountered, where the 3′ end opens completely with G14 and G15 leaving the G-tetrad planes, which rearrange to form two G:G:G planes (17). To prove the intrinsic stability of the G-triplex, the TBA oligonucleotide was shortened by four residues at the 3′ end (truncated TBA, 11-mer-3′-t-TBA hereafter) and its structural features were investigated in solution. The analysis of the NMR spectra showed the presence of a predominant folded structure in equilibrium with the unfolded strand, and with the structured form highly favored at low temperature. In particular, the presence of four well-defined exchangeable proton signals in the 11.0–12.5 ppm region, which are typical of DNA structures with Hoogsteen-like hydrogen bonds (4), were observed in the spectra (Figure 1). Moreover, a network of NOE (Nuclear Overhauser Effect) interactions between imino protons was consistent with the presence of two G-triads, thus supporting the predictions of molecular dynamics simulations. An important point to address is whether other TBA truncations are capable of yielding a stable G-triplex structure. To this end, a total of eight different constructs lacking from one to four bases at either the 3′ or the 5′ end have been screened by NMR and circular dichroism (CD) spectroscopies. Only the 11-mer-3′-t-TBA was found to be a single G-triplex species with a relatively high stability.
Figure 1.

Imino protons region of the 1H NMR spectrum, acquired at 900 MHz (proton Larmor frequency) and 274 K.
An experimental structure of 11-mer-3′-t-TBA at atomic detail is needed at this point to answer further questions raised by metadynamics prediction. Among them, it is important to clarify how the absence of the four bases at 3′ end of the guanine-rich stretch of TBA, which prevents the formation of a normal G-quadruplex structure, affects the G-triplex structure predicted starting from the whole TBA (Figure 2). The experimentally validated Hoogsteen-like hydrogen bonds among G-triads in the G-triplex folding predicted by metadynamics calculations are shown in Figure 3 (17).
Figure 2.

Representation of the 3D structure of the TBA DNA G-quadruplex (A, B) (PDB ID: 1QDF) and of the G-triplex conformation of the 11-mer-3′-t-TBA fragment predicted by metadynamics calculation (C, D) (17). The four GG tracts of TBA are displayed with different color: G1-G2 in blue, G5-G6 in green, G10-G11 in red and G14-G15 (for TBA only) in yellow.
Figure 3.
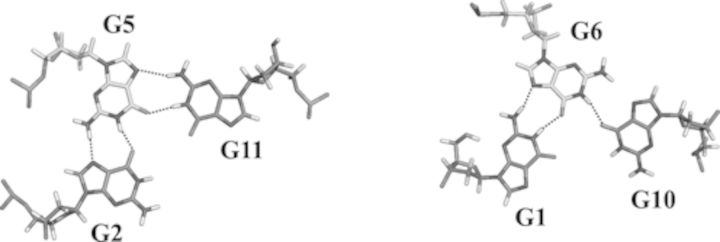
Top view of the two G-triad planes with the experimentally detected hydrogen-bond network.
A well-known issue, encountered in the structure calculation of nucleic acids, is the low proton density and the lack of long-range NOE interactions. Therefore, the quality of experimental structures based on classical restraints such as NOE and J-couplings may not be high enough to permit an analysis at atomic resolution and to solve possible structural ambiguities. Residual dipolar couplings (RDCs) can greatly improve the quality of the calculated structures, providing long-range information and increasing their precision and accuracy (19–21). Moreover, since RDCs depend on the relative orientations of individual bond vectors in the molecule, they can improve the local and global folding also in small DNA/RNA constructs (22). Therefore, we set to solve the fully experimental solution structure of the 3′ TBA-truncated 11-mer oligonucleotide (11-mer-3′-t-TBA) by using both the traditional solution NMR restraints (NOEs and torsional angles) and one-bond RDCs. This is the first experimentally determined structure of a G-triplex oligonucleotide arrangement, and the first entry of this type in the protein data bank (PDB codes: 2MKM and 2MKO). The overall folding predicted by metadynamics simulations is confirmed by the experimental structure. However, few differences are observed.
MATERIALS AND METHODS
Sample preparation
All oligonucleotides were synthesized using standard protocol. The oligomers were detached from the support and deprotected by treatment with concentrated aqueous ammonia at 328 K for 12 h. The combined filtrates and washings were concentrated under reduced pressure, redissolved in H2O, analyzed and purified by high-performance liquid chromatography on a Nucleogel SAX column (Macherey–Nagel, 1000–8/46); using buffer A: 20 mM KH2PO4/K2HPO4 aqueous solution (pH 7.0), containing 20% (v/v) CH3CN; buffer B: 1 M KCl, 20 mM KH2PO4/K2HPO4 aqueous solution (pH 7.0), containing 20% (v/v) CH3CN; a linear gradient from 0% to 100% B for 30 min and flow rate 1 ml/min were used. The fractions of the oligomers were collected and successively desalted by Sep-pak cartridges (C-18). The isolated oligomers proved to be >99% pure by NMR.
NMR spectroscopy
The truncated TBA sequences were characterized, after the thermal annealing, by 1D 1H-NMR experiments carried out with a Varian Unity-Inova three-channel 700-MHz spectrometer at 278 and 298 K. Homonuclear and heteronuclear 2D NMR experiments (2D 1H-1H COSY, 2D 1H-1H TOCSY, 2D 1H-1H NOESY (at mixing times of 100 and 300 ms) and JR-HMBC), performed to characterize in detail the construct 5′-GGTGTGGTTGG-3′, were also carried out at 298 K with Bruker spectrometers operating at 500, 600 and 800 MHz, equipped with triple resonance cryo-probes.
In the case of 11-mer-3′-t-TBA, all experiments were collected on oligomer samples at the concentration of 0.7–0.8 mM in potassium phosphate buffer (70 mM KCl, 10 mM KH2PO4, 0.2 mM EDTA, pH 7).
Resonance assignments and 304 proton–proton distance restraints were collected on 11-mer-3′-t-TBA as previously described (17). NMR spectra to gather additional restraints were performed at 274 K on Bruker spectrometers operating at 500 and 600 MHz, equipped with triple-resonance cryo-probes. 2D 1H-1H E.COSY (23) experiments to determine 1H-1H three-bond couplings were acquired at 500 MHz. For the determination of the dihedral angles of the backbone, phosphorus-based NMR experiments were collected at 600 MHz on a 11-mer-3′-t-TBA sample dissolved in 2H2O. 31P chemical shifts were referenced to an external standard of H3PO4 at 85% and are reported in Table 1. 2D 31P-1H HSQC (24) experiments were performed using 1968 scan in the t2 dimension and 80 t1 increments, whereas three-bond 1H-31P couplings were obtained from the analysis of a constant-time 2D 31P-1H COSY (25) acquired with 2720 scans and 50 increments in the t2 dimension. The delays used for the 1H-31P polarization transfer in these experiments ranged from 12 to 16 ms.
Table 1. 31P chemical shifts for 11-mer-3′-t-TBA in the presence of 80 mM of K+ at 274 K.
| Residue | 31P (ppm) |
|---|---|
| G1 | |
| G2 | −0.622 |
| T3 | −2.152 |
| T4 | −1.576 |
| G5 | −0.538 |
| G6 | −0.901 |
| T7 | −1.146 |
| G8 | −1.376 |
| T9 | |
| G10 | −0.029 |
| G11 | −0.862 |
The chemical shifts have been referenced to a standard of 85% H3PO4 in H2O at 298 K. The guanines in the syn conformation for the glycosidic torsion angle have been highlighted in bold.
2D 13C-1H HSQC spectra, centered on the aliphatic and aromatic regions, were collected on a Bruker spectrometer operating at 800 MHz and 277 K in natural isotopic abundance, and allowed the assignment of 13C resonances. RDCs (DHC) were measured using the InPhase AntiPhase (IPAP) method (26) from non-decoupled 2D 1H-13C HSQC spectra in the direct dimension. These spectra were recorded in 100% 2H2O, phosphate buffered solution of 11-mer-3′-t-TBA on a Bruker spectrometer operating at 950 MHz equipped with a cryo-probe. The RDCs were calculated as the difference in the splitting of the one-bond 1H-13C couplings in partially aligned and isotropic samples. Two different alignment media have been used to determine RDCs in G-triplex: (i) bicelles, containing C8E5 glycol and n-octanol (r = 0.87), at a concentration of C8E5/water equal to 5% w/v (27); (ii) filamentous phages Pf1 (28,29) provided by ASLA biotech at the concentration of 18 mg/ml. For both the isotropic and anisotropic samples, one experiment with the INEPT (Insensitive Nuclei Enanched by Polarization) delay matching the sugar JCH-coupling (1/4J = 1.72 ms) and a second experiment matching the aromatic JCH-coupling (1/4J = 1.51 ms) were collected at 950 MHz and 277 K. For each set, 1741 (1H), 448 and 256 (13C) complex points were recorded for spectral widths of 11 433 Hz (1H) and 20 310 Hz (13C) for the aliphatic and 3345 Hz (13C) for the aromatic spectrum, respectively.A total of 21 and 14 13C-1H RDCs were collected from the spectra acquired in the presence of bicelles and filamentous phages Pf1, respectively (see Supplementary Table S1 in supplementary data).
All spectra were processed with the Bruker TOPSPIN software packages and analyzed by the program CARA (Computer Aided Resonance Assignment, ETH Zurich) (30).
Circular dichroism spectroscopy
CD spectroscopy has been used to further investigate the structure adopted by the truncated TBA sequences in solution. Oligonucleotide samples were prepared by using the same buffer solution used for NMR experiments. CD spectra and CD melting curves of oligonucleotide samples were recorded by using a Jasco J-715 spectropolarimeter equipped with a Jasco JPT-423-S temperature controller using 1-mm path-length cuvettes. CD spectra were averaged over three scans at 273 K, which were recorded in the wavelength range of 200–360 nm at a scan rate of 100 nm/min with a response time of 1 s and a bandwidth of 2 nm. Buffer baseline was subtracted from each spectrum. The DNA concentration (as single strand) was 40 μM. CD melting and annealing curves were recorded as a function of temperature in the range 273–363 K with a scan rate of 0.5 K/min by following CD signal changes at the wavelength corresponding to maximum absorbance variation upon folding of each sample.
Structure calculations
The structure of 11-mer-3′-t-TBA was calculated including the experimental NMR restraints in the web service XPLOR-NIH running on the weNMR community portal (https://www.wenmr.eu/) (31). The distances between non-exchangeable protons were estimated based on the NOE cross-peak volumes at a mixing time of 100 ms using the distance H2′-H2′ (1.85 Å) as reference for the volume calibration. Exchangeable proton-based restraints were obtained from 2D 1H-1H NOESY spectra acquired with a mixing time of 300 ms. The upper distance limits for all the restraints derived from these spectra were all set to 6.50 Å to take into account possible spin diffusion effects. A total of 304 NOEs restraints were used in the calculations (188 intraresidue, 67 sequential and 49 interesidue) (Supplementary Table S2). Atoms participating in hydrogen bonds in the G-triad planes were restrained with upper and lower distance limits corresponding to ideal hydrogen bonds. For each individual hydrogen bond, heavy atom–heavy atom and heavy atom–proton distance restraints have been imposed (Supplementary Table S3). A total of 67 dihedral restraints were implemented from the analysis of the intraresidue NOEs, the 1H-31P couplings, and phosphorus chemical shifts (Supplementary Table S4). In XPLOR-NIH, a random conformation was first generated for the single-stranded 11-mer-3′-t-TBA sequence and then used to produce a family of 400 structures. The experimentally determined distance restraints, G-triad hydrogen-bonding restraints and dihedral angles were included in structure calculations. All ambiguous distance restraints were specified with the SUM averaging option in XPLOR-NIH. The force constants were scaled to 50 kcal mol−1Å−2 for NOE and hydrogen-bond distance restraints. The force constants of dihedral angle restraints were 200 kcal mol−1 rad−2 in the simulated annealing protocol.
The 400 structures calculated using the NOEs and dihedral angles restraints were refined against the two sets of RDCs, introduced simultaneously (32). A harmonic potential with a flat bottom was used as the dipolar coupling energy term (33) and the RDC potential was turned on only in the cooling stage of the simulated annealing protocol. The initial tensors of the two set of RDCs were estimated with the program FANTALIN implemented in XPLOR-NIH (34,35). The starting structures were either the model of 11-mer-3′-t-TBA predicted by metadynamics calculations (17) or the initial experimental structure obtained without the inclusion of RDC. In both cases the subsequent calculations converged to virtually the same RDC tensors. At the first stage of refinement protocol with RDCs, during the tensor convergence, the weight of these restraints was kept at 0.1 kcal mol−1 Hz−2. The final axial and rhombic parameters of the tensors were found to converge at the value of 0.023 × 10−32 and −0.012 × 10−32 m3, respectively, for the bicelles, while for the filamentous phages the two parameters were found to converge at the value of 0.023 × 10−32 and −0.010 × 10−32 m3, respectively (see Table 2). These values have been used in the final structure calculation protocol, where the weight of RDCs was increased to 1 kcal mol−1 Hz−2. As expected, the orientation of the tensor axes in bicelles and phages is sizable different (Supplementary Figure S1).
Table 2. Starting and final converged tensor parameters for the two external orienting media.
| Metadynamics predicted model | Converged | Experimental structure (NOEs, H-bonds and dihedrals) | Converged | |
|---|---|---|---|---|
| A) Bicelle | ||||
| Starting values | Final values | Starting values | Final values | |
| Δχax | 0.011 × 10−32 m3 | 0.023×10−32 m3 | 0.017 × 10−32 m3 | 0.023 × 10−32 m3 |
| Δχrh | −0.004 × 10−32 m3 | -0.012 × 10−32 m3 | −0.010 × 10−32 m3 | -0.012×10−32 m3 |
| B) Filamentous phages, Pf1 | ||||
| Starting values | Final values | Starting values | Final values | |
| Δχax | 0.016 × 10−32 m3 | 0.023 × 10−32 m3 | 0.021 × 10−32 m3 | 0.022 × 10−32 m3 |
| Δχrh | −0.010 × 10−32 m3 | -0.010 × 10−32 m3 | −0.010 × 10−32 m3 | -0.010 × 10−32 m3 |
The initial tensor parameters were obtained by either using the metadynamics-predicted model or the experimental structure calculated from the NOEs and dihedral angles. The member of the family closest to the mean structure was used as experimental structure in tensor analysis.
Restrained molecular dynamics calculation and structure validation
The 10 lowest energy structures were then subjected to restrained molecular dynamics (rMD) using the AMBER 12.0 (36) package available through the WeNMR web portal (http://py-enmr.cerm.unifi.it/access/index). The ff99sb force field and specific parameters for nucleic acids (DNA_CI.lib) were used in the AMBER calculations (37). NOE and H-bond restraints were applied with force constants of 20 kcal mol−1 Å−2, while sugar and backbone dihedral angles restraints were used with force constants of 200 kcal mol−1 rad−2. RDCs restraints were applied with a force constant of 0.3 kcal mol−1 Hz−2. The DNA was placed in a box of TIP3P water, with 10 Å distance from the end of the box. Conversely, the reciprocal orientation of guanines forming the G-triads and the presence of G-triad planes was determined only by experimental restraints and not imposed (38). The structures were initially minimized by fixing the DNA atoms and leaving free the TIP3P waters. Then, a second minimization step was performed without any spatial restraints and using only NMR restraints. The rMD calculations were performed using a simulated annealing protocol, all structures were warmed up from 0 to 600 K in 10 ps, maintained at 600 K for 15 ps, and then cooled to 0 K in 10 ps. A time step of 1.0 fs and a cutoff value of 15 Å were used for the evaluation of non-bonded interactions. The final structures were minimized using the steepest descent and conjugate gradient methods. The same protocol was used also to minimize the structures bearing a potassium ion in the cavity between the two G-triad planes. The potassium ion was placed manually in the cavity of the empty G-triplex structures previously calculated with XPLOR-NIH by using the program PYMOL. Distances between the potassium ion and the carbonyl oxygen of the guanines forming the G-triads were restrained according to the experimental values observed in the quadruplex structures (39).
Structural parameters were evaluated using X3DNA (40) and quality assessments were made with Molprobity (41).
Data deposition
The coordinates for the G-triplex (5′-GGTTGGTGTGG-3′) have been deposited in the Protein Data Bank (accession codes 2MKM and 2MKO).
RESULTS
Analysis of the truncated TBA sequences
The removal of a single guanine at the 3′ end of TBA to provide the strand 5′-GGTTGGTGTGGTTG-3′ already destabilizes the G-quadruplex structure. The several aromatic signals observed in the 1D 1H NMR spectra indicate a conformational heterogeneity of the construct, with six weak signals corresponding to imino protons still present in the region between 12.5 and 11 ppm (Figure 4B). The removal of a second guanine at the 3′ end seems to increase the conformational fluctuations and the heterogeneity of the construct with the predominance of broad signals in the aromatic and imino protons regions (Figure 4C). Conversely, the signals of the G-triplex conformation are already unequivocally present in the spectra of the construct lacking the last three bases at the 3′ end (starred signals in Figure 4D). Although conformational heterogeneity is still present in this construct, a slow exchange equilibrium shifted toward the G-triplex folding begins to be present in solution. Figure 4E shows the already described 11-mer-3′-t-TBA (17). Collectively, these results underline the stabilizing effects of the two guanines at the 3′ end on the G-quadruplex folding, but more important they show the critical role of the two thymines forming the loop in the TBA, and here observed to destabilize the G-triplex conformation. The destabilizing effect of the truncations at the 5′ end on the G-quadruplex structure of TBA is immediately apparent from the 1D 1H NMR spectra of the corresponding constructs (Figure 5B–D). More importantly, the construct obtained by the removal of the four bases at the 5′ end of TBA (Figure 5E), which would correspond to a possible structural analog of 11-mer-3′-t-TBA, is affected by conformational heterogeneity. 2D spectra suggest the presence of two distinct species. The full NMR characterization of the latter to prove or disprove the possible presence of G-triplex folding will be the subject of a subsequent work (see Supplementary Data).
Figure 4.

1D 1H NMR spectrum of TBA (panel A), and of its truncated sequences at the 3′ end (panels B–E) collected at 278 K.
Figure 5.

1D 1H NMR spectrum of TBA (panel A), and of its truncated sequences at the 5′ end (panels B–E) collected at 298 K.
The CD spectrum of 11-mer-3′-t-TBA shows two positive bands at 289 nm and 253 nm, respectively, and a negative band at 265 nm (17). This spectrum resembles that of TBA (with two positive bands at 294 and 247 nm, respectively, and a negative band at 268 nm). These CD profiles are indicative of a homopolar stacking of the nucleobases (42). However, in the 11-mer-3′-t-TBA construct both positive and negative bands are slightly shifted, thus suggesting that the stacking of the bases is not identical to that present in TBA.
The CD melting profile of 11-mer-3′-t-TBA shows almost superimposable heating and cooling curves (17), therefore the melting profile is reversible, with no significant hysteresis. For the 11-mer-3′-t-TBA the analysis of the CD melting profiles have provided a melting temperature, Tm, of 307(±1) K, and a van't Hoff enthalpy change (ΔH°vH) of 145(±15) kJ mol−1. Furthermore, DSC experiments were carried out to characterize the denaturation thermodynamics of 11-mer-3′-t-TBA with a model-independent analysis method (17). The integration of the denaturation peaks provided a ΔH°cal of 135(±5) kJmol−1, which is almost identical to the van't Hoff enthalpy calculated from DSC curves, and very close to that calculated from CD. This indicates that the transition of 11-mer-3′-t-TBA is a two-state process.
To investigate the conformational heterogeneity of the truncated sequences at 3′ and 5′ ends of TBA, we have also acquired CD spectra, and CD melting and annealing curves, for all the constructs. The analysis shows that most of the melting and annealing CD experiments are characterized by hysteresis. Moreover, the CD spectra, before and after denaturation, are not superimposable (see Supplementary Data section, Supplementary Figures S12 and S13). This indicates that most truncated forms are characterized by conformational heterogeneity. These observations are in good agreement with the 1D 1H-NMR experiments. Only in the case of 11-mer-5′-t-TBA the CD spectrum, characterized by two positive bands at 292 and 244 nm, respectively, and a negative band at 263 nm, indicate the prevalence of a main conformation. As reported for the 11-mer-3′-t-TBA, also for the main conformation of the 11-mer-5′-t-TBA, the small differences observed in the CD spectrum with respect to the TBA construct can reasonably be explained by a slightly different arrangement of the stacking between the two G-triads.
Glycosidic torsion angle determination and sugar and backbone conformations of the 11-mer-3′-t-TBA
The proton resonance assignment has been previously reported (17) and it was performed through the analysis of 2D 1H-1H NOESY, 2D 1H-1H TOCSY, 2D 1H-1H DQF COSY and JR-HMBC NMR spectra.
Structural information on 11-mer-3′-t-TBA could be retrieved by both 2D 1H-1H NOESY and 2D 1H-1H COSY experiments. In particular, according to the H8-H1′ and H8-H2′/H2″ NOE relative intensities in the 2D 1H-1H NOESY spectrum of 11-mer-3′-t-TBA, three guanine residues (G1, G5, G10) were classified in the syn conformation, and the four other guanine residues (G2, G6, G8, G11) in the anti conformation. The glycosidic torsion angle χ was thus restricted in the structure calculation to 60°± 40° and to 240° ± 40° for the two different classes of residues, respectively. The range 240° ± 70° was used to restraint the glycosidic torsion angle χ of the thymine bases belonging to the loops.
The prevalent conformation of the sugar was determined from the analysis of the 2D 1H-1H DQF-COSY and 2D 1H-1H E.COSY spectra. In the 2D 1H-1H DQF-COSY spectrum strong H1′-H2′ cross-peaks are observed for G2, T3, T4, G5, G6, T7, T9, G10. Conversely, stronger H1′-H2′ cross-peaks are detected for G1 and G8. At the same time, G11 displays H1′-H2′ and H1′-H2′ 2D COSY cross-peaks of equal intensity. All the H3′-H4′ cross-peaks, except for G11, are missing in this spectrum, due to the very weak coupling between these two protons. These evidences indicate that all nucleotides, but G11, adopt predominantly a S-type, C2′-endo, sugar conformation, while G11 possibly interconverts between the C3′-endo and C2′-endo conformations. As previously observed in the 2D 1H-1H DQF-COSY, also in the 2D 1H-1H E.COSY spectrum, the cross-peaks related to H3′-H4′ coupling are absent, further suggesting that the two protons are staggered by 90°, as it occurs in the C2′-endo conformation. The 3J couplings evaluated from the 2D 1H-1H E.COSY spectrum in the H1′-H2′/2′ region are in agreement with a predominance of the S-type sugar conformation (3J1′2′ is in the range between 7 and 11 Hz). Therefore, in order to maintain the C2′-endo conformation of the sugars in XPLOR-NIH calculation, the dihedral angles ν1 and ν2 for all nucleotides, but G11, were restricted to 30° ± 5° and −27.5° ± 5°, respectively. Furthermore, a value of −90° ± 5° was imposed to the pseudo-dihedral angle H3′-C3′-C4′-H4′ to maintain the H3′ and H4′ protons turned by 90°, as indicated by the experimental data. In AMBER calculations, the range for the pseudo-rotation phase angle P typical of the C2′-endo conformation (171° ± 9°) was directly imposed.
The values of the backbone dihedral angles have been determined (i) from the analysis of the relative peak intensities of the 2D 1H-1H NOESY spectrum, (ii) from the qualitative analysis of the 31P-NMR spectra and (iii) from the evaluation of the 31P-1H coupling constants.
The ranges for the γ backbone torsion angles have been derived from the analysis of the intensity of the NOESY cross-peaks between aromatic and sugar protons and sugar–sugar protons (43). No cross-peaks or very weak cross-peaks were found from H6/H8 to any H5′/H5″ protons; moreover, H2′-H5′/H5′ cross-peaks are weaker than H2′-H1′ cross-peaks for all the residues but for G5 and G10, where the relative intensity of the two cross-peaks is similar. These spectral features allowed us to restrict the backbone γ angle (of all residues but G5 and G10) in the range 60° ± 40°, which results in a swinging of the geminal H5′/H5′ protons away from the core of the DNA helix into the solvent and far from the aromatic and H2′ protons (43). These findings are further supported by the phosphorus-based NMR experiments.
The 2D 31P-1H HSQC (24) and 2D 31P-1H COSY (25) spectra (Figure 6) enabled us to assign the phosphorus resonances of 11-mer-3′-t-TBA. The analysis of the chemical shifts shows that most of the 31P resonances are clustered in the region −1 ± 0.5 ppm, as reported for regular right-handed B-DNAs. A second group of signals corresponding to T3, T4 and G10 fall outside this region (Table 1). These experimental evidences support the absence of a helical structure for the T3-T4 region and suggest a higher flexibility of this loop. Also the non-canonical value of the 31P chemical shift of G10 could be explained by an increased flexibility due to the lack of the fourth strand in the G-triplex structure.
Figure 6.

31P-1H HSQC (top) and 31P-1H COSY (bottom) of 11-mer-3′-t-TBA at 600 MHz (proton Larmor frequency), and 274K.
Additional information on the backbone torsion angles were obtained from the analysis of the 2D 31P-1H HSQC and 2D 31P-1H COSY spectra. In the 2D 31P-1H HSQC some 31P nuclei provide strong cross-peaks with either the H3′ sugar proton of the 5′-adjacent nucleotide, or with the H4′ of the 3′-adjacent nucleotide. Therefore, the backbone torsion angle γ was restrained according to the presence or the absence of the cross-peak between 31P and H4′, as detailed below. It is also interesting to note that all the nucleotides with syn conformation for the glycosidic torsion angle exhibit the cross-peak between 31P and H3′, while those in the anti conformation show a cross-peak between 31P and H4′, although the different cross-peak pattern is a consequence of both sugar and backbone conformational preferences. More in detail, a strong cross-peak between the phosphorus and the H3′ sugar proton of the previous nucleotide is observed for G5 and G10, while a cross-peak between the phosphorus and the H4′ proton of its own sugar is present for G2, G6 and G11. Moreover, in the 2D 31P-1H HSQC, both 31P cross-peaks with the adjacent sugar protons H5′ and H5″ are absent for all the guanines. Conversely, in the case of T3, T4 and T7, correlations among 31P with either both H5′-H5′ or only H5′ are present. The cross-peak corresponding to the phosphorus of T9 could not be assigned and therefore no angular restraint was applied to T9 in structure calculations. The 2D 31P-1H COSY was acquired with a smaller coupling constant for the 1H-31P polarization transfer (16 instead of 20 Hz used in the 2D 31P-1H HSQC). In this case, besides the correlations observed in the 2D 31P-1H HSQC, the cross-peaks between the phosphorus atoms of all the nucleotides with the H3′ sugar–proton of the antecedent residues were identified. Few weak cross-peaks between the phosphorus and the proximal H5′/H5′ protons (i.e. only in the case of G2 and G6) were detected. Although 4JH4′-P, 3JH5′-P and 3JH5″-P cannot be accurately measured, the presence or absence of the corresponding cross-peaks provides qualitative information about the conformation of the backbone. In particular, the absence or very weak intensity of both the H5′-P and H5′-P signals indicates that the β backbone torsion angle is in the canonical trans conformation (44). Therefore, in the structure calculation the β torsion angle of the following residues, G2, G5, G6, G8, G10, G11, was restricted to the range 180° ± 50°. Moreover, in the case of the guanines in the anti conformation, the 4JH4′-P coupling constants are large enough to give rise to cross-peaks in the spectrum; this occurs only when β/γ fall in the trans/g+ region (44,45). The unambiguous presence of all H4′-P cross-peaks confirms that for G2, G6 and G11 β is in trans and γ is g+ conformation. Therefore, also the analysis of the 2D 31P-1H spectra, as well as of the NOESY peak intensities, confirmed the range 60° ± 40° for the γ torsion angle.
Determination of the 11-mer-3′-t-TBA folding topology
The folding topology of 11-mer-3′-t-TBA, from the analysis of characteristic NOEs between imino and H8 protons, was already proposed to be consistent with an intra-molecular G-triplex involving two G-triads, G1:G6:G10 and G2:G5:G11. The presence of the two G-triads is further supported by additional NOEs among the H1 protons and of the loop protons with the two G-triads (Supplementary Figure S2). The hydrogen-bond directionalities of the two putative G-triads are clockwise and anti-clockwise, respectively (Figure 3). The glycosidic conformations of guanines are syn-anti-syn around the first G-triad and anti-syn-anti around the second G-triad, as it occurs in the corresponding G-quadruplex. These glycosidic conformations are consistent with an antiparallel-stranded G-triplex core, containing three G-tracts (G1-G2, G5-G6, G10-G11) with alternating anti-parallel orientations. As in TBA, the TT and TGT tracts exhibit a peculiar pattern of NOEs and form loops at opposite ends of the G-triplex that span the narrow and the wide grooves, respectively. In the proposed G-triplex fold, the imino protons of G1, G6, G5 and G11 are involved in the formation of Hoogsten-like hydrogen bonds forming the G-triads. Therefore, these protons are the most protected from the exchange with water. On the other hand, imino protons of G2 and G10 are not involved in any hydrogen bond and exchange quickly with the solvent. This picture is consistent with the presence of only four sharp signals in the region of the imino protons of the 1H NMR spectrum (Figure 1).
NMR structure determination and structure statistics of 11-mer-3′-t-TBA
The ‘ab initio’ solution structure of 11-mer-3′-t-TBA was calculated with XPLOR-NIH using a simulated annealing protocol, starting from a single-stranded linear DNA structure. All the experimental restraints including distance restraints, glycosidic torsion angles, sugar and backbone conformations, and subsequently RDCs as described in the Materials and Methods section, were used in the calculations. NOE distance restraints were obtained from the analysis of 2D 1H-1H NOESY spectra acquired with different mixing times (100 and 300 ms). On average 28 NOE-derived distance restraints and six angle restraints per residue were used for the structure calculation (Table 3). Hydrogen-bond restraints between G5-G2, G11-G5, G1-G6 and G6-G10, respectively, were also included in the structure-calculation protocol. These hydrogen bonds were identified by the presence and the typical chemical shifts of the signals corresponding to the imino and amino exchangeable protons of the bases. The presence of three Hoogsteen-like base parings (G5-G2, G11-G5, G1-G6) is also confirmed by the NOEs between the imino protons and the aromatic protons of the interacting bases. Also, the downfield shifts of the H21 protons for G1, G5, G11 agree with the presence of a hydrogen bond involving the amino proton and N7 of the neighboring base. In the case of G6, the formation of the H-bond involving its own imino proton and O6 of G10 is demonstrated by the presence in the spectra of a sharp peak corresponding to H1 of G6. Conversely, the absence of a downfield shift for the H21 proton of G6 and the absence of NOEs of H1 and H21 of G6 with H8 of G10 did not allow us to establish the presence of a hydrogen bond between H21 of G6 and N7 of G10. Therefore, this particular interaction was not implemented as a restraint in the calculation.
Table 3. NMR distance, dihedral angles and RDC restraints used in the structure calculations.
| NMR distance and dihedral angle restraints | |||
|---|---|---|---|
| Distance restraints | |||
| Total number of NOEs | 304 | ||
| Intraresidue [i = j] | 188 | ||
| Interresidue | 115 | ||
| Sequential [|i − j| = 1] | 67 | ||
| Non-sequential | 49 | ||
| Hydrogen bonds | 22 | ||
| Dihedral restraints | |||
| Total number of dihedral angles | 67 | ||
| Backbone dihedral angles: | 39 | ||
| β | 6 | ||
| γ | 9 | ||
| χ | 22 | ||
| Sugar dihedral angles | 30 | ||
| Residual dipolar couplings | |||
| Bicelles | |||
| Aliphatic | 13 | ||
| Aromatic | 8 | ||
| Phages Pf1 | |||
| Aliphatic | 6 | ||
| Aromatic | 8 | ||
The 10 lowest energy structures calculated using only short-range interactions (NOEs, H-bonds and dihedral angles) (Supplementary Figure S3) were selected for further analysis. This family already shows a well-defined structure (Root Mean Square Deviation, RMSD 0.616) and a good local structure statistics with no NOE violation >0.5 Å and no torsion angle violation >10°. However, visual inspection of the structures shows that the two G-triad planes are quite distorted in all the members of the family. The non-planarity of the two G-triads is easily attributable to the lack of experimental restraints. To verify this hypothesis and to improve the quality of the experimental structure, RDCs measured in the presence of two different orienting media (bicelles and filamentous phages Pf1) have been included in the refinement of the G-triplex structure.
The experimental RDCs used in XPLOR-NIH included the C1′–H1′, C4′–H4′ and the aromatic C-H groups. The C3′-H3′, C2′-H2′ and C2′-H2″ RDCs have not been considered for the refinement because of their proximity to the signal of water, and the sensitivity to the conformational equilibrium of the deoxyribose ring, respectively. The family of 10 structures, refined with RDC and with the lowest XPLOR-NIH energy function, exhibited again good structural statistics, a good agreement with the experimental RDCs, and also an improved planarity of G-triads. The local and global folding is well defined with an RMSD of 0.537. This family of 10 structures was minimized in AMBER through a molecular dynamics protocol. The final family of minimized structures (see Figure 7) displays a total RMSD of 0.522. The structure minimization in AMBER was performed both in the presence and in the absence of a potassium ion, placed in the cavity between the two G-triad planes. The quality of the structural statistics for these two families of minimized structures is similarly good, and is even better when the potassium ion is introduced in the middle of the G-triad planes (Table 4) (Figure 7).
Figure 7.
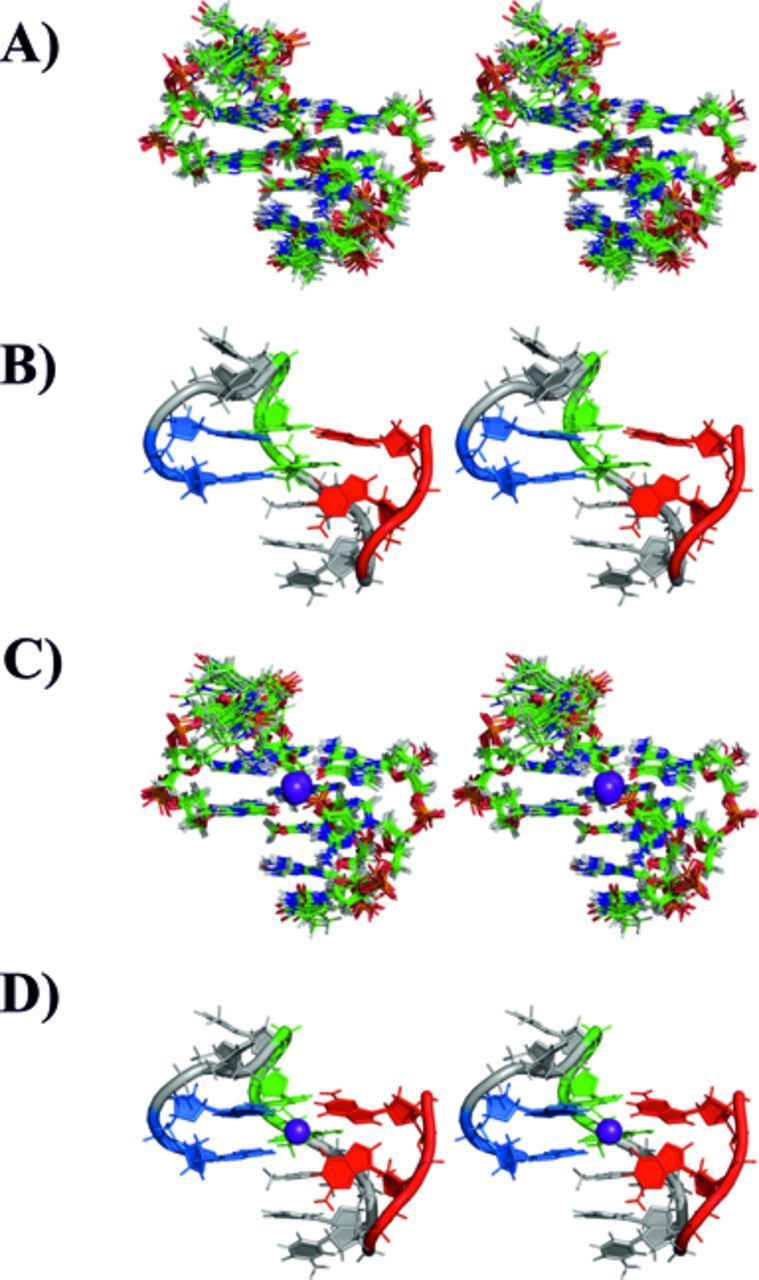
Stereo view representation (crossed eyes) of the family of 10 structures of 11-mer-3′-t-TBA with lowest XPLOR-NIH energy, and after AMBER rMDs (A). The member of the family closest to the mean structure is shown in panel (B) as cartoon. The corresponding family of structures minimized in the presence of the potassium ion is shown in panel (C). The member of the family bearing the potassium ion and closest to the mean structure is shown in panel (D) as cartoon. The potassium ion is shown as a violet sphere.
Table 4. Distance, dihedral angles and RDC statistics of the structure calculations.
| XPLOR-NIH (NOEs, H-bonds and dihedral angles) | XPLOR-NIH (NOEs, H-bonds, dihedral angles and RDCs) | AMBER (NOEs, H-bonds, dihedral angles and RDCs) | AMBER (NOEs, H-bonds, dihedral angles, RDCs, with potassium) | |
|---|---|---|---|---|
| Number of violations | ||||
| NOEs (>0.5) | 0.0 | 0.0 | 0.0 | 0.0 |
| Dihedral (>10°) | 0.0 | 0.01 | 0.01 | 0.02 |
| RDC Q factor | 0.66 ± 0.08 | 0.21 ± 0.01 | 0.13 ± 0.02 | 0.13 ± 0.01 |
| RMSD of the family of 10 lowest energy structures | 0.616 | 0.537 | 0.522 | 0.456 |
The values are averages for the family of 10 structures. The RMSD are reported with respect to the mean structure.
Effect of monovalent cations on G-triplex structure
The 11-mer-3′-t-TBA construct has been studied also in the presence of monovalent cations other than potassium, such as sodium and ammonium ions. As already observed for TBA, sodium ions do not stabilize the G-triplex structure. In the presence of sodium ions (80 mM) and at room temperature (298 K), the large number of signals in the aromatic region and the absence of imino protons indicate the presence of unfolded species. Folded species start to appear only at low temperature (i.e. 274 K) (Supplementary Figure S4).A more detailed analysis has been carried out in the presence of ammonium ions. The titration of the G-triplex with ammonium ions suggests an affinity similar to that reported for the G-quadruplex TBA (16). The addition of ammonium to a solution of 11-mer-3′-t-TBA increases the amount of a folded species that is present in small percentage in solution at low temperature, even in the absence of any monovalent cation (Supplementary Figure S5). The established interaction between 11-mer-3′-t-TBA and ammonium is demonstrated by the presence of one signal in the 2D 15N-1H HSQC apart from the signal of bulk ammonium. A second weak cross-peak in 2D 15N-1H HSQC starts to appear by increasing the concentration of ammonium. The analysis of the 2D 1H-1H NOESY spectrum of the predominant species present in solution reveals the same pattern of NOEs observed in K+-11-mer-3′-t-TBA, and the same alternation of syn-anti bases. However, even by increasing the concentration of the ammonium ion, not all the unfolded oligonucleotide is converted into the G-triplex conformation (Supplementary Figures S6 and S7).
DISCUSSION
The pioneering studies of Bock et al. (10) on libraries of guanine-rich oligonucleotides led to the identification of the TBA. The structure of TBA has been extensively characterized by NMR and X-ray crystallography. In TBA, there are only two G-tetrads forming a total of 16 Hoogsteen-like hydrogen bonds (with each guanine involved in four hydrogen bonds) between the imino protons H1 and oxygen O6 and between amino protons H21 and nitrogen N7. In all deposited TBA structures, the two G-tetrad planes are separated by less than 4 Å (18,46,47). TBA is stable over a wide range of temperatures, either in the presence of K+, or NH4+, or of divalent cations such as Sr2+ and Pb2+. Interestingly, multiple species are reported to be present in solution with Na+ (16).
The analysis of the minimized family of structures of 11-mer-3′-t-TBA shows that the G-triplex consists of two G-triads linked with two parallel G-strands interleaved with an anti-parallel G-strand (G1-G2, G5-G6, G10-G11), which are connected together by two side loops (T3-T4, T7-G8-T9). The G-triplex of 11-mer-3′-t-TBA maintains a structure that recalls the chair-like, antiparallel structure of TBA also in the absence of the fourth missing strand (Figure 8). From the structure of 11-mer-3′-t-TBA it is apparent that one of the bases in the TGT loop (T7) forms stacking interactions with the above G1:G6:G10 plane. As in the G-quadruplex (48), this interaction may have a stabilizing effect. The TGT loop presents many crucial and intense NOEs with the imino protons of the guanines of the neighboring plane. In particular, the methyl group of T7 shows strong correlations with G1-NH, G6-NH and G10-NH. Further NOEs, between the imino proton of G8 and the imino protons of the bases of the G1:G6:G10 plane, have been also identified. Therefore, the TGT loop (in particular bases T7 and G8) is very close to, and quite rigidly held to, the G-stem plane. On the other hand, the TT loop appears more flexible and with a less defined structure. However, some NOEs of G11-HN with T3 and T4 are maintained, indicating that this loop is anyway kept close to the adjacent plane.
Figure 8.

Representation of the 3D structure of the TBA DNA G-quadruplex (A, B) (PDB ID: 1QDF) and of the experimental structure of the 11-mer-3′-t-TBA fragment (C, D) (present work). The four GG tracts of TBA are displayed with different color: G1-G2 in blue, G5-G6 in green, G10-G11 in red and G14-G15 (for TBA only) in yellow.
The comparison of the TBA structure with that of 11-mer-3′-t-TBA shows that the lack of the four bases at the 3′ end in the G-triplex does not cause an extensive structural rearrangement. The stacking bases G1-G2 and G10-G11 roughly remain in place and do not form new hydrogen bonds. This is experimentally proven by the loss in the 1H NMR spectra of the signals corresponding to the imino protons of G2 and G10. Such signals can be detected in the spectra only if the imino protons are involved in hydrogen bonds. This finding supports the observation that the Hoogsteen-like hydrogen bonds involving G14 and G15 in TBA are not replaced by new interactions involving G2:G11 and G1:G10. The reduction of Hoogsteen-like hydrogen bonds accounts for the reduced stability of 11-mer-3′-t-TBA with respect to the TBA structure (Figure 8). Also the similarity between the CD spectrum of TBA and that of 11-mer-3′-t-TBA, as well as the shifting of both positive and negative bands, suggests a similar, though not identical, homopolar stacking of the nucleobases (17,42,49,50). For 11-mer-3′-t-TBA, the presence of a unimolecular folding was already demonstrated by the CD melting curve and by the DSC analysis (17). In this respect, the possible replacement of G14 and G15 in 11-mer-3′-t-TBA by a GG motif of a different strand has been further investigated. The NMR titrations of the 11-mer-3′-t-TBA with the dinucleotide GG and the oligonucleotide TGGT demonstrate that the two GG motifs do not bind to the G-triplex construct.
Overall, the present experimental ‘ab initio’ solution structure of 11-mer-3′-t-TBA is largely superimposable to the conformation of the same oligonucleotide stretch as part of the TBA construct along its unfolding pathway predicted by metadynamics (17). The chair-like folding of the backbone is conserved as far as the two G-triads planes are concerned. However, it is interesting to note that the structure of 11-mer-3′-t-TBA in solution shows some differences if compared with the in-silico prediction. This could also be due to intrinsic differences between 11-mer-3′-t-TBA and TBA arising from the absence of the four last bases in the former. The superimposition of the two structures shows a good agreement for the G-triad G2:G5:G11 that is well defined and rather planar also in the experimental structure. Also in the G-triad G1:G6:G10 bases G1 and G6 maintain the same arrangement, while the conformation of G10 could not be defined by the experimental restraints. The aromatic signal of this base is indeed broader than the others, suggesting the presence of a possible conformational exchange involving G10. Moreover, the lack of RDCs and NOEs for G10 has also prevented the identification of an averaged position. Therefore, the obtained orientation for this base is mainly determined by the XPLOR-NIH and AMBER force fields, and thus probably selected because it is energetically more favored in the absence of experimental restraints. In this regard, it should be mentioned that no violations of the experimental restraints were observed when the planarity was imposed to G10 in a calculation performed to evaluate the conformation of this base. On the other hand, it is remarkable that the out-of-plane orientation of G10 is perfectly compatible with the structure of 11-mer-3′-t-TBA calculated by imposing the presence of a potassium ion, where O6 of G10 appears perfectly poised to coordinate the potassium ion itself.
While the G-triads conformation predicted by metadynamics is largely confirmed by the experiments, structural differences are present in the TGT and TT loops. As described above, in the solution structure of 11-mer-3′-t-TBA, the G-triad plane formed by G1, G6 and G10 is stabilized by T7, which forms stacking interactions, in particular, with G6. Conversely, in the G-triplex structure predicted by metadynamics, the slightly different arrangement of the TGT loop causes the stacked base T7 to turn away and to be replaced by G8 in its interaction with the same G-triad plane. However, both these residues remain very close to the G-triad plane, suggesting a possible switching in the position of these two residues as a consequence of the absence of the four additional nucleotides at the 3′ end of the 11-mer-3′-t-TBA. In the experimental structure of 11-mer-3′-t-TBA the proximity of G8 to the adjacent G-triad is maintained, although G8 appears slightly translated because of the insertion of T7. Other differences occur at the level of the TT loop. In the structure predicted by metadynamics, T3 and T4 are stacked under the G-triad, although molecular dynamics and metadynamics simulations showed some flexibility of this loop. This arrangement is not present in the experimental structure, where the orientation of these bases is different. In particular, the position of T3 is well determined by many restraints, while that of T4 is less precisely defined, suggesting a more pronounced flexibility of this residue. Therefore, while the loop TGT seems to have a stabilizing effect toward the G-triplex structure, apparently the TT loop does not contribute to stabilize the G-triplex structure and, as in TBA, could be prone to interact with a target (48).
Of particular relevance are the results obtained by the analysis of the NMR spectra collected on the truncated sequences of TBA. In fact, these data reinforce the findings of the metadynamic simulations which indicate the prevalent occurrence of the opening and closing at the 3′ end in the folding/unfolding pathway of TBA. Moreover, the different stability of the truncated sequences of TBA, and their heterogeneity demonstrated by the CD melting curves, underlines the critical effect of the length of TT loops on the stabilization of the G-stem (48). Only in the case of the 11-mer-5′-t-TBA, the prevalence of a main conformation has been observed.
This new, detailed, structural information, and the extensive biophysical characterization previously performed on the G-triplex, shed light on the folding mechanism of TBA. The calorimetric and van't Hoff enthalpies of 11-mer-3′-t-TBA, previously evaluated from DSC experiments, pointed out that the melting transition occurs in a two-state process, since the value of the two quantities is almost identical (17).
Collectively, our experimental data reinforce the idea that the folding/unfolding process of TBA can proceed as an apparent two-state mechanism. In our conditions the temperature needed to detach G14 and G15 from the G-quadruplex structure is, apparently, high enough to immediately unfold the just formed G-triplex species. Therefore, it is plausible that the folding/unfolding process of the unimolecular G-quadruplex DNA structures occurs through a very fast process, where the G-triplex intermediate is not detectable with the available experimental techniques.
Concerning the effect of the different cations on the folding stability of the G-triplex, our results show that the 11-mer-3′-t-TBA largely behaves like the other G-quadruplex-forming DNA sequences (51,52). The stabilizing effect observed for K+ and NH4+ relies on their ionic radius and low dehydration energy. The role of the ionic radius and dehydration energy in the stabilization of the G-quadruplex folding of TBA by K+, NH4+, Rb+ has been well characterized in the past by CD spectra and melting profiles (52).
In conclusion, the experimental structure of 11-mer-3′-t-TBA proves the presence and the stability of the G-triplex folding predicted by metadynamics calculations. The differences between the experimental and predicted structures do not affect the folding of the G-triplex, which is globally maintained. Collectively, the experimental structure here presented is consistent with the possibility that G-triplex folding intermediates may play a role in human genome, and therefore its detailed structural features constitute a useful tool to analyze its interactions with relevant biological partners.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Constructive criticisms and several very helpful suggestions by Vladimir Sklenar are gratefully acknowledged. We are also thankful to Janez Plavec for his helpful comments and to Teresa Carlomagno for several suggestions on how to best strengthen the message of the paper.
FUNDING
MIUR PRIN [PRIN-2012 SK7ASN_001]; Ente Cassa di Risparmio di Firenze, EU Bio-NMR [261863]; ITN - pNMR [317127]; EU ESFRI Instruct Core Centre CERM, Italy, AIRC IG 2013 [AIRC # 14150]; EU WeNMR [261572]. European Union [ERC-2009-AdG-247075]; Gabriele Charitable Foundation. Funding for open access charge: MIUR PRIN [PRIN-2012 SK7ASN_001]; AIRC IG 2013 [AIRC # 14150].
Conflict of interest statement. None declared.
REFERENCES
- 1.Broyde S., Wang L., Zhang L., Rechkoblit O., Geacintov N.E., Patel D.J. DNA adduct structure-function relationships: comparing solution with polymerase structures. Chem. Res. Toxicol. 2008;21:45–52. doi: 10.1021/tx700193x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phan A.T. Human telomeric G-quadruplex: structures of DNA and RNA sequences. FEBS J. 2010;277:1107–1117. doi: 10.1111/j.1742-4658.2009.07464.x. [DOI] [PubMed] [Google Scholar]
- 3.Smith F.W. Quadruplex structure of Oxytricha telomeric DNA oligonucleotides. Nature. 1992;356:164–168. doi: 10.1038/356164a0. [DOI] [PubMed] [Google Scholar]
- 4.Macaya R.F., Schultze P., Smith F.W., Roe J.A., Feigon J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hud N.V., Schultze P., Sklenár V., Feigon J. Binding sites and dynamics of ammonium ions in a telomere repeat DNA quadruplex. J. Mol. Biol. 1999;285:233–243. doi: 10.1006/jmbi.1998.2327. [DOI] [PubMed] [Google Scholar]
- 6.Parkinson G.N., Lee M.P.H., Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 7.Biffi G., Tannahill D., McCafferty J., Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013;5:182–186. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wells R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007;32:271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 9.McLuckie K.I.E., Di Antonio M., Zecchini H., Xian J., Caldas C., Krippendorff B.-F., Tannahill D., Lowe C., Balasubramanian S. G-Quadruplex DNA as a Molecular target for induced synthetic lethality in cancer cells. J. Am. Chem. Soc. 2013;135:9640–9643. doi: 10.1021/ja404868t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bock L.C., Griffin L.C., Latham J.A., Vermaas E.H., Toole J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 11.Crnugelj M., Hud N.V., Plavec J. The solution structure of d(G(4)T(4)G(3))(2): a bimolecular G-quadruplex with a novel fold. J. Mol. Biol. 2002;320:911–924. doi: 10.1016/s0022-2836(02)00569-7. [DOI] [PubMed] [Google Scholar]
- 12.Sket P., Plavec J. Tetramolecular DNA quadruplexes in solution: insights into structural diversity and cation movement. J. Am. Chem. Soc. 2011;132:12724–12732. doi: 10.1021/ja104889t. [DOI] [PubMed] [Google Scholar]
- 13.Ambrus A., Chen D., Dai J., Bialis T., Jones R.A., Yang D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z., Dai J., Veliath E., Jones R.A., Yang D. Structure of a two-G-tetrad intramolecular G-quadruplex formed by a variant human telomeric sequence in K+ solution: insights into the interconversion of human telomeric G-quadruplex structures. Nucleic Acids Res. 2010;38:1009–1021. doi: 10.1093/nar/gkp1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray R.D., Trent J.O., Chaires J.B. Folding and unfolding pathways of the human telomeric G-quadruplex. J. Mol. Biol. 2014;426:1629–1650. doi: 10.1016/j.jmb.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trajkovski M., Šket P., Plavec J. Cation localization and movement within DNA thrombin binding aptamer in solution. Org. Biomol. Chem. 2009;7:4677–4684. doi: 10.1039/b914783g. [DOI] [PubMed] [Google Scholar]
- 17.Limongelli V., De Tito S., Cerofolini L., Fragai M., Pagano B., Trotta R., Cosconati S., Marinelli L., Novellino E., Bertini I., et al. The g-triplex DNA. Angew. Chem. Int. Ed. Engl. 2013;52:2269–2273. doi: 10.1002/anie.201206522. [DOI] [PubMed] [Google Scholar]
- 18.Marathias V.M., Wang K.Y., Kumar S., Pham T.Q., Swaminathan S., Bolton P.H. Determination of the number and location of the manganese binding sites of DNA quadruplexes in solution by EPR and NMR in the presence and absence of thrombin. J. Mol. Biol. 1996;260:378–394. doi: 10.1006/jmbi.1996.0408. [DOI] [PubMed] [Google Scholar]
- 19.Zídek L., Padrta P., Chmelík J., Jr, Sklenár V. Internal consistency of NMR data obtained in partially aligned biomacromolecules. J. Magn. Reson. 2003;162:385–395. doi: 10.1016/s1090-7807(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 20.Zídek L., Stefl R., Sklenár V. NMR methodology for the study of nucleic acids. Curr. Opin. Struct. Biol. 2001;11:275–281. doi: 10.1016/s0959-440x(00)00218-9. [DOI] [PubMed] [Google Scholar]
- 21.Carlomagno T. NMR in natural products: understanding conformation, configuration and receptor interactions. Nat. Prod. Rep. 2012;29:536–554. doi: 10.1039/c2np00098a. [DOI] [PubMed] [Google Scholar]
- 22.Vermeulen A., Zhou H., Pardi A. Determining DNA global structure and DNA bending by application of NMR residual dipolar couplings. J. Am. Chem. Soc. 2000;122:9638–9647. [Google Scholar]
- 23.Griesinger C., Sorensen O.W. ECOSY, avance-version (04/01/15);complementary E.COSY program for KcMAX = 3; (number of relevant spins = 3);phase sensitive. J. Magn. Reson. 1987;75:474–492. [Google Scholar]
- 24.Luy B., Marino J.P. (1)H-(31)P CPMG-correlated experiments for the assignment of nucleic acids. J. Am. Chem. Soc. 2001;123:11306–11307. doi: 10.1021/ja0166747. [DOI] [PubMed] [Google Scholar]
- 25.Carlomagno T., Hennig M., Williamson J.R. A novel PH-CT-COSY methodology for measuring JPH coupling constants in unlabeled nucleic acids. Application to HIV-2 TAR RNA. J. Biomol. NMR. 2002;22:65–81. doi: 10.1023/a:1013811631477. [DOI] [PubMed] [Google Scholar]
- 26.Enthart A., Freudenberger J.C., Furrer J., Kessler H., Luy B. The CLIP/CLAP-HSQC: pure absorptive spectra for the measurement of one-bond couplings. J. Magn. Reson. 2008;192:314–322. doi: 10.1016/j.jmr.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Rückert M., Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 2000;122:7793–7797. [Google Scholar]
- 28.Hansen M.R., Hanson P., Pardi A. Filamentous bacteriophage for aligning RNA, DNA, and proteins for measurement of nuclear magnetic resonance dipolar coupling interactions. Meth. Enzymol. 2000;317:220–240. doi: 10.1016/s0076-6879(00)17017-x. [DOI] [PubMed] [Google Scholar]
- 29.Stefl R., Wu H., Ravindranathan S., Sklenár V., Feigon J. DNA A-tract bending in three dimensions: solving the dA4T4 vs. dT4A4 conundrum. Proc. Natl. Acad. Sci. USA. 2004;101:1177–1182. doi: 10.1073/pnas.0308143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keller R. The Computer Aided Resonance Tutorial. Goldau, Switzerland: CANTINA Verlag; 2004. [Google Scholar]
- 31.Schwieters C.D., Kuszewski J.J., Tjandra N., Clore G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 32.Banci L., Bertini I., Cavallaro G., Giachetti A., Luchinat C., Parigi G. Paramagnetism-based restraints for Xplor-NIH. J. Biomol. NMR. 2004;28:249–261. doi: 10.1023/B:JNMR.0000013703.30623.f7. [DOI] [PubMed] [Google Scholar]
- 33.Tjandra N., Omichinski J.G., Gronenborn A.M., Clore G.M., Bax A. Use of dipolar 1H–15N and 1H–13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Mol. Biol. 1997;4:732–738. doi: 10.1038/nsb0997-732. [DOI] [PubMed] [Google Scholar]
- 34.Banci L., Bertini I., Bren K.L., Cremonini M.A., Gray H.B., Luchinat C., Turano P. The use of pseudocontact shifts to refine solution structures of paramagnetic metalloproteins: Met80Ala cyano-cytochrome c as an example. JBIC. 1996;1:117–126. [Google Scholar]
- 35.Banci L., Bertini I., Huber J.G., Luchinat C., Rosato A. Partial orientation of oxidized and reduced cytochrome b5 at high magnetic fields: magnetic susceptibility anisotropy contributions and consequences for protein solution structure determination. J. Am. Chem. Soc. 1998;120:12903–12909. [Google Scholar]
- 36.Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 37.Perez A., Marchan I., Svozil D., Sponer J., Cheatham T.E., Laughton C.A., Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tolbert B.S., Miyazaki Y., Barton S., Kinde B., Starck P., Singh R., Bax A., Case D.A., Summers M.F. Major groove width variations in RNA structures determined by NMR and impact of 13C residual chemical shift anisotropy and 1H-13C residual dipolar coupling on refinement. J. Biomol. NMR. 2010;47:205–219. doi: 10.1007/s10858-010-9424-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russo Krauss I., Merlino A., Randazzo A., Novellino E., Mazzarella L., Sica F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012;40:8119–8128. doi: 10.1093/nar/gks512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu X.-J., Olson W.K. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masiero S., Trotta R., Pieraccini S., De Tito S., Perone R., Randazzo A., Spada G.P. A non-empirical chromophoric interpretation of CD spectra of DNA G-quadruplex structures. Org. Biomol. Chem. 2010;8:2683–2692. doi: 10.1039/c003428b. [DOI] [PubMed] [Google Scholar]
- 43.Kim S.-G., Lin L.-J., Reid B.R. Determination of nucleic acid backbone conformation by proton NMR. Biochemistry. 1992;31:3564–3574. doi: 10.1021/bi00129a003. [DOI] [PubMed] [Google Scholar]
- 44.Gorenstein D.G. Conformation and dynamics of DNA and protein-DNA complexes by 31P NMR. Chem. Rev. 1994;94:1315–1338. [Google Scholar]
- 45.Markley J.L., Bax A., Arata Y., Hilbers C.W., Kaptein R., Sykes B.D., Wright P.E., Wüthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids (IUPAC Recommendations 1998) Pure Appl. Chem. 1998;70:117–142. doi: 10.1006/jmbi.1998.1852. [DOI] [PubMed] [Google Scholar]
- 46.Schultze P., Macaya R.F., Feigon J. Three-dimensional solution structure of the thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG) J. Mol. Biol. 1994;235:1532–1547. doi: 10.1006/jmbi.1994.1105. [DOI] [PubMed] [Google Scholar]
- 47.Mao X., Marky L.A., Gmeiner W.H. NMR structure of the thrombin-binding DNA aptamer stabilized by Sr2+ J. Biomol. Struct. Dyn. 2004;22:25–33. doi: 10.1080/07391102.2004.10506977. [DOI] [PubMed] [Google Scholar]
- 48.Reshetnikov R., Golovin A., Spiridonova V., Kopylov A., Šponer J. Structural dynamics of thrombin-binding DNA aptamer d(GGTTGGTGTGGTTGG) quadruplex DNA studied by large-scale explicit solvent simulations. J. Chem. Theory Comput. 2010;6:3003–3014. doi: 10.1021/ct100253m. [DOI] [PubMed] [Google Scholar]
- 49.Karsisiotis A.I., Hessari N.M., Novellino E., Spada G.P., Randazzo A., Webba da Silva M. Topological characterization of nucleic acid G-quadruplexes by UV absorption and circular dichroism. Angew. Chem. 2011;123:10833–10836. doi: 10.1002/anie.201105193. [DOI] [PubMed] [Google Scholar]
- 50.Randazzo A., Spada G.P., da Silva M.W. Circular dichroism of quadruplex structures. Top. Curr. Chem. 2013;330:67–86. doi: 10.1007/128_2012_331. [DOI] [PubMed] [Google Scholar]
- 51.Hud N.V., Smith F.W., Anet F.A., Feigon J. The selectivity for K +versus Na+ in DNA quadruplexes is dominated by relative free energies of hydration: a thermodynamic analysis by 1H NMR. Biochemistry. 1996;35:15383–15390. doi: 10.1021/bi9620565. [DOI] [PubMed] [Google Scholar]
- 52.Kankia B.I., Marky L.A. Folding of the thrombin aptamer into a G-Quadruplex with Sr2+: stability, heat, and hydration. J. Am. Chem. Soc. 2001;123:10799–10804. doi: 10.1021/ja010008o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.