


Abstract
The positive role of PARP1 in regulation of various nuclear DNA transactions is well established. Although a mitochondrial localization of PARP1 has been suggested, its role in the maintenance of the mitochondrial DNA is currently unknown. Here we investigated the role of PARP1 in the repair of the mitochondrial DNA in the baseline and oxidative stress conditions. We used wild-type A549 cells or cells depleted of PARP1. Our data show that intra-mitochondrial PARP1 interacts with a key mitochondrial-specific DNA base excision repair (BER) enzymes, namely EXOG and DNA polymerase gamma (Polγ), which under oxidative stress become poly(ADP-ribose)lated (PARylated). Interaction between mitochondrial BER enzymes was significantly affected in the presence of PARP1. Moreover, the repair of the oxidative-induced damage to the mitochondrial DNA in PARP1-depleted cells was found to be more robust compared to control counterpart. In addition, mitochondrial biogenesis was enhanced in PARP1-depleted cells, including mitochondrial DNA copy number and mitochondrial membrane potential. This observation was further confirmed by analysis of lung tissue isolated from WT and PARP1 KO mice. In summary, we conclude that mitochondrial PARP1, in opposite to nuclear PARP1, exerts a negative effect on several mitochondrial-specific transactions including the repair of the mitochondrial DNA.
INTRODUCTION
Poly(ADP-ribose) polymerase-1 (PARP1), a major member of the PARP family, is generally viewed as a ubiquitous nuclear protein involved in chromatin remodeling and the promotion of DNA repair (1–4). In contrast to the well-established roles of PARP1 in regulating nuclear processes, less is known about the role of PARP1 in the regulation of mitochondrial functions. Intra-mitochondrial PARylation and mitochondrial dysfunction linked to PARP1 hyperactivation during oxidative stress have previously been proposed (5–11). Furthermore, the potential role of PARP1 as a nuclear epigenetic regulator for the maintenance of mitochondrial DNA integrity has been suggested (5,6,12). We have shown earlier that cells depleted from PARP1 possess higher cellular bioenergetics parameters (13). However, the role of PARP1 in mitochondrial DNA repair remained unclear. In the current study we investigated role of the PARP1 in the maintenance of the mitochondrial DNA integrity. In contrast to its known positive role in the nucleus, the data in the current report show that PARP1 is a negative regulator of several mitochondrial DNA transactions, including DNA repair.
MATERIALS AND METHODS
Cell culture
A549 was obtained from ATCC. A549 stable lentiviral silencing of PARP1 (shPARP1) and scrambled (shCTR) lines were generated as described (14). Cells were maintained in RPMI 1640 media supplemented with 10% heat-inactivated fetal bovine, 50 units/ml penicillin, 50 μg/ml streptomycin and 1.5 μg/ml of puromycin (for stable depleted cells), and cultured at 37°C, 5% CO2.
Quantification of the mitochondrial and nuclear DNA damage
Integrity (the level of the DNA damage) of the nuclear and the mitochondrial DNA was analyzed by semi-quantitative, long-amplicon polymerase chain reaction (PCR) assays (LA-PCR) using LongAmp Taq DNA Polymerase (New England BioLabs, Ipswich, MA, USA) (15,16). Total DNA was isolated using DNase Blood and Tissue Kit (QIAGEN, Hilden, Germany). Briefly, damage to nuclear DNA was estimated by quantification of the PCR amplification of the 10-kb nuclear-specific DNA fragment using PicoGreen fluorescent dye to detect amplified double-stranded DNA (Quant-iT™ PicoGreen; Life Technologies, Carlsbad, CA, USA). Damage to the mitochondrial DNA was estimated by quantification of the PCR amplification of the 8.9-kb mitochondrial-specific DNA fragment using PicoGreen staining. Obtained data were normalized by the secondary PCR amplification of 221-bp mitochondrial genome-specific fragment for correction of the multiple copies of the mitochondrial DNA.
Real-time PCR (qPCR)
qPCR was performed as described (17). Briefly, 1 μg of total RNA from control and PARP1-depleted A549 cells isolated using TRIzol Reagent (Life Technologies; Carlsbad, CA) was used to synthesized cDNA using the High-Capacity cDNA Reverse Transcription kit (Life Technologies). The 50 x diluted cDNA was used for qPCR using the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific) and CFX96 TouchTM Real-Time PCR Detection System (Bio-Rad). The primers used are as follows. PARP1: 5′-GCT CCT GAA CAA TGC AGA CA-3′, 5′-CAT TGT GTG TGG TTG CAT GA-3′; β-actin 5′-GAC CCA GAT CAT GTT TGA GAC C-3′, 5′-CAT CAC GAT GCC AGT GGT AC-3′; mtDNA: 5′-CCC CAC AAA CCC CAT TAC TAA ACC CA-3′, 5′ TTT CAT CAT GCG GAG ATG TTG GAT GG-3′; EXOG: 5′-GCT CAG TAT CTAC CGA ACC ACT-3′, 5′-AAA CAC CAG TCC TGA CAA CTT C-3′; Polγ: 5′-AGC GCA GTC TGT GGA TAG C-3′, 5′-CTG GAA GTT CTC ACG AAT GTC C-3′; Lig3: 5′-TCA CTG GCG TGA TGT AAG ACA-3′, 5′-CCT GGA ATG ATA GAA CAG GCT TT-3′; Polβ: 5′-CCA GTG GTG ACA TGG ATG TTC-3′, 5′-TGC TCC ACA ACC TGA TGT AAC-3′. The expression of the DNA repair enzymes was normalized by the relative expression of β-actin.
Intracellular localization of PARP1
A549 cells were seeded in Lab-Tek II chamber coverglass system (Nalgen) and incubated at 37°C and 5% CO2 humidified incubator overnight. Next, cells were fixed and permeabilized with 4% paraformaldehyde, 0.2% Tween-20 in phosphate buffered saline (PBS) for 20 min at room temperature. After washing with PBS, cells were incubated with 1% bovine serum albumin (BSA) in PBS containing 0.25% Triton X-100 for 30 min at room temperature and probed with primary antibodies (1:200 dilution) overnight at 4°C on a rocking platform. After washing three times with PBS, cells were probed with mouse and rabbit-specific fluorescent-labeled secondary antibodies (1:200, Alexa Fluor 488 and 546, both from Life Technologies) for 2 h at room temperature. Slides were washed with PBS, air-dried and mounted with DAPI-containing solution. Fluorescent signal was visualized using the Nikon eclipse 80i inverted microscope as above.
Analysis of the total DNA repair assay in the mitochondrial extracts
The repair of tetrahydrofuran- (THF) containing oligo duplexes was assayed using oligo sequence and procedure as before (18–20). Briefly, 20 μl reaction assay contains 20 μM each of four unlabeled dNTPs, four μCi [α-32P]dATP and 5 μg of the mitochondrial protein extract. After incubation at 37°C for 60 min, the reaction was terminated by addition of 10 μl of 70% formamide and the final product was separated from the repair intermediates by electrophoresis in a 20% acrylamide/7M urea gel. The radioactivity in the product band was quantitated by PhosphorImager (Molecular Dynamics, Sunnyvale, CA, USA) using ImageQuant software. Preliminary enzyme assays were carried out to ensure the linearity of the reaction with respect to both time and the amount of extract.
Preparation of mitochondrial extract and western analysis
The mitochondrial extracts were prepared as described (19). Protein concentration was determined with Bradford reagent (Bio-Rad, Hercules, CA, USA). Western analysis was performed as described (21) with the membranes sequentially probed using antibodies against PARP1, HRP-conjugated actin (Santa Cruz, Dallas TX, USA), FEN1, APE1, Polγcat, Polγacc, Lig3, Polβ (Gene Tex, Irvine, CA, USA), PAR (BD Pharmingen, San Jose, CA, USA), GAPDH (Cell Signaling, Danvers, MA, USA), the 56 kDa subunit of ATP synthase (Molecular Probe-Invitrogen, Carlsbad, CA, USA), FeS subunit of mitochondrial complex III, subunit of complex I (NDUFS3), (Abcam, Cambridge, MA, USA), EXOG and FLAG-HRP conjugated (Sigma).
Proximity ligation assay
Proximity ligation assay (PLA) was performed using a pair of primary antibodies (listed in western analysis section) raised in two different species (as indicated in the figures) according to the manufacturer's recommendations (Olink Bioscience, Uppsala, Sweden) as described (22). Briefly, cells were fixed and permeabilized with 4% paraformaldehyde, 0.2% Tween-20 in PBS for 20 min at room temperature. After blocking for 1 h at room temperature, cells were probed with primary antibodies (at 1:200 dilution) overnight at 4°C. Signal detection and amplification was performed per manufacturer's instructions. Images were captured using Nikon eclipse 80i inverted fluorescent microscope with Photometric CoolSNAP HQ2 camera and NIS-Elements BR 3.10 software.
Immunoprecipitation
Isolated mitochondria were lysed in non-denaturating lysis buffer containing 1% Triton X-100, 50 mM Tris–HCl pH 7.4, 300 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 0.02% sodium azide and complete protease inhibitor cocktail. FLAG-specific immunoprecipitation was performed similarly as in (18) using 70 μl of ANTI-FLAG M2-Agarose (Sigma), washed twice with Tris-buffered saline (TBS) buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.5) and incubated with 1 mg mitochondrial extract isolated from A549 cells 48 h post-transfection with human EXOG cDNA cloned into p3x-FLAG-CMV-14 vector (Sigma) followed by incubation with 0.1 U/ml of GOx for 1 h, and for 4 h at 4°C in a tube rotator. To remove non-specific proteins, four washes with TBS buffer were performed, and the final pellet was resuspended in SDS Sample buffer for western analysis.
Mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured using the TMRE-Mitochondrial Membrane Potential Assay kit (Abcam, Cambridge, MA, USA). 5×104 cells/well in a 96-well plate were used for fluorescence measurement in microplate reader. As a control, 20 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) was used.
MTT assay
Cell viability was analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay that was conducted as described (20,38).
Measurement of the citrate synthase
Citrate synthase, the initial enzyme of TCA cycle and marker of the mitochondrial matrix frequently (47) used as a normalization factor for various mitochondrial-specific activities, was measured with a Citrate Synthase Assay Kit according to manufacturer's recommendations (Sigma).
Transient depletion of PARP1 by siRNA
A549 cells obtained from (ATCC) were transfected with 40 nM siRNA specific for PARP1 (at#4390825; Life Technologies) or control siRNA using Lipofectamine 2000 (Invitrogen) per the manufacturer's protocol. To optimize the siRNA concentration for maximal target depletion, preliminary transfections were carried out with 10–100 nM for each siRNA. The level of depletion was calculated by densitometric analysis of western blots and normalized to the respective loading control of three independent experiments. Cells with 80–90% of PARP1 depletion measured 48 h post-transfection were used in subsequent experiments.
Statistical analysis
Data are shown as means±SD standard deviation (SD). Student's t-test was used to detect differences between groups; *P < 0.05 and **P < 0.01 represent statistically significant differences from control.
RESULTS
PARP1 negatively regulates the integrity of the mitochondrial DNA
It is generally accepted that PARP1 positively regulates nuclear DNA repair by recognizing DNA damage (particularly single-strand breaks, SSBs), followed by the promotion of the recruitment of DNA repair enzymes to the site of DNA damage (23–25). Here we investigated the effect of stable PARP1 depletion on the integrity of the mitochondrial and nuclear DNA in resting state and during oxidative stress (the integrity of the DNA reflects the relative level of the DNA damage). In A549 cells with more than 90% depletion of PARP1 (shown by western analysis in Figure 1A), the semi-quantitative PCR of long DNA fragments (LA-PCR) revealed that the mitochondrial DNA of PARP1-deficient cells contains 60% less damage (meaning higher DNA integrity) than that of wild-type cells under basal conditions (Figure 1A). The amount of DNA damage in the nuclear DNA in wild-type and PARP1-depleted cells was comparable under baseline conditions (Figure 1A), in line with previous reports (26,27). It should be emphasized that the LA-PCR detects mostly SSBs (one of the most frequently occurring type of DNA damage induced by oxidative stress) and other types of the damage that halt progression of the DNA polymerase and allow concurrent monitoring level of DNA damage of both mitochondrial and nuclear DNA, without prior separation of mitochondrial DNA (15,16).
Figure 1.

Effect of PARP1 depletion and PARP1 inhibition on the integrity and repair of the mitochondrial DNA. (A) Integrity of the mitochondrial and nuclear DNA in baseline conditions of A549 shCTR and A549 shPARP1 cells was determined by LA-PCR. The protein level of PARP1 was analyzed by western blot and is shown in the inset. (B) Sensitivity of the mitochondrial DNA to increasing levels of oxidative stress, generated by increasing concentrations of glucose oxidase (GOx) measured by LA-PCR after 1 h of GOx treatment. (C) Repair of the mitochondrial DNA integrity in a control (shCTR) and PARP1-depleted (shPARP1) A549 cells. (D) Repair of the mitochondrial DNA integrity in parental A549 cells transiently transfected with scrambled (siCTR) or PARP1-specific (siPARP1) siRNA. (E) Repair of the mitochondrial DNA integrity in A549 cells preincubated with the PARP inhibitor PJ34 (10 μM) for 30 min. The graphs represent means±SD calculated based on at least two independent experiments run in triplicates (*P < 0.05, **P < 0.01). For (B) in order to directly compare sensitivity of the mitochondrial DNA in control and PARP1-depleted A549 cells, the integrity of mitochondrial DNA in both untreated groups was normalized as 100%. For (C-E) in order to directly compare repair rate in control and PARP1-depleted A549 cells, the initial level of mitochondrial DNA damage induced by GOx was normalized as 1.
In order to generate low and steady levels of oxidative stress, we used various concentrations of glucose oxidase (GOx), which in the presence of glucose in culture medium generates hydrogen peroxide (H2O2). Comparison of the amount of H2O2 generated by GOx with various concentrations of H2O2 is shown in Supplementary Figure S1. We monitored the amount of DNA damage (expressed as decreasing of the DNA integrity) in the nuclear and mitochondrial DNA after 1 h of GOx exposure using LA-PCR. No significant differences were detected between wild-type and PARP1-depleted cell types in the sensitivity of the mitochondrial DNA to oxidative stress (Figure 1B). It needs to be mentioned that this treatment did not induce any visible cell death. However, when we monitored the repair of the mitochondrial DNA integrity (after reducing mitochondrial DNA integrity by approximately 80% with exposure to 0.25 U/ml GOx for 1 h, followed by removal of GOx from the culture medium), we detected a 2-fold faster repair of the mitochondrial DNA in PARP1-depleted cells, than in the wild-type controls (Figure 1C). Similar to the cells with permanent PARP1 depletion, cells transiently depleted of PARP1 by siRNA exhibited faster repair of the mitochondrial DNA than the corresponding wild-type controls (Figure 1D). In order to distinguish between the catalytic activity of PARP1 and its physical presence, we monitored repair of the mitochondrial DNA after oxidative challenge in the presence of the PARP inhibitor PJ34 (28). In contrast to PARP1 depletion (but similar to the effect of PARP inhibition on nuclear DNA repair), 10 μM of the PJ34 impaired the rate of the mitochondrial DNA repair (Figure 1E). In control experiments we detected no effect of PJ34 treatment on the repair of the mitochondrial DNA integrity in PARP1-depleted cells (Supplementary Figure S2). In order to exclude the possible side effect of PARP1 depletion on the expression level of DNA repair enzymes, we have quantified the expression levels of several key DNA repair enzymes such as DNA polymerase γ (Polγ), EXOG, DNA ligase 3 (Lig3) and Polβ. No statistically significant difference was detected between control and PARP1-depleted A549 cells in the expression levels of these enzymes (Supplementary Figure S3). In a set of control experiments, in line with prior studies (26,27) we have also confirmed the higher sensitivity of the nuclear DNA to oxidative stress in PARP1-depleted cells (Figure 2A), while the repair efficiency of nuclear DNA was similar in wild-type and PARP1-depleted cells (Figure 2B). As expected, inhibition of PARP activity by 10 μM PJ34 impaired the repair rate of the nuclear DNA (Figure 3C). Taken together, the data presented above provide evidence that the presence of PARP1 negatively affects the maintenance of the mitochondrial DNA integrity, in contrast to its positive role in maintaining the integrity of the nuclear DNA.
Figure 2.
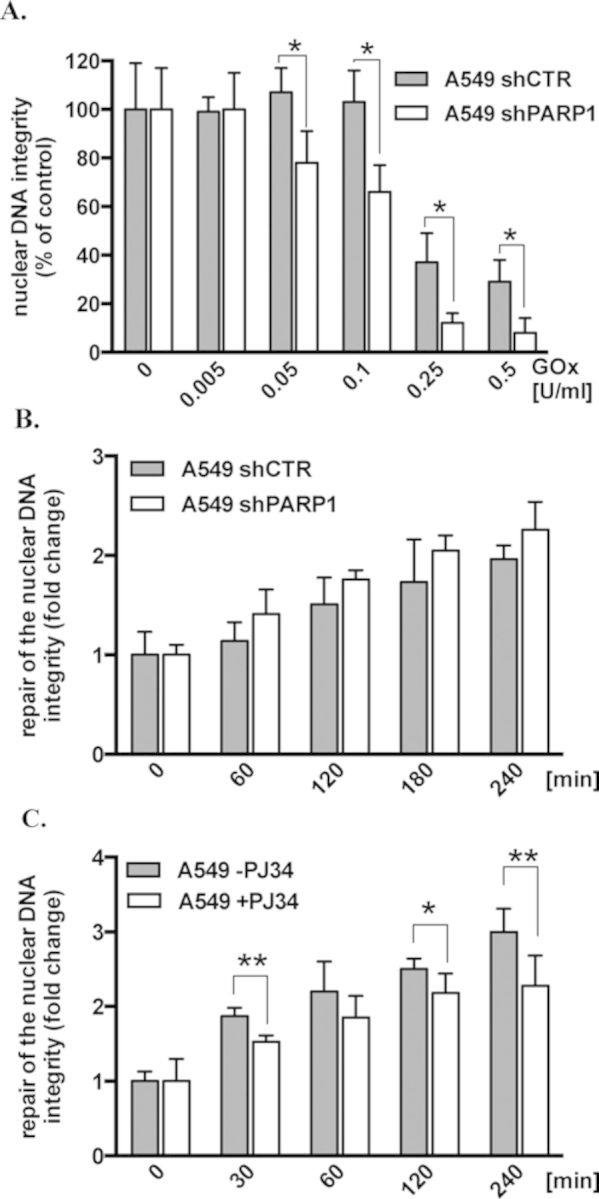
Effect of PARP1 depletion and PARP inhibition on the integrity and the repair of the nuclear DNA. (A) Sensitivity of the nuclear DNA to increasing concentration of GOx. (B) Repair of the nuclear DNA integrity in control (shCTR) and PARP1-depleted (shPARP1) A549 cells. (C) Repair of the nuclear DNA integrity in A549 cells preincubated with the PARP inhibitor PJ34 (10 μM). The graphs represent means±SD calculated based on at least two independent experiments run in triplicates (*P < 0.05, **P < 0.01). For (A) in order to directly compare sensitivity of the nuclear DNA in control and PARP1-depleted A549 cells, the integrity of the nuclear DNA in both untreated groups was normalized as 100%. For (B and C) in order to directly compare repair rate in control and PARP1-depleted A549 cells, the initial level of nuclear DNA damage induced by GOx was normalized as 1.
Figure 3.

PARP1 interacts with mitochondrial DNA repair enzymes in resting and oxidatively stressed cells. (A) The interaction of PARP1 with mitochondrial DNA repair enzymes, EXOG and Polγ, was investigated using PLA in A549 cells in resting cells. The primary antibody of two different species (m, mouse; r, rabbit), the detection reagent (Texas Red) and the mounting medium containing DAPI staining were used as described in ‘Materials and Methods’. The specificity of the interaction was monitored by the interaction between PARP1 and the 56 kDa subunit of mitochondrial ATP synthase (56 kDa CV). (B) The interaction between PARP1 and EXOG localized within the mitochondria, as indicated by co-stain with the mitochondrial-specific subunit of NADH dehydrogenase, NDUFS3 (subunit of complex I), and analyzed with fluorescent microscopy in wild-type A549 cells. Representative images of at least two independent experiments are shown.
Mitochondrial PARP1 interacts with mitochondrial-specific DNA repair enzymes
In line with previous studies, the majority of PARP1 localized to the nucleus, but a distinct fraction was also detected in the mitochondria (Supplementary Figure S4). To provide direct evidence for the functional role of PARP1 in the regulation of mitochondrial DNA integrity, we used the PLA, which allows the detection of close proximity/interaction between two proteins in cellulo. There are only two known mitochondrial-specific DNA repair enzymes: Polγ and EXOG; the rest is shared by the nucleus and mitochondria (29–32). Using PLA we detected interactions between PARP1 and EXOG, as well as between PARP1 and catalytic subunit of Polγ (Polγcat) in resting wild-type A549 cells (Figure 3A). We have not detected any interaction between the 56 kDa subunit of ATP synthase and PARP1 (Figure 3A), indicative of the specificity of the assay (i.e. confirming that PLA signal comes only from co-localized mitochondrial PARP1 with specific mitochondrial proteins). To directly show mitochondrial localization of the obtained PLA signal, we further stained the slide using an antibody against subunit of NADH dehydrogenase (NDUFS3); both signals significantly overlap, suggesting mitochondrial localization of the interaction between EXOG and PARP1 (Figure 3B). As a confirmatory approach, we detected the presence of PARP1 in EXOG IP and EXOG in PARP1 IP (Supplementary Figure S5). The perinuclear localization of EXOG-PARP1 interaction was further confirmed by confocal microscopy (Supplementary Figure S6). Next, we compared the interaction of PARP1 with DNA repair enzymes in control (shCTR) and PARP1-depleted (shPARP1) A549 cells subjected to a low concentration of GOx (0.1 U/ml), which is known to induce damage preferentially to the mitochondrial DNA (Figures 1 and 2). Oxidative stress induced a significant increase of the interaction of PARP1 with EXOG and Polγcat in control A549 cells (left panels of Figure 4A). As expected, the interaction between PARP1 and mitochondrial DNA repair enzymes was markedly reduced in PARP1-depleted A549 cells (right panels of Figure 4A). In control experiments we also monitored the interaction of PARP1 with nuclear-specific DNA repair enzymes, namely, Polβ and FEN1 (33), in resting and oxidatively stressed (0.25U GOx/ml) A549 cells. Interestingly we could not detect any interaction between PARP1 and the nuclear DNA repair enzymes in both condition and cell types (Figure 4B). Taken together, the data presented above demonstrate a specific interaction between PARP1 and key mitochondrial DNA repair enzymes, an effect, which is further enhanced by oxidative stress.
Figure 4.

Mitochondrial-specific DNA repair enzymes interact with PARP1. (A) Interaction between PARP1 and mitochondrial-specific DNA repair enzymes, EXOG and Polγcat (catalytic subunit of Polγ), in resting and oxidatively stressed A549 shCTR and shPARP1 cells was analyzed by PLA. (B) Interaction of PARP1 and nuclear-specific DNA repair enzymes, FEN1 and Polβ, in resting and oxidatively stressed A549 shCTR and shPARP1 cells was analyzed by PLA. Representative images of at least two independent experiments are shown.
Oxidative stress triggers PARylation of the mitochondrial DNA repair enzymes
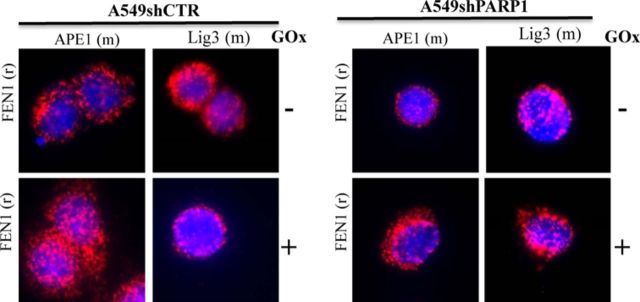
Although the role of PARP1 in nuclear DNA damage recognition and repair is widely accepted, XRCC1 is the only known BER enzyme shown to be PARylated by PARP1 (34,35): this enzyme facilitates nuclear DNA repair complex assembly; however, it is not directly involved in the process of DNA repair. Here we investigated whether the mitochondrial DNA repair enzymes undergo PARylation using PLA, which technique also allows monitoring of post-translational modification of the target protein. In agreement with the previous data, we detected PARylation of both EXOG and Polγcat in response to oxidative stress (0.1 U/ml of GOx) in A549 shCTR, but not in A549 shPARP1 cells (Figure 5A). The increase in the perinuclear signal observed is consistent with the interpretation that mitochondrial DNA-specific damage occurred under these experimental conditions. To further confirm that PARylation of the mitochondrial-specific DNA repair enzymes occurs in oxidative stress conditions, we immunoprecipitated (IP) EXOG from crude mitochondrial extracts isolated from oxidatively stressed A549 cells transfected with an EXOG-FLAG construct. Western analysis showed the presence of PAR groups on EXOG-IP's (Figure 5B). We also investigated the PARylation of nuclear-specific BER enzymes, namely, FEN1 and Polβ, in basal and oxidative stress conditions in control A549 cells. To induce damage in the nuclear DNA, we employed a higher concentration (0.25 U/ml) of GOx, which induces a significant amount of SSBs in the nuclear DNA (Figure 2). In contrast to the mitochondrial BER enzymes, nuclear Polβ and FEN1 were not PARylated, either in resting conditions or during oxidative stress (Figure 5C). Taken together, the data presented above show that mitochondrial-specific DNA repair enzymes (but not nuclear DNA repair enzymes) are PARylated by PARP1 in response to oxidative stress.
Figure 5.

Mitochondrial DNA repair enzymes are PARylated under oxidative stress conditions. (A) PARylation of mitochondrial-specific DNA repair enzymes, EXOG and Polγcat, is induced by oxidative stress in control but not in PARP1-depleted A549 cells. (B) EXOG, the mitochondrial-specific DNA repair enzyme is PARylated in oxidatively stressed cells. A549 cells were exposed to 0.1 U/ml GOx to induce oxidative stress. Immunoprecipitation (IP) of EXOG from mitochondrial fraction was performed as described in ‘Materials and Methods’. FLAG-specific antibody was used to ensure specificity of the transfection. EXOG and PAR-specific antibodies were used for western analysis. (C) In cellulo PARylation of nuclear-specific BER enzymes, FEN1 and Polβ, in resting and oxidatively stressed of control and PARP1-depleted A549 cells was analyzed by PLA. Representative images of at least two independent experiments are shown.
Interaction between mitochondrial DNA repair enzymes is affected upon oxidative stress in the presence of PARP1
The results presented above indicated that in the control A549 cells, PARP1 interacts with and PARylates a set of mitochondrial-specific DNA repair enzymes. Next, we have investigated the effect of PARP1 on binary interaction between several mitochondrial DNA repair enzymes. We detected low level of the interaction between EXOG and APE1 or Lig3, Polγcat and Lig3, and Polγacc (an accessory subunit of Polγ) and EXOG or APE1, in control A549 cells, which was unaffected by oxidative stress (upper panels of Figure 6A). In contrast, in a similar study in A549 cells depleted of PARP1, we observed a marked enhancement of the same set of the analyzed binary interactions in response to oxidative stress (lower panels of Figure 6A). The functional consequence of this unexpected finding was further investigated by analysis of the total DNA repair capacity in mitochondrial extracts isolated from control and PARP1-depleted A549 cells. A significantly higher DNA repair activity (that was further enhanced by the oxidative stress) was detected in mitochondrial extracts of cells depleted from PARP1, as evidenced by the analysis of the incorporation of 32P-dATP into THF containing 52-nt long oligo duplex (Figure 6B).
Figure 6.

The presence of PARP1 affects interaction between mitochondrial DNA repair enzymes. (A) Binary interactions between three mitochondrial-specific DNA repair enzymes (EXOG, Polγcat and Polγacc (catalytic and accessory subunit of DNA Polγ) and other DNA repair enzymes were analyzed by PLA in control and PARP1-depleted A549 cells under baseline and oxidative stress conditions. (B) Representative autoradiogram of the total DNA repair activity of control and PARP1-depleted A549 cells of mitochondrial extracts isolated from resting and oxidatively stressed cells. Incorporation of 32P-dATP into THF containing 52-nt long oligo duplex is shown. The graph represents quantification of the total mitochondrial DNA repair expressed as means±SD calculated based on three independent experiments (*P < 0.05).
In control experiments, we have analyzed the effect of PARP1 on the binary interaction between nuclear FEN1 and APE1 or Lig3 in resting and oxidatively stressed A549 cells. In contrast to the mitochondria, PARP1 did not affect the interaction between these DNA repair enzymes in the nucleus (Figure 7). Taken together, our data clearly show the negative role of PARP1 in the interaction between mitochondrial-specific DNA repair, which results in a decrease in DNA repair capacity within the mitochondria. Although we have shown that—in contrast to the known positive role of PARP1 in the nucleus—the presence of the PARP1 in mitochondria negatively regulates several mitochondrial DNA transactions, the effect of PARP1 on other mitochondrial functions remains to be investigated in future studies.
Figure 7.
PARP1 does not affect interaction between nuclear DNA repair enzymes. The binary interaction between nuclear-specific DNA repair enzyme, FEN1, and other DNA repair enzyme in resting and oxidatively stressed control and PARP1-depleted A549 cells was analyzed by PLA. Representative images of at least two independent experiments are shown.
Depletion of PARP1 enhances mitochondrial biogenesis
We have shown earlier that depletion of the PARP1 in cultured cells significantly increases various bioenergetic parameters, including basal respiration and respiratory reserve capacity (7). Our data so far indicated that PARP1 negatively affects the integrity of the mitochondrial DNA by decreasing the efficiency of its repair. Because the loss of mitochondrial DNA integrity has been proposed to adversely affect mitochondrial protein transcription and overall mitochondrial homeostasis (36,37), next we investigated whether PARP1 depletion affects mitochondrial functions under our baseline experimental conditions. We detected an ∼30% increase in citrate synthase activity, a known mitochondrial matrix-specific enzyme, of A549 cells depleted from PARP1 (Figure 8A). This was associated with an ∼50% increase of the mitochondrial DNA copy number (Figure 8B), with elevated levels of multiple mitochondrial DNA-encoded proteins (56 kDa subunit of ATP synthase and FeS subunit of ubiquinol-cytochrome c reductase, complex V and III) (Figure 8C), and with an increased (∼40%) MMP (Figure 8D). To test the functional consequence of increased mitochondrial biogenesis in PARP1-depleted cells, we treated both A549 cell types with increasing concentration of GOx, and measured conversion of MTT into formazan by mitochondrial dehydrogenases (38). Significantly higher resistance to oxidative stress was observed in A549 cells depleted of PARP1 (Figure 8E). Taken together, from the data presented above we conclude that cells depleted from PARP1 exhibit improved mitochondrial homeostasis, as well as a resistance to oxidative stress.
Figure 8.

PARP1 depletion increases mitochondrial biogenesis. Effect of PARP1 depletion on: (A) citrate synthase, mitochondrial matrix marker analyzed by the measurement of citrate synthase activity; (B) mitochondrial DNA copies, as analyzed by qPCR using set of mitochondrial DNA-specific primers; (C) expression of the α subunit of the ATP synthase (complex V, CV) and FeS subunit of ubiquinol-cytochrome c reductase (complex III, CIII), as analyzed by western blotting of total cell extracts. The level of PARP1 and GAPDH (as a loading control) are also shown. Representative images of at least two independent experiments are shown. (D) Effect of PARP1 depletion on MMP. (E) Effect of PARP1 depletion on reduction of MTT. Experiments were conducted in A549 control (shCTR) and PARP1-depleted (shPARP1) cells. The graphs represent means±SD calculated based on two independent experiments run in triplicates (*P < 0.05, **P < 0.01).
Lower level of the mitochondrial DNA damage and increased mitochondrial biogenesis in the lungs of PARP1−/− mice
Next we investigated the effect of PARP1 deficiency by evaluating the amount of DNA damage in the mitochondrial DNA of wild-type and PARP1−/− mice (Figure 9A). In agreement with our in vitro data, in lung extracts of PARP1−/− mice we detected significantly less (∼40%) SSBs, while nuclear DNA damage of the PARP1−/− tissue was comparable to wild-type controls (Figure 9B). This was associated with ∼60% higher amount of the citrate synthase activity (marker of the mitochondrial matrix) (Figure 9C) and ∼60% higher mitochondrial DNA copy number (Figure 9D). Taken together, the data presented above confirm and extend our in vitro results showing that mitochondrial-specific PARP1 has negative effect of several mitochondrial functions.
Figure 9.

Lung tissue from PARP1−/− mice exhibits increased mitochondrial DNA integrity and higher mitochondrial function. (A) Protein level of PARP1; (B) the integrity of the mitochondrial and nuclear DNA; (C) mitochondrial matrix marker (citrate synthase); and (D) the number of the mitochondrial DNA copies; all analyzed in lung tissue homogenates from wild-type and PARP1−/− mice. The graphs represent means±SD calculated based on n = 4 animals for WT and KO run in triplicates (*P < 0.05, **P < 0.01).
DISCUSSION
The data presented here show that—in contrast to the previously reported positive regulatory role of the PARP1 in the regulation of nuclear DNA integrity—the presence of the PARP1 in the mitochondria leads to a pronounced inhibitory effect on mitochondrial DNA damage repair. This effect is likely related to the fact that PARP1 directly interacts with and PARylates mitochondrial-specific DNA repair enzymes: EXOG and Polγ. Oxidative stress induced a marked increase in the PARylation of the mitochondrial but not nuclear BER enzymes. Finally, we present data that PARP1 negatively affects mitochondrial biogenesis and mitochondrial function both in cultured cells and in vivo.A single mammalian cell contains up to several thousands copies of duplex, circular 16.5-kb mitochondrial DNA within 80–700 mitochondria (39). The mutation rate of human mitochondrial DNA, due to the close proximity to reactive oxygen species generation from the mitochondrial electron transport chain, and due to its unchromatinized nature, is 20–100-fold higher than that in the nuclear counterpart (40,41). The mitochondrial DNA is constantly damaged by reactive oxidants, resulting in the generation of oxidized bases, abasic sites and SSBs, all of which are physiologically repaired by a robust mitochondrial DNA base excision repair (BER). Mammalian mitochondria are proficient for both BER sub-pathways, namely the single nucleotide-BER (42) and the long-patch-BER (18,43,44). We have previously shown that EXOG plays a key role in mitochondrial long-patch-BER (32). Moreover, we have also demonstrated that accumulation of SSBs in the mitochondrial DNA, on its own, is sufficient to trigger cell death (32), a finding that points to the importance of the maintenance of the mitochondrial DNA integrity.
Although the majority of the BER enzymes are shared by the nucleus and the mitochondria, there are exclusively nuclear enzymes (such as Polβ and FEN1) and exclusively mitochondrial enzymes (such as EXOG and Polγ) as well. Although it was suggested that the small fraction of FEN1 is localized in the mitochondria, we and others were unable to detect a direct role of the FEN1 in the repair of the mitochondrial DNA (18,32,43). FEN1 is generally viewed as a nuclear DNA repair enzyme (48), a counterpart of the mitochondrial-specific EXOG. The present results demonstrate that PARP1 interacts with mitochondrial (but not nuclear) BER enzymes. This interaction is already present in baseline conditions, but it is markedly enhanced by oxidative mitochondrial DNA damage (Figure 4). We speculate that the baseline interaction is due to a constant, low-level oxidant production within the mitochondria, which is a byproduct of oxidative phosphorylation. We hypothesize that in the mitochondria (where both the DNA itself and the DNA repair processes are different from the nuclear processes), PARylation (i.e. the placement of negatively charged PAR groups) on DNA repair enzymes either inhibits their catalytic activity, or it delays the assembly of mitochondrial DNA repair complexes. Both hypotheses are supported by our data; the interaction between mitochondrial DNA repair enzymes and mitochondrial DNA repair capacity is affected by the presence of PARP1 (Figure 6). We speculate that the addition of PAR groups on mitochondrial DNA repair enzymes may reduce their affinity to DNA and thus may attenuate the rate of the mitochondrial DNA repair, but this hypothesis requires further investigation. From an evolutionary standpoint, it is likely that the interaction of PARP1 with DNA repair enzymes has been fine-tuned and optimized for the nuclear compartment (where PARP1 PARylates various chromatin-associated proteins, in order to promote a relaxed chromatin conformation and to recruit DNA repair enzymes, thereby facilitating DNA repair) (45). In contrast, in the mitochondrial compartment, where the milieu is markedly different (lack of chromatin, circular form of DNA, small size of mitochondrial genome, different set of DNA repair enzymes), the actions of PARP1 may result in a markedly different, perhaps less then optimal consequences in terms of DNA repair efficiency. Although it seems counterintuitive, it is also conceivable that by negatively regulating mitochondrial DNA integrity under baseline conditions, PARP1 offers the opportunity for a regulated increase in mitochondrial biogenesis/mitochondrial function under conditions when such response may be beneficial for the cell.
Data presented in the present report also indicate a fundamental difference in the formation of the DNA repair proficient complexes—the existence of which has previously been shown in the nucleus (46). The lack of enhancement of the interaction between nuclear DNA repair enzymes in response to oxidative stress (Figure 7) suggests the existence of pre-formed DNA repair complexes in the nucleus. In contrast, the increased interaction of mitochondrial DNA repair enzymes in response to oxidative stress (Figure 6) clearly indicates a different mechanism for the repair of the mitochondrial DNA. Taking into account that damage to the mitochondrial DNA alone is sufficient to induce cell death (32) and that the formation of repair proficient complexes is genotoxic agent dependent, the current findings open new opportunities to investigate mitochondrial DNA repair as a novel therapeutic target.
As indicated earlier, it must also be noted that the effect of PARP1 depletion and PARP1 inhibition has different outcomes for the repair of the nuclear DNA; inhibition will trap PARP1 on SSBs inhibiting repair process, while lack of PARP1 does not change kinetics of the repair rate (26,27). Our data on nuclear DNA repair (Figure 2) are agreement with this concept. Moreover (as previously shown for the repair of the nuclear DNA), we have observed that PARP inhibition (in contrast to PARP1 depletion) significantly decreases the repair rate of the mitochondrial DNA (Figure 1E), presumably via mechanisms that are similar to the events that occur in the nucleus (i.e. trapping of PARP1 at the strand break) (26,27).
Taken together, the current report demonstrates that the presence of PARP1 in the mitochondria (as opposed to the situation in the nucleus) negatively affects mitochondrial DNA repair. As mitochondrial DNA integrity is known to affect mitochondrial protein transcription, and, consequently mitochondrial homeostasis (including electron transport, cellular bioenergetics, mitochondrial biogenesis and the response to increased oxidative injury) (36,37), the current study links PARP1 to these functional mitochondrial effects through the regulation of mitochondrial DNA integrity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R01GM056687 to C.S., R01CA53791 to S.M.]. Funding for open access charge: National Institutes of Health [R01GM056687 to C.S., R01CA53791 to S.M.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hassa P.O., Hottiger M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008;13:3046–3082. doi: 10.2741/2909. [DOI] [PubMed] [Google Scholar]
- 2.Krishnakumar R., Kraus W.L. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim M.Y., Zhang T., Kraus W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 4.Bouchard V.J., Rouleau M., Poirier G.G. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Hematol. 2003;31:446–454. doi: 10.1016/s0301-472x(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 5.Du L., Zhang X., Hann Y.Y., Burke N.A., Kochanek P.M., Watkins S.C., Graham S.H., Carcillo J.A., Szabo C., Clark R.S. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- 6.Cipriani G., Rapizzi E., Vannacci A., Rizzuto R., Moroni F., Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J. Biol. Chem. 2005;280:17227–17234. doi: 10.1074/jbc.M414526200. [DOI] [PubMed] [Google Scholar]
- 7.Pankotai E., Lacza Z., Muranyi M., Szabo C. Intra-mitochondrial poly(ADP-ribosyl)ation: potential role for alpha-ketoglutarate dehydrogenase. Mitochondrion. 2009;9:159–164. doi: 10.1016/j.mito.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Scovassi A.I. Mitochondrial poly(ADP-ribosylation): from old data to new perspectives. FASEB J. 2004;18:1487–1488. doi: 10.1096/fj.04-1841rev. [DOI] [PubMed] [Google Scholar]
- 9.Formentini L., Macchiarulo A., Cipriani G., Camaioni E., Rappizzi E., Pellicciari R., Moroni F., Chiarugi A. Poly(ADP-ribose) catabolism triggers AMP-dependent mitochondrial energy failure. J. Biol. Chem. 2009;284:17668–17676. doi: 10.1074/jbc.M109.002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai Y., Chen Y., Watkins S.C., Nathaniel P.D., Guo F., Kochanek P.M., Jenkins L.W., Szabo C., Clark R.S. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 2008;104:1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- 11.Rossi M.N., Carbone M., Mostocotto C., Mancone C., Tripodi M., Maione R., Amati P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lapucci A., Pittelli M., Rapizzi E., Felikci R., Moroni F., Chiarugi A. Poly(ADP-ribose) polymerase-1 is a nuclear epigenetic regulator of mitochondrial DNA repair and transcription. Mol. Pharmacol. 2011;79:932–940. doi: 10.1124/mol.110.070110. [DOI] [PubMed] [Google Scholar]
- 13.Modis K., Gero D., Erdelyi K., Szoleczky P., DeWitt D., Szabo C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem. Pharmacol. 2012;83:633–643. doi: 10.1016/j.bcp.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erdelyi K., Bai P., Kocacs I., Szabo E., Mocsar G., Kakuk A., Szabo C., Gergely P., Virag L. Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. FASEB J. 2009;23:3553–3563. doi: 10.1096/fj.09-133264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furda A.M., Bess A.S., Meyer J.N., Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 2012;920:111–132. doi: 10.1007/978-1-61779-998-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos J.H., Meyer J.N., Mandavilli B.S., Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 17.Bai P., Canto C., Oudart H., Brunyanszki A., Cen Y., Thomas C., Yamamoto H., Huber A., Kiss B., Houtkooper R.H., et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metabol. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szczesny B., Tann A.W., Longley M.J., Copeland W.C., Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008;283:26349–26356. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szczesny B., Tann A.W., Mitra S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: susceptibility of skeletal muscles to oxidative injury. Mech. Ageing Dev. 2010;131:330–337. doi: 10.1016/j.mad.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szczesny B., Olah G., Walker D.K., Volpi E., Rasmussen B.B., Szabo C., Mitra S. Deficiency in repair of the mitochondrial genome sensitizes proliferating myoblasts to oxidative damage. PloS One. 2013;8:e75201. doi: 10.1371/journal.pone.0075201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramana C.V., Boldogh I., Izumi T., Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc. Natl Acad. Sci. U.S.A. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fredriksson S., Gullberg M., Jarvius J., Olsson C., Pietras K., Gustafsdottir S.M., Ostman A., Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 23.Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 24.de Murcia J.M., Niedergang C., Trucco C., Ricoul M., Dutrillaux B., Mark M., Oliver F.J., Masson M., Dierich A., LeMeur M., et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl Acad. Sci. U.S.A. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trucco C., Oliver F.J., de Murcia G., Menissier-de-Murcia J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998;26:2644–2649. doi: 10.1093/nar/26.11.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godon C., Cordelieres F.P., Biard D., Giocanti N., Megnin-Chanet F., Hall J., Favaudon V. PARP inhibition versus PARP-1 silencing: different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res. 2008;36:4454–4464. doi: 10.1093/nar/gkn403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strom C.E., Johansson F., Uhlen M., Szigyarto C.A., Erixon K., Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39:3166–3175. doi: 10.1093/nar/gkq1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jagtap P., Soriano F.G., Virag L., Liaudet L., Mabley J., Szabo E., Hasko G., Marton A., Lorigados C.B., Gallyas F., Jr, et al. Novel phenanthridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit. Care Med. 2002;30:1071–1082. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 29.Graziewicz M.A., Longley M.J., Copeland W.C. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem. Rev. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 30.Cymerman I.A., Chung I., Beckmann B.M., Bujnicki J.M., Meiss G. EXOG, a novel paralog of Endonuclease G in higher eukaryotes. Nucleic Acids Res. 2008;36:1369–1379. doi: 10.1093/nar/gkm1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kieper J., Lauber C., Gimadutdinow O., Urbanska A., Cymerman I., Ghosh M., Szczesny B., Meiss G. Production and characterization of recombinant protein preparations of endonuclease G-homologs from yeast, C. elegans and humans. Protein Expr. Purif. 2010;73:99–106. doi: 10.1016/j.pep.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Tann A.W., Boldogh I., Meiss G., Qian W., Van Houten B., Mitra S., Szczesny B. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of exog (5′-exo/endonuclease) in their repair. J. Biol. Chem. 2011;286:31975–31983. doi: 10.1074/jbc.M110.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hegde M.L., Mantha A.K., Hazra T.K., Bhakat K.K., Mitra S., Szczesny B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 2012;133:157–168. doi: 10.1016/j.mad.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masson M., Niedergag C., Schreiber V., Muller S., Menissier-de-Murcia J., de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Khamisy S.F., Masutani M., Suzuki H., Caldecott K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cline S.D. Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta. 2012;1819:979–991. doi: 10.1016/j.bbagrm.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berneburg M., Kamenisch Y., Krutmann J., Rocken M. ‘To repair or not to repair—no longer a question’: repair of mitochondrial DNA shielding against age and cancer. Exp. Dermatol. 2006;15:1005–1015. doi: 10.1111/j.1600-0625.2006.00508.x. [DOI] [PubMed] [Google Scholar]
- 38.van Meerloo J., Kaspers G.J., Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol. Biol. 2011;731:237–245. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- 39.Miller F.J., Rosenfeldt F.L., Zhang C., Linnane A.W., Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cadenas E., Davies K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 41.Pesole G., Gissi C., De Chirico A., Saccone C. Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol. 1999;48:427–434. doi: 10.1007/pl00006487. [DOI] [PubMed] [Google Scholar]
- 42.Stierum R.H., Dianov G.L., Bohr V.A. Single-nucleotide patch base excision repair of uracil in DNA by mitochondrial protein extracts. Nucleic Acids Res. 1999;27:3712–3719. doi: 10.1093/nar/27.18.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akbari M., Visnes T., Krokan H.E., Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair. 2008;7:605–616. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Liu P., Qian L., Sung J.S., de Souze-Pinto N.C., Zheng L., Bogenhagen D.F., Bohr V.A., Wilson D.M., III, Shen B., Demple B. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol. Cell. Biol. 2008;28:4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okano S., Lan L., Caldecott K.W., Mori T., Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003;23:3974–3981. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanssen-Bauer A., Solvang-Garten K., Sundheim O., Pena-Diaz J., Anderson S., Slupphaug G., Krokan H.E., Wilson D.M., III, Akbari M., Otterlei M. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ. Mol. Mutagen. 2011;52:623–635. doi: 10.1002/em.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holloszy J.O., Oscai L.B., Don I.J., Mole P.A. Mitochondrial citric acid cycle and related enzymes: adaptive response to exercise. Biochem. Biophys. Res. Commun. 1970;40:1368–1373. doi: 10.1016/0006-291x(70)90017-3. [DOI] [PubMed] [Google Scholar]
- 48.Balakrishnan L., Bambara R.A. Flap endonuclease 1. Annu. Rev. Biochem. 2013;82:119–138. doi: 10.1146/annurev-biochem-072511-122603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.