


Abstract
In Escherichia coli, an increase in the ATP bound form of the DnaA initiator protein results in hyperinitiation and inviability. Here, we show that such replication stress is tolerated during anaerobic growth. In hyperinitiating cells, a shift from anaerobic to aerobic growth resulted in appearance of fragmented chromosomes and a decrease in terminus concentration, leading to a dramatic increase in ori/ter ratio and cessation of cell growth. Aerobic viability was restored by reducing the level of reactive oxygen species (ROS) or by deleting mutM (Fpg glycosylase). The double-strand breaks observed in hyperinitiating cells therefore results from replication forks encountering single-stranded DNA lesions generated while removing oxidized bases, primarily 8-oxoG, from the DNA. We conclude that there is a delicate balance between chromosome replication and ROS inflicted DNA damage so the number of replication forks can only increase when ROS formation is reduced or when the pertinent repair is compromised.
INTRODUCTION
Most bacterial chromosomes carry a single origin of replication, oriC, where replication starts. The oriC region is characterized by the presence of an AT-rich region and multiple binding sites for the DnaA initiator protein (1). DnaA belong to the AAA+ (ATPases Associated with diverse Activities) proteins, and the Escherichia coli DnaA protein binds ATP and ADP with similar affinities. However, only the ATP bound form is active in initiation (2). The current model for replication initiation is derived from work on E. coli and proposes that one or more right-handed DnaAATP helices are formed on multiple DnaA binding sites of the origin, which leads to duplex opening in the AT-rich region, i.e. open complex formation (1,2). Thereafter, DnaA loads the helicase DnaB onto the single-stranded DNA of the open complex, which promotes further duplex opening and assembly of the replisome.
Replication initiation is a highly regulated step in E. coli that commences virtually simultaneously at all cellular origins and only once per cell cycle (3). This tight control is mainly ensured by a fluctuation in the DnaAATP/DnaAADP ratio over the cell cycle (4) along with a temporal inactivation of newly replicated origins by the Dam/SeqA system (5,6).
Initiation takes place when the cellular DnaAATP/DnaAADP ratio is high (4). Following initiation, two processes converts DnaAATP to DnaAADP. First, RIDA (Regulatory Inactivation of DnaA) is executed by the Hda protein in association with DNA-loaded DnaN (the β-clamp) which activates the intrinsic ATPase activity of DnaA thereby turning DnaAATP into DnaAADP and lowering the DnaAATP/DnaAADP ratio (7,8). Second, DDAH (datA-dependent DnaAATP hydrolysis) is a process where Integration Host Factor (IHF)-dependent DnaAATP hydrolysis takes place at the datA locus (9).
Overall, RIDA seems more important than DDAH in lowering the DnaAATP/DnaAADP ratio to prevent reinitiation; RIDA deficient cells (i.e. hda mutants) overinitiate replication, are severely compromised for growth (8) and acquire second site suppressor mutations rapidly (10,11), whereas this is not the case for DDAH compromised (datA deleted) cells (12). It is likely that lethality resulting from loss of Hda is similar to what was observed for overinitiation in the dnaAcos mutant where hyperinitiation leads to fork collapse and DNA strand breaks (13), i.e. replication stress. Before a new round of initiation can take place, the DnaAATP level must increase past a critical level. This is accomplished by de novo synthesis of DnaA which by and large will be ATP bound because ATP is much more abundant than ADP within the cell, and by rejuvenation of DnaAADP into DnaAATP at DARS loci (14) and possibly at the interface of the cellular membrane and cytosol (15).
When growing aerobically, E. coli cells use oxygen as the terminal electron acceptor. This allows for a more efficient energy production in comparison to anaerobic respiration and fermentation. However, reactive oxygen species (ROS) are derived from the metabolism of molecular oxygen and the major sources of endogenous ROS are hydrogen peroxide (H2O2) and superoxide anion (O2−), which are formed when flavoenzymes accidentally pass electrons to oxygen (16). ROS can react with DNA to generate a number of base modifications (17). Relative to other nucleobases, oxidation of guanine to 8-oxo-7,8 dihydroguanine (8-oxoG (GO)) appears most readily because of its low redox potential (18). When incorporated into DNA, 8-oxoG can base pair with adenine leading to G to T transversions. In E. coli three enzymes named MutT, MutM and MutY protect the cell from the mutagenic action of 8-oxoG (19). MutT is a nucleotide sanitizer which hydrolyzes 8-oxo-dGTP to 8-oxodeoxyguanosine monophosphate (dGMP) to prevent incorporation into DNA (19). When present in the DNA, 8-oxoG is primarily excised by the formamidopyrimidine DNA glycosylase (Fpg) which is the product of the mutM gene of the GO system (18), and Fpg is the primary enzyme that removes not only oxidized purines but also pyrimidines in vivo (20), thereby reducing the accumulation of mutations. MutY is a glycosylase that removes adenines incorporated opposite 8-oxoG, i.e. the product of replication past 8-oxoG (19). This allows for insertion of a C opposite the lesion which is subsequently subject to Fpg-dependent repair. Repair of 8-oxoG lesions may result in double-strand DNA breaks if these are closely spaced, or if they are encountered by a replication fork while being repaired.
In this work, we demonstrate that otherwise lethal overinitiation is tolerated under anaerobic conditions and we report that cells deficient in Hda can be maintained that way without selection for suppressor mutations. We also show that aerobic survival of Hda-deficient cells can be promoted by neutralizing ROS or by deletion of mutM of the GO system. These data suggest that overinitiating cells lose their fitness when grown aerobically because of an increasing number of replication forks encountering a single-stranded repair intermediary generated during the removal of oxidized bases form the DNA. Such encounters will lead to double-strand breaks (DSB) which, when frequent, can result in cell death.
MATERIALS AND METHODS
Growth conditions
Cells were grown in Luria–Bertani (LB) medium (or AB minimal medium (21)) supplemented with 0.2% glucose or 0.4% glycerol, 0.5% casamino acids and 10 μg/ml thiamine. When indicated for anaerobic growth purposes, LB medium was supplemented with 0.2% glucose and buffered with A-salts. AB minimal medium used for anaerobic growth was supplemented with 1% glucose and 1% casamino acids. Unless specified, all cells were cultured at 37°C. When necessary, antibiotic selection was maintained at the following concentrations: kanamycin 50 μg/ml; chloramphenicol, 20 μg/ml; ampicillin, 150 μg/ml; tetracycline, 10 μg/ml. Anaerobic growth condition was maintained using anaerobic atmosphere generation bags (Sigma-Aldrich 68061) in an anaerobic jar for growth on plates. For liquid cultures, the growth medium was de-gazed under vacuum prior to cell inoculation, and placed with anaerobic atmosphere generating bags in a container. The container was placed on an orbital shaker at 37°C. Cells were inoculated and serially diluted in order to obtain cultures at OD450 ∼0.1 about 24 h after inoculation in anaerobic conditions. When indicated, glutathione (Sigma-Aldrich G4251) was supplemented at a final concentration of 10 mM.
Bacterial strains and plasmids
All strains used for analysis are derivatives of MG1655. Strains are listed in Supplementary Table S1. Construction of strains and plasmids is described in Supplementary Material.
Whole-genome sequencing
Whole-genome sequencing was performed at the SNP&SEQ Technology Platform of Uppsala University on a HiSeq2000 (Illumina) platform. A total of 8.7 million paired-end reads were generated, with an average read length of 100 nucleotides.
Flow cytometry
Flow cytometry was performed as described previously (22) using an Apogee A40 instrument. For each sample, 40 000–200 000 cells were analysed. Numbers of origins per cell and relative cell mass were determined as described previously (22).
Determination of ROS using hydroxyphenyl fluorescein (HPF) was by adding 5 μM HPF to the cell culture 1 h before analysis by flow cytometry using an Apogee A40 instrument with excitation wavelength set at 488 nm and fluorescence collected between 515 and 545 nm. Samples were analysed either with or without washing once in growth medium. Determination using dihydrorhodamine was done according to (23).
Pulsed field gel electrophoresis
Sample preparation was performed essentially as described in (24). Cells were pelleted washed twice in SE buffer (75 mM NaCl, 25 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4) and resuspended in CSB buffer (100 mM Tris, 100 mM EDTA, pH 7.5). Plugs were prepared by mixing an equal volume cells and low-melting agarose 2% (BioRad 161–3100). Plugs were first incubated for 2 h at 37°C in a buffer containing lysozyme and RNAse (lysozyme 0.1 mg/ml, RNAse 30 μg/ml, Sarcosyl 1%, EDTA 100 mM pH 9.0). The plugs were then incubated in a proteinase K buffer (proteinase K 1 mg/ml, Sarcosyl 1%, EDTA 500 mM pH 9.0) overnight at 56°C. The plugs were finally washed three times in CSB buffer and stored at 4°C prior to loading. 1% agarose gels were run for 24 h at 6 V/cm in 0.5 × TBE at 14°C: initial switching time 60 s and final switching time 120 s. The gels were stained using SybrGold for 1 h prior to imaging. Plugs containing the chromosomes of the yeast Saccharomyces cerevisiae were used as molecular weight standards (Bio-Rad 170-3605).
Microscopy
All samples for microscopy were kept on ice for 4–8 h with frequent whirly mixing prior to visualization. This was done in order to ‘oxygenate’ the samples allowing for folding of the Green Fluorescent Protein (GFP) and mCherry chromophores in anaerobic grown cells. Cells were then deposited on a 1% AB medium agarose pad. Microscope analyses were done using an AxioImager Z1 microscope (Carl Zeiss MicroImaging, Inc). The microscope pictures were processed and analysed with Volocity (PerkinElmer), ImageJ and Adobe Illustrator software.
Quantitative polymerase chain reaction (qPCR)
Samples were prepared by spinning down 1 ml of culture for 5 min at 15 000 × g. Cells were re-suspended in 100 ul 10 mM tris pH 7,4 and kept at −20°C and diluted 50 times in DNA/RNA free water prior to analysis. The qPCR was performed as previously reported (10) using Takara SYBR Premix Ex Taq II (RR820A) in a BioRAD CFX96 (95°C 30 s, 39 × (95°C 5 s + 60°C 30 s), 95°C 15 s, 60°C 60 s). All ori/ter ratios were normalized to the ori/ter ratio of MG1655 in late phase corresponding to an ori/ter of one. Primers are listed in Supplementary Material.
RESULTS
Hda-deficient cells are viable in the absence of oxygen
Cells deficient in Hda were previously shown to be either inviable or severely compromised for growth (8,10). Therefore, introduction of an hda deletion into wild-type (wt) cells results in a delayed appearance of small, heterogeneous colonies (10) (Figure 1A). However, with time the accumulation of suppressor mutations (termed hsm; hda suppressor mutation) arise resulting in colonies which remain homogeneous upon re-streaking and which are often large. Several hsm mutations have been identified, including hsm-2 which is a mutation in dnaA resulting in replacement of phenylalanine with valine at position 349 of the DnaA protein (11). As expected the introduction of an hda deletion into hsm-2 cells immediately resulted in big colonies of homogeneous size (10) (Figure 1A).
Figure 1.

Lethal overinitiation is suppressed by the absence of oxygen. (A) The hda::cat allele was introduced into wt and hsm-2 cells by bacteriophage P1 transduction. Plates were incubated for 16 h at 37°C on LB plates supplemented with 0.2% glucose and chloramphenicol aerobically and anaerobically. (B) dnaAcos mutant cells were streaked on LB plates supplemented with 0.2% glucose followed by incubation aerobically at either 42°C (permissive temperature) or 30°C (non-permissive temperature) and anaerobically at 30°C. (C) Wild-type hsm-2 cells were transformed anaerobically with plasmid pBR322-DARS2. Cells from the resultant colonies were subsequently re-streaked aerobically and anaerobically on LB plates supplemented with 0.2% glucose and incubated at 37°C for 16 h.
When an hda deletion was introduced into wild-type and hsm-2 cells by bacteriophage P1, and transductants incubated under anaerobic conditions the resultant colonies were homogeneous and similar in size for both recipients suggesting that the loss of Hda is not lethal under these conditions (Figure 1A). We determined the genome sequence for one Δhda transductant in otherwise wild-type cells, and found no mutations or genomic rearrangement except for the introduced Δhda::cat mutation. We can therefore conclude that hda mutant cells are viable and not severely growth compromised in absence of oxygen.
Hyperinitiation caused by the dnaAcos mutation or extra DARS2 copies is also tolerated in the absence of oxygen
It was reported that the Hda protein may have functions other than in RIDA, which results in cold sensitivity (25). It is therefore possible that the inviability of hda mutant cells during aerobic growth is related to processes different from DNA replication. We therefore determined the viability of other mutations/conditions resulting in a dramatic and lethal overinitiation of replication in the absence of oxygen. The dnaAcos mutant results in cold sensitivity due to hyperactivity of the DnaA protein at non-permissive temperature. As expected, the dnaAcos mutant grows at 42°C but not at 30°C under aerobic conditions (Figure 1B). In contrast, the dnaAcos mutant was viable when incubated anaerobically at 30°C; the otherwise non-permissive temperature (Figure 1B).
The DARS2 sequence is instrumental in regeneration of DnaAATP from DnaAADP. When present on a multicopy plasmid, DARS2 results in an elevated DnaAATP level, overinitiation from oriC and inviability (14). We cloned the DARS2 sequence in the high copy number plasmid pBR322, using a host strain that initiate replication independent of dnaA and oriC (i.e. carrying the dnaA::cat, rnhA-373 mutations). When the resultant plasmid, pBR322-DARS2, was transformed into wild-type cells, colonies obtained under aerobic conditions were mostly small and heterogeneous, whereas those obtained without oxygen were uniform. Transformation of pBR322-DARS2 into hsm-2 cells resulted in homogeneous colonies both in the presence and absence of oxygen (not shown). When re-streaked, wild-type cells containing pBR322-DARS2 formed colonies under anaerobic but not aerobic conditions, whereas hsm-2 cells containing the same plasmid formed colonies in the presence and absence of oxygen (Figure 1C). Altogether, these data suggest that inviability resulting from replication overinitiation can be alleviated in the absence of oxygen. The data also suggest that the DnaAF349V protein, resulting from the hsm-2 mutation affect the ability of DnaA to bind and/or hydrolyse ATP, in a manner similar to the nearby dnaAA345S mutation which also suppresses the loss of Hda (26).
Chromosome replication during anaerobic growth
Wild-type cells were grown exponentially in minimal medium supplemented with glucose and casamino acids under aerobic and anaerobic conditions. The doubling times of the cultures were 33 and 49 min, respectively (Table 1). Replication initiation took place in synchrony in aerobic as well as anaerobic cells (Figure 2B). However, cells grown in the absence of oxygen were slightly smaller than aerobically grown cells and contained fewer origins (Table 1; Figure 2B) as would be expected due to their slow growth. The origin concentration (ori/mass) was the same for aerobic and anaerobic grown cells, indicating that the accumulation of active DnaA protein which ensures replication initiation at a specific cell mass per chromosomal origin is mostly unaffected by the absence of oxygen. Because the DnaA concentration was found to be the same during anaerobic and aerobic growth (Supplementary Figure S1), it seems likely that there is no gross difference in the activity of the protein, i.e. the DnaAATP/DnaAADP ratio is similar for the two growth conditions.
Table 1.
| Strain | Growth conditiona | Origins/cellb | Cell massc | Origins/massd | Doubling time (min) |
|---|---|---|---|---|---|
| wt | aerobic | 4.7 | 1 | 1 | 33 |
| wt | anaerobic | 4.0 | 0.9 | 1 | 49 |
| wt | 1 h after shift to aerobic growth | 4.7 | 1 | 1 | 33 |
| wt | 2 h after shift to aerobic growth | 4.7 | 1 | 1 | 33 |
| Δhda | anaerobic | 5.9 | 1 | 1.3 | 52 |
| Δhda | 1 h after shift to aerobic growth | 8.8 | 1 | 2 | NR |
| Δhda | 2 h after shift to aerobic growth | 9.6 | 1.2 | 1.8 | NR |
| Δhda | 4 h after shift to aerobic growth | 9.5 | 1.3 | 1.6 | NR |
| wt/ pBR322-DARS2 | anaerobic | 7.4 | 0.9 | 1.7 | 52 |
| wt/pBR322-DARS2 | 3 h after shift to aerobic growth | 11.8 | 1.3 | 2 | NR |
aGrowth was in minimal medium supplemented with glucose and casamino acids.
bDetermined as average fluorescence from flow cytometric analysis.
cDetermined as average light scatter from flow cytometric analysis.
dAverage fluorescence/average light scatter. Numbers are normalized to 1 for wt grown aerobic.
NR, not relevant.
Figure 2.
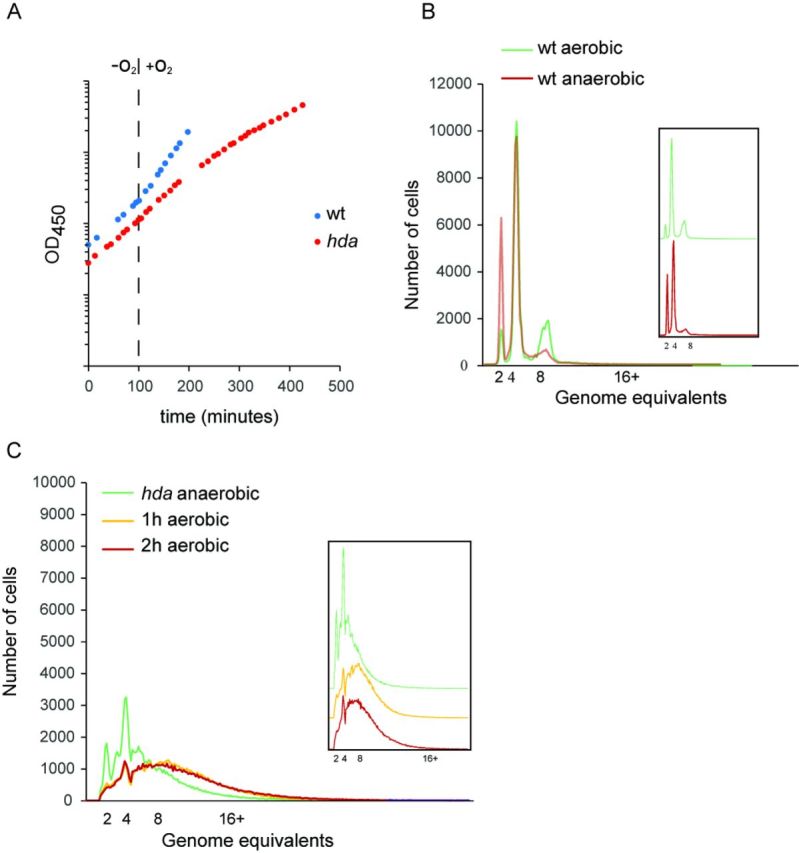
Overinitiation of hda mutant cells at anaerobic and aerobic growth. (A) Growth of wild-type and hda mutant cells under anaerobic conditions and shifted to aerobic growth. Cultures were followed by measuring OD450. (B) Wild-type cells were grown aerobically or anaerobically at 37°C, and treated with rifampicin and cephalexin prior to flow cytometric analysis. Insert displays the overlaid plots one above the other. (C) hda mutant cells were under anaerobic conditions prior to shifting to an aerobic environment. Samples taken at times indicated were treated with rifampicin and cephalexin prior to flow cytometric analysis. Inserts displays the overlaid plots one above the other.
Anaerobically grown Δhda cells had a doubling time close to that of wild-type cells (52 min versus 49 min; Table 1), yet cells were very different. Initiations no longer occurred in synchrony and Δhda cells contained all integral numbers of replication origins (up to >10; Figure 2C), i.e. initiations from each replication origin was not limited to once only each generation. The average number of origins per cell as measured by flow cytometry was 5.9 compared to 4.0 for wild-type cells (Table 1). However, due to incomplete replication run-out in the presence of rifampicin and cephalexin this is probably an underestimate. A similar result was obtained following 8 h (∼12 mass doublings) of Hda depletion during aerobic growth (Supplementary Figure S2). The size of hda mutant cells was increased, consistent with previous observations (27), and the origin concentration (ori/mass) was increased by about 30% relative to wild-type cells (Table 1). The same observations were made for Δhda cells grown in LB medium supplemented with glucose which allows for faster growth anaerobically (Supplementary Table S2). Therefore, Hda mutant cells are viable despite of overinitiation during anaerobic growth and suppression of the Hda phenotype does not result from a reduced growth rate.
The ori/ter ratio increases in Hda-deficient cells in the presence of oxygen
A large body of evidence suggests that loss of Hda is associated with severe growth inhibition during aerobic conditions (8,10,11), whereas our data suggests that this is not the case in the absence of oxygen. We consequently decided to follow cells during a shift from anaerobic to aerobic growth conditions. Wild-type cells immediately grew faster, i.e. the doubling time decreased from 49 to about 33 min (Figure 2A). Replication initiation remained synchronous, and the cellular origin content increased from an average of 4.0 in the absence of oxygen to 4.7, whereas there was no significant change in the origins/mass ratio because cell size also increased (Figure 2B; Table 1). The ori/ter ratio, determined by qPCR analysis, increased slightly but still remained around two throughout the experiment (Figure 3A).
Figure 3.
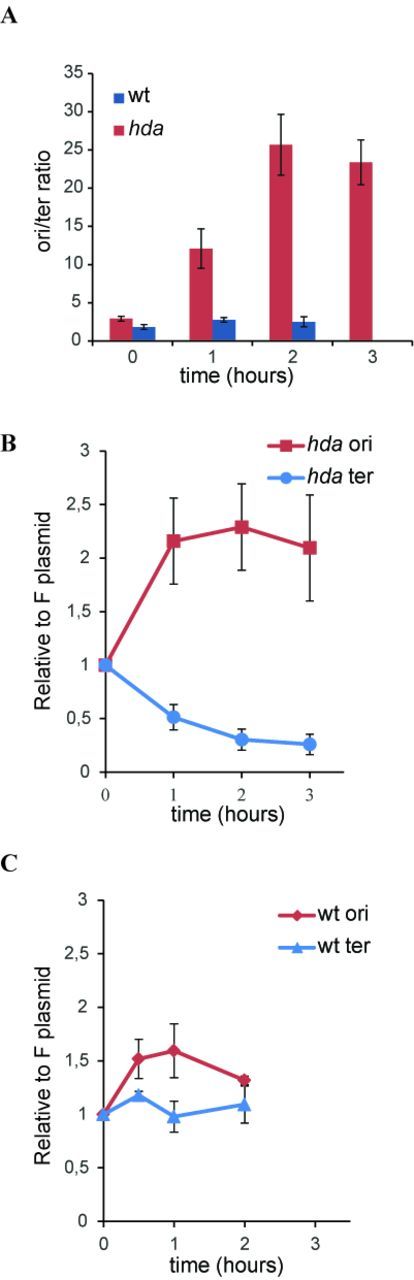
Abortive chromosome replication in aerobically grown hda cells. The ori/ter ratio of wild-type (blue) or hda (red) cells was determined by qPCR from anaerobically grown cells (T = 0) or cells at indicated times after a shift to aerobic conditions (A).
The ori/F ratio (red) or ter/F ratio (blue) was determined by qPCR from anaerobically grown cells or following a shift to aerobic conditions in hda (B) or wild-type cells (C).
Hda-deficient cells behaved quite different when shifted to aerobic growth. The doubling time gradually increased (Figure 2A), confirming that Hda is essential under these conditions. The average number of origins, determined by flow cytometry, increased from 5.9 to ∼9 after 1 h (Figure 2C; Table 1), and remained close to that level (Table 1). Again the absolute number or origins/cell was difficult to assess due to incomplete replication run-out. Cell size increased to a lesser extent, resulting in a 23% increase in origin concentration 4 h following the shift. The ori/ter ratio was ∼3 during anaerobic growth and this increased rapidly to >20 two hours after the shift and remained at that level (Figure 3A). We also followed wild-type cells transformed with plasmid pBR322-DARS2 during a shift from anaerobic to aerobic growth conditions (Supplementary Figure S3). Overall, the phenotype of wild-type cells containing plasmid pBR322-DARS2 resembled that of Hda-deficient cells but was more severe, i.e. the ori/ter increased to a higher level (∼35) when shifted to aerobic growth. This probably reflects the fact that in cells containing a multicopy DARS2 plasmid, DnaAADP is constantly regenerated into DnaAATP resulting in the vast majority of DnaA being ATP bound (14). Loss of Hda may have a less severe effect on the DnaAATP/DnaAADP ratio as the DDAH pathway to convert DnaAATP to DnaAADP is still functional (9).
Replication forks collapse in Hda-deficient cells during aerobic growth
This increased ori/ter ratio in Hda-deficient cells in the presence of oxygen could result from either an increase in initiation frequency or a reduced ability of forks already started to reach the terminus or both. In order to discriminate between these scenarios we transformed the F-derived plasmid pALO277 (28); simply referred to as F into wild-type and hda cells. The F plasmid replication is controlled by the plasmid encoded RepE protein and is not limited by DnaA availability or activity (29). The F plasmid copy number per mass was found to stay constant over a wide range of growth rates (30) or decrease slightly at fast growth (31). We determined the number of origins and termini relative to plasmid F by qPCR analysis during a shift from anaerobic to aerobic growth. For wild-type cells, both ori/F and ter/F ratios increased slightly (Figure 3C) and this explains why only a modest increase in ori/ter ratio was observed for these cells (Figure 3A). In Hda-deficient cells the ori/F increased about 2-fold one hour following the shift and remained at that level. On the other hand, the ter/F ratio decreased 3- to 4-fold following the shift (Figure 3B). Together, this explains the dramatic increase in ori/ter ratio increase after the shift (Figure 3A). Because ter is more affected than ori these data show that the dramatic increase in ori/ter ratio displayed by hda cells during aerobic growth largely results from an inability of replication forks to reach the terminus and only to a lesser degree from more initiations from oriC.
Morphology of Hda-deficient cells
To visualize the distribution of origins and termini by microscopy we labelled each locus in vivo as described previously (32). We inserted the P1 parS sequence close to the origin of replication and the pMT parS sequence close to the terminus (32). Co-expression of mCherry-labelled pMT-ParB and GFP-labelled P1-ParB (33) from a construct inserted at the attTN7 chromosomal locus allowed us to visualize origins as GFP foci and termini as mCherry foci in the same cells.
Anaerobically grown wild-type cells mainly had two or four origin foci and usually only one terminus focus (Figure 4A; Supplementary Table S3). We only observed a second terminus focus in cells about to divide. On average, cells contained 3.0 origin foci and had an ori/ter foci ratio of 2.5. However, one should bear in mind that foci numbers underestimate the actual numbers of both origins and termini due to the limited resolution of light microscopy. When shifted to aerobic growth, wild-type cells became larger with an increased number of cellular origin foci (3.9) and an ori/ter foci ratio of 2.8.
Figure 4.
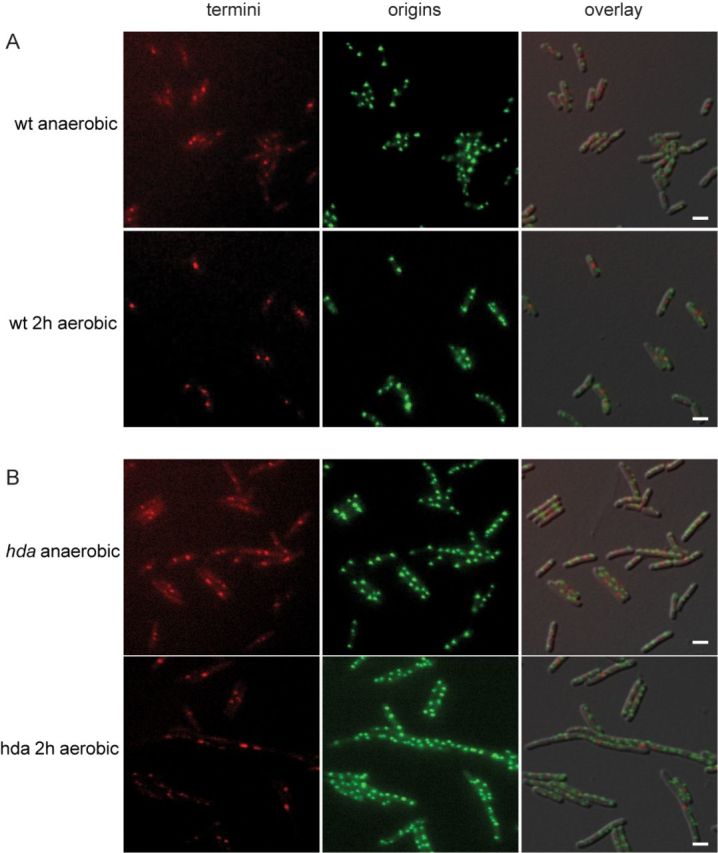
Localization of ori and ter in hda mutant cells. Wild-type (A) and hda mutant (B) cells were grown anaerobically and shifted to aerobic growth. At the times indicated, cells were spotted on an AB medium agarose pad. Scale bar is 2 μm.
An hda mutant grown anaerobically had on average 3.7 origin foci per cells (Figure 4B; Supplementary Table S3) and an ori/ter foci ratio of 2.4. The cells were bigger than wild-type in agreement with data from flow cytometry. When shifted to aerobic growth, cells gradually increased in size and became heterogeneous (Figure 4B and Supplementary Figure S4) indicating that cell division was perturbed. Small DNA-less cells also started appearing (Supplementary Figures S4 and S5, asterisks). The cellular location of the chromosome was also affected as some cells contained large areas devoid of DNA at the tips of the cells while the nucleoid covered the central part of the cell only (Supplementary Figure S5, arrows). The average number of origin foci increased from 3.7–8.3 but there appeared to be proportionality between cell size and number of foci. This was not the case for ter foci where cells mainly contained one or two foci in the middle of the cells resulting in an average of 1.5 at anaerobic growth. The number of foci did not change when shifted to aerobic growth despite of the increase in cell size (Supplementary Table S3). Hence, there was poor correlation between cell size and number of ter foci, which is exemplified by large cells containing one ter focus only, and very long filaments possessing more than 20 origins and only one or two visible termini stuck in the middle of the cell (Supplementary Figure S4). Therefore, replication initiation from oriC seems little affected by the shift and initiation frequency remains coupled to mass growth. However, an increase in number of origins is not accompanied by an increase in termini. This supports the conclusion derived from the qPCR analysis (Figure 3) that not all forks reach the terminus. To determine whether replication forks collapses in Hda-deficient cells, resulting in formation of DSB, we analysed genomic DNA of Δhda cells during shift to aerobic conditions by pulsed field gel electrophoresis (PFGE). As a control, we included DNA from wild-type cells treated with ciprofloxacin, a fluoroquinolone drug that trap a covalent type II topoisomerase-DNA complex, leading to DSB (34). As expected, treatment with ciprofloxacin resulted in a time-dependent appearance of low molecular weight DNA species characteristic of DSB (Figure 5A). Low molecular weight DNA also appeared in a time-dependent manner in Δhda cells upon a shift to aerobic growth (Figure 5B), demonstrating the presence of DSB in these cells.
Figure 5.
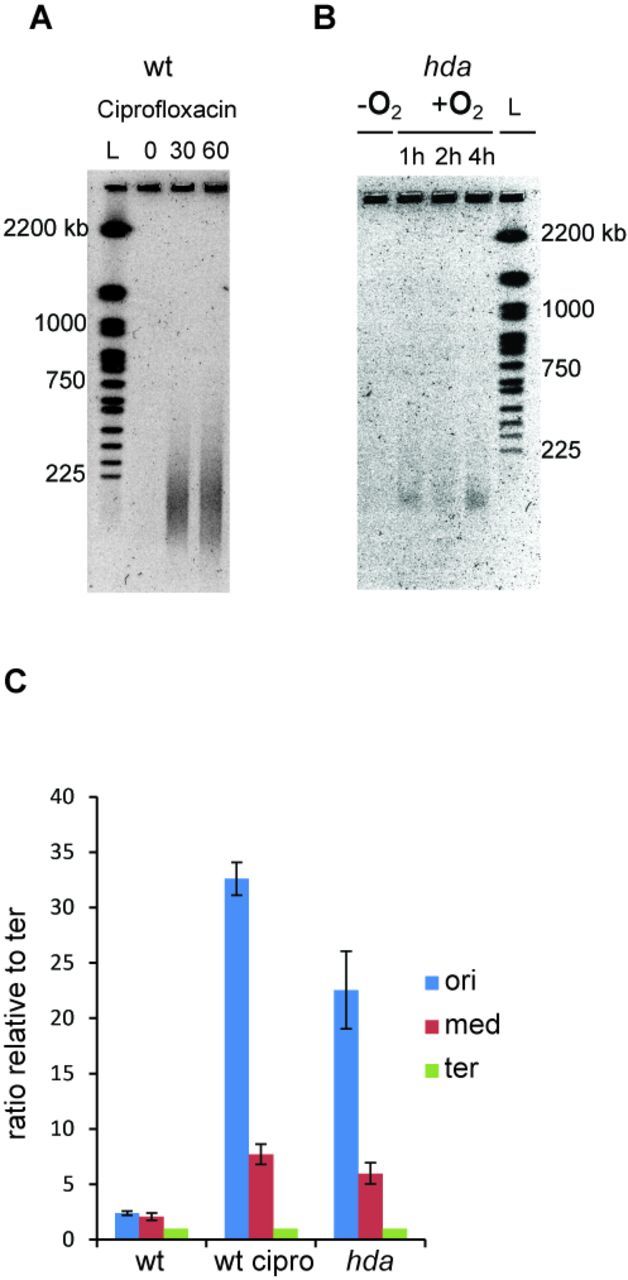
DSBs in the chromosomal DNA of hda mutant cells during aerobic growth. DSB are visualized by PGFE. (A) Wild-type cells treated with ciprofloxacin. At time T = 0 ciprofloxacin was added to a final concentration of 2 μg/ml. (B) A culture of hda mutant cells was grown under anaerobic conditions. At time T = 0 the culture was shifted to aerobic growth. (C) The frequencies of ori, middle and ter regions were determined by qPCR from aerobically grown wild-type cells, wild-type cells treated for 30 min with 2 μg/ml ciprofloxacin and from hda cells 2 h following a shift from anaerobic to aerobic conditions.
A qPCR analysis of three chromosomal loci, ori, middle (stpA) and ter demonstrated that more breaks occurred between ori and stpA than between stpA and ter for both ciprofloxacin-treated wild-type cells and aerobic hda cells (Figure 5C)
Oxidative damage underlies the aerobic inviability of Hda-deficient cells
When growing aerobically, E. coli uses oxygen as the terminal electron acceptor. However, ROS come as a byproduct from the metabolism of molecular oxygen and can generate lesions in the DNA. To test whether the aerobic growth defect of hda mutant cells resulted from an inability to cope with the oxidizing environment, we decided to delete hda in the presence of the ROS scavenger reduced glutathione (GSH). Δhda cells isolated under anaerobic conditions were re-streaked on plates with or without glutathione in the presence of oxygen. The addition of GSH resulted in improved growth and homogeneous colonies of Hda-deficient cells (Figure 6A), indicating that ROS-mediated oxidation is indeed the cause of the growth defect of hda cells during aerobic growth.
Figure 6.
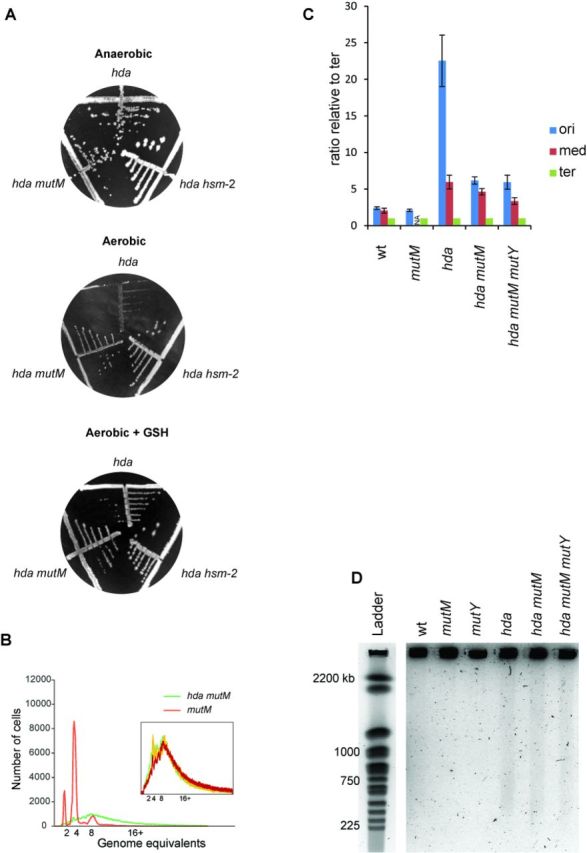
The aerobic growth defect of hda mutant cells is suppressed by addition of GSH or by deletion of mutM. (A) The hda::cat allele was introduced into wt, mutM and hsm-2 cells by phage P1 transduction under anaerobic conditions. Transductants were restreaked on LB plates supplemented with 0.2% glucose and chloramphenicol aerobically or anaerobically. When indicated, reduced glutathione (GSH) was added to the plates to a final concentration of 10 mM. (B) mutM and mutM hda cells were grown under aerobic conditions. Cells were treated with rifampicin and cephalexin prior to flow cytometric analysis. Insert displays the overlaid histograms of three independent mutM hda clones. (C) The frequencies of ori, middle and ter regions were determined by qPCR from aerobically grown wild-type, hda mutM and hda mutM mutY cells as well as hda cells 2 h following a shift from anaerobic to aerobic conditions. NA, not available. (D) Wild-type, mutM, mutY, hda mutM and hda mutM mutY cells were grown aerobically in AB minimal medium supplemented with 0,2% glucose and 1% casamino acids. Samples were taken and processed for PFGE as described (Materials and Methods). The hda single mutant was grown anaerobically in the same medium and the sample for PFGE was taken 2 h following a shift to aerobic conditions.
We could not determine any difference in ROS levels by measuring hydroxyl radical formation between wild-type and hda cells using two different reporters (23,35), and therefore conclude that excess replication from oriC does not result in an increased cellular ROS level and presumably not an increased frequency of ROS-inflicted DNA damage. Therefore, it is unrepaired damage by ROS that cause inviability of hyperreplicating cells as we show below.
Double-strand DNA breaks in Hda-deficient cells result from repair of oxidized bases
DNA damage resulting from ROS includes a number of base modifications, most notably 8-oxoG. When present in DNA, 8-oxoG is primarily excised by the Fpg glycosylase (mutM gene product) of the GO system (19). Cells deficient in both Hda and Fpg formed small but homogeneous colonies under aerobic conditions demonstrating that loss of Fpg suppressed the growth defect of Δhda cells (Figure 6A). We subjected several Δhda ΔmutM clones to a flow cytometric analysis. These cells grew slower than wild-type (52 min versus 33 min). They all had similar profiles after treatment with rifampicin and cephalexin with an increased number of cellular origins and poor run-out profile compared to wild-type cell (Figure 6B; Supplementary Table S4), resembling the profile obtained for Hda-deficient cells grown aerobically (Figure 2). Loss of MutM is therefore not likely to suppress Hda deficiency by reducing initiations from oriC and in agreement with this we found mutM cells to have a similar origin concentration (origins/mass) as wild-type cells (Supplementary Table S4).A qPCR analysis of the ori, middle (stpA) and ter loci indicated that the ori/ter ratio of Δhda ΔmutM was significantly lowered (∼6) compared to that of aerobically growing Δhda cells (>20; Figure 6C). Therefore, loss of Fpg must allow more replication forks to proceed to the terminus. A further deletion of mutY (Δhda ΔmutM ΔmutY) did not lower the ori/ter ratio further (Figure 6C), demonstrating that the residual DSBs in Δhda ΔmutM cells are not caused by the action of MutY. A knockout of mutY of the GO system was not found to suppress Δhda cells (Supplementary Figure S6). This is in agreement with the MutY glycosylase playing a secondary role to Fpg in the repair of 8-oxoG lesions (18).
Some DSB persist in the absence of MutM in Hda-deficient cells
Genomic DNA from wild-type, hda, ΔmutM, ΔmutY and Δhda ΔmutM and Δhda ΔmutM ΔmutY cells was subjected to pulse field gel electrophoresis. This revealed the presence of DNA breaks in Δhda, Δhda ΔmutM and Δhda ΔmutM ΔmutY mutant cells (Figure 6D). Although hard to quantify, the amount of DSB in Δhda single mutant cells seemed somewhat higher than for double or triple mutant cells (Figure 6D). This is in agreement with the observations that deletion of mutM or mutM and mutY only partly restores the ori/ter ratio of Hda-deficient cells in comparison to anaerobic grown cells (Figure 6C). The persistence of DSB in Δhda ΔmutM cells is also consistent with the flow cytometry analysis (Figure 6B), and is expected because ROS-dependent DNA damage is not limited to lesions repaired by Fpg (17). However, loss of Fpg may reduce the amount of DSB to a level which allows survival despite of overinitiation from oriC.
DISCUSSION
In E. coli severe overinitiation is observed when the activity of the DnaA protein is increased. This results in replication stress and cell death. Such an increase in DnaA activity can result from mutations in dnaA itself (13,36), from mutations in Hda which is instrumental for RIDA-dependent conversion of DnaAATP to DnaAADP (8) or from increasing the dosage of the DARS sequences that promote rejuvenation of DnaAADP to DnaAATP (14). Inviability caused by hyperinitiation is known to be associated with a reduced rate of replication and DSB (13,22,37). It has been proposed that stalled forks are being caught by other forks ‘coming from behind’ and the collision results in DSB (13).
Here, we present the first mechanistic evidence as to how hyperinitiation results in formation of DSB. Our results indicate that DNA breaks are caused by replication forks encountering lesions generated by enzymes removing oxidized bases from the DNA duplex. These lesions are normally repaired in timely fashion but when the interval between replication forks is diminished, the probability of a fork meeting a lesion is increased.
Hyperinitiation is tolerated during anaerobic growth
Loss of the Hda protein results in inviability or severe growth inhibition (8,10,11), and consequently suppressor mutations arise with high frequency. In vivo studies of replication initiation in the absence of Hda have therefore previously been carried out using a temperature-sensitive allele of hda (27) or in cells that contain additional compensatory mutations (10,11). In other cases it is not clear whether suppressor mutations were present or not (38). Deletion of hda was tolerated in the absence of oxygen as whole-genome sequencing revealed that suppressor mutations were not present. Similarly, the dnaAcos mutation or increased copies of DARS2 was tolerated during anaerobic growth.
We found no evidences for timing of initiation being different in the presence or absence of oxygen in wild-type cells. The levels of DnaA was similar, suggesting that the overall level of ATP bound DnaA may also be quite similar between these growth conditions. This agrees well with observations that neither the cellular ATP level nor the ATP/ADP ratio is decreased during anaerobic relative to aerobic growth (39,40) but poorly with an older study showing a reduced ATP level and ATP/ADP ratio under anaerobic growth (41). Anaerobic condition is therefore not likely to restore viability of hyperinitiating cells by lowering the frequency at which DNA replication starts to a tolerable level.
Chromosome breakage during aerobic growth
Inviability associated with replication stress was only observed during aerobic growth, and coincided with a dramatic increase in ori/ter ratio. The increase primarily resulted from a decrease in terminus concentration, whereas oriC was less affected. Therefore, replication forks started at oriC frequently collapse before reaching the terminus and this explains the appearance of double-strand DNA breaks in these cells. We propose that these DNA breaks are the reason for inviability associated with hyperinitiation.
Aerobic growth of hda mutants could be restored by addition of the ROS scavenger GSH, indicating that a significant fraction of the strand breaks observed resulted from replication forks encountering oxidative lesions in the DNA. A deletion of mutM also suppressed loss of Hda under aerobic conditions indicating that the action of the Fpg glycosylase is instrumental in DSB formation.ROS is formed as a result of aerobic respiration, and oxidative damage to DNA is quite frequent and the steady-state frequency of 8-oxoG was estimated to be 2.5 lesions per 105 dG residues during exponential growth (42) corresponding to one lesion per 160 kb of genomic DNA; about 100-fold higher than observed for eukaryotic cells (43). A separate study reports a somewhat lower density of lesions, in this case less than 1 per 170 kb of DNA (44). Given that 8-oxoG lesions are efficiently repaired by the GO system (19), these steady-state levels suggest that the lesions form frequently in the bacterial DNA. The Fpg glycosylase (MutM) excises a number of oxidized bases from DNA but its primary activity is against 8-oxoG, the most common lesion derived from ROS. The Fpg enzyme possesses both a DNA glycosylase activity, that excises oxidized base lesions, and an intrinsic lyase activity, cleaving the DNA (βδ elimination) at the AP site to produce both 5′ and 3′ ends containing phosphomonoesther nucleotides (45). As this strand break cannot be immediately processed by PolI it may persist for some time. If encountered by a replication fork while undergoing repair, the result will be a DSB. In E. coli a single DSB is pontentally lethal (46).
Because we did not observe any increase in ROS levels between wild-type and hda mutant cells we consider the frequency of oxidative lesions the same. We suggest that an increased initiation frequency results in more ongoing replication forks, which raises the likelyhood of forks encountering Fpg repair intermediaries leading to DSB formation. The generation of DSB by replication forks encountering base-excision nicks have been described previously (47). All replication forks originate at oriC, and will collapse at the first lesion encountered irrespective of this being generated by repair of 8-oxoG, ultraviolet irradiation or a genotoxic agent, such as ciprofloxacin. If multiple lesions of either type are present and evenly distributed along the chromosome, the net result will be that replication forks predominantly collapse in oriC proximal regions, such as observed here and previously (13,37,47,48). Although we assume an unchanged level of oxidative damage between wild-type and Hda-deficient cells, we cannot rule out that hyperinitiation indirectly (altered gene regulation, suppression of repair processes or altered energy metabolism), may result in an increased frequency of oxidative damage in general or primarily in newly replicated DNA, i.e. near oriC, and that this contributes to an increased ori/ter ratio and inviability. The generation of DSBs by overinitiation also explains why cells that only overinitiate replication slightly become dependent on homologous recombination for survival (49). Certain hda mutants are viable despite the presence of a functional RIDA system. It was suggested that Hda also plays a role in stabilizing PolIII at the replication fork and controlling access of the trans-lesion polymerases PolII and Pol IV to the β-clamp (25). It is tempting to speculate that these Hda mutant proteins may assist trans-lesion polymerases in replicating across oxidative lesions in the DNA. In the absence of Fpg, the MutY glycosylase will excise the A that is normally incorporated opposite of 8-oxoG. MutY is a monofunctional glycosylase (50) whose action results in an AP site that is processed directly by AP endonucleases Nfo and XthA encoding Exonuclease III and endonuclease IV, respectively (51), and finally Polymerase I (PolI) and DNA ligase. We speculate that in the MutY repair process the nicked DNA strand persists shorter than one promoted by Fpg and that this could explain why loss of MutY did not suppress Hda deficiency during aerobic growth. We also observed DSB in cells deficient in both Hda and Fpg, albeit at a lower level. There could be several reasons for this. First, oxidative damage results in a large number of lesions to the DNA and only a subset of these are subject to Fpg repair. Repair of other oxidized bases may therefore contribute to the DSB observed. It has also been reported that the bifunctional DNA glycosylases nth (endonuclease III), nei (endonuclease VIII) or KsgA may process some of the 8-oxoG lesions in the absence of Fpg (52,53) and DSB could be generated in the repair process. Finally, a replication fork encountering an 8-oxoG may stall or pause for a while, which could result in collision from behind by another replication fork (13).
Interestingly, repair of 8-oxoG lesions by the GO system has also been implicated in sensitizing bacteria to fluoroquinolone-induced replication stress. Strains deficient in both MutM and MutY survive norfloxacin exposure better than wild-type cells (54). This suggests an additive effect of 8-oxoG repair to the DSB-induced death by fluoroquinolone exposure. Fluoroquinolone resistant clinical isolates of E. coli frequently carry mutations in mutM (55).
Heterogeneity of Hda-deficient cells
Hda mutant cells became heterogeneous following a shift to aerobic growth, with both filament and minicell formation. Because DNA replication stopped or stalled before reaching the terminus, cell division was likely to be blocked, whereas cell mass continued to increase. In E. coli there are no mechanisms (checkpoints) that block replication initiation due to fork stalling and/or DNA damage. Therefore, new rounds of replication takes place concurrent with the increase in cell mass, and the result is large, near-polyploid cells with a centrally located nucleoid containing multiple copies of oriC and only one or two copies of the terminus (Figure 4). Cell filamentation was independent of SOS induction as we saw no significant increase in sulA expression (not shown). This is in agreement with earlier data demonstrating that cells overinitiating DNA replication due to the cold sensitive mutations hdaCs or dnaAcos fails to divide in an SOS-independent manner when grown at non-permissive temperature (27,56). Midcell trapping of the nucleoid inactivates the cell division protein FtsZ, independently of SOS induction or SlmA (57) and results in frequent division near the termini of filaments, i.e. minicell formation (Figure 4, Supplementary Figures S4 and S5).
Multiple ways to counteract lethal overinitiation in bacteria
Suppressor mutations that counteract lethal overinitiation in bacteria have previously been isolated in many laboratories and fallen into two groups. The first group of suppressors affects the DnaA initiator protein itself and includes mutations in dnaA to lower the activity of the resultant DnaA protein (11,26) or as recently discovered for C. crescentus second site mutations that lower the amount of DnaA protein by accelerating its degradation (58). The second group of suppressors affects the oriC region and includes mutations in oriC itself that lowers its ability to initiate replication (59) and mutations affecting the dam/seqA system to increase sequestration of oriC and thereby restricting initiations (11,60). Common to overinitiation suppressors affecting oriC or DnaA is that they reduce initiation frequency from oriC, thereby fully or partly restoring origin concentration and origin/terminus ratio of the suppressed cells to near wild-type levels.
Here, we demonstrate a third way E. coli can cope with replication stress resulting from hyperinitiation. Lethality is normally caused from an elevated number of replication forks encountering intermediates in the repair of oxidized bases, primarily 8-oxoG and thereby causing DSB. Removing the cause of oxidative damage, i.e. anaerobic growth or addition of an antioxidant (GSH), or loss of mutM encoding Fpg, the main player in repair of oxidative lesions, restores viability of hyperinitiating cells without reducing initiations from oriC. This also implies that DNA replication is not normally limited by the availability of precursors or precursor synthesis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Strain MS5 and MS158 were generously provided by Mathieu Stouf. Plasmid pRN010 was obtained from D. Chattoraj. We thank Leif Kirsebom, Uppsala University for help with the whole genome sequencing and Martin Marinus providing strains and for helpful discussions.
FUNDING
European Union [PIRG05-GA-2009-247241]; Danish Research Council for Natural sciences [09-064250/FNU]; Lundbeck Foundation; Novo Nordisk Foundation. Funding for open access charge: University of Copenhagen.
Conflict of interest statement. None declared.
REFERENCES
- 1.Leonard A.C., Mechali M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013;5:a010116. doi: 10.1101/cshperspect.a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skarstad K., Katayama T. Regulating DNA replication in bacteria. Cold Spring Harb. Perspect. Biol. 2013;5:a012922. doi: 10.1101/cshperspect.a012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boye E., Løbner-Olesen A., Skarstad K. Limiting DNA replication to once and only once. EMBO Rep. 2000;1:384–974. doi: 10.1093/embo-reports/kvd116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurokawa K., Nishida S., Emoto A., Sekimizu K., Katayama T. Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. EMBO J. 1999;18:6642–6652. doi: 10.1093/emboj/18.23.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boye E., Løbner-Olesen A. The role of Dam methyltransferase in the control of DNA replication in E. coli. Cell. 1990;62:981–989. doi: 10.1016/0092-8674(90)90272-g. [DOI] [PubMed] [Google Scholar]
- 6.Lu M., Campbell J.L., Boye E., Kleckner N. SeqA: a negative modulator of replication initiation in E. coli. Cell. 1994;77:413–426. doi: 10.1016/0092-8674(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 7.Katayama T., Kubota T., Kurokawa K., Crooke E., Sekimizu K. The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosomal replicase. Cell. 1998;94:61–71. doi: 10.1016/s0092-8674(00)81222-2. [DOI] [PubMed] [Google Scholar]
- 8.Kato J., Katayama T. Hda, a novel DnaA-related protein, regulates the replication cycle in Escherichia coli. EMBO J. 2001;20:4253–4262. doi: 10.1093/emboj/20.15.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasho K., Katayama T. DnaA binding locus datA promotes DnaA-ATP hydrolysis to enable cell cycle-coordinated replication initiation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:936–941. doi: 10.1073/pnas.1212070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riber L., Olsson J.A., Jensen R.B., Skovgaard O., Dasgupta S., Marinus M.G., Lobner-Olesen A. Hda-mediated inactivation of the DnaA protein and dnaA gene autoregulation act in concert to ensure homeostatic maintenance of the Escherichia coli chromosome. Genes Dev. 2006;20:2121–2134. doi: 10.1101/gad.379506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charbon G., Riber L., Cohen M., Skovgaard O., Fujimitsu K., Katayama T., Lobner-Olesen A. Suppressors of DnaA(ATP) imposed overinitiation in Escherichia coli. Mol. Microbiol. 2011;79:914–928. doi: 10.1111/j.1365-2958.2010.07493.x. [DOI] [PubMed] [Google Scholar]
- 12.Kitagawa R., Ozaki T., Moriya S., Ogawa T. Negative control of replication initiation by a novel chromosomal locus exhibiting exceptional affinity for Escherichia coli DnaA protein. Genes Dev. 1998;12:3032–3043. doi: 10.1101/gad.12.19.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simmons L.A., Breier A.M., Cozzarelli N.R., Kaguni J.M. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol. Microbiol. 2004;51:349–358. doi: 10.1046/j.1365-2958.2003.03842.x. [DOI] [PubMed] [Google Scholar]
- 14.Fujimitsu K., Senriuchi T., Katayama T. Specific genomic sequences of E. coli promote replicational initiation by directly reactivating ADP-DnaA. Genes Dev. 2009;23:1221–1233. doi: 10.1101/gad.1775809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng W., Li Z., Skarstad K., Crooke E. Mutations in DnaA protein suppress the growth arrest of acidic phospholipid-deficient Escherichia coli cells. EMBO J. 2001;20:1164–1172. doi: 10.1093/emboj/20.5.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imlay J.A. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 17.Bjelland S., Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Neeley W.L., Essigmann J.M. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 19.Michaels M.L., Miller J.H. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7, 8-dihydro-8-oxoguanine) J. Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schalow B.J., Courcelle C.T., Courcelle J. Escherichia coli Fpg glycosylase is nonrendundant and required for the rapid global repair of oxidized purine and pyrimidine damage in vivo. J. Mol. Biol. 2011;410:183–193. doi: 10.1016/j.jmb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark D.J., Maaløe O. DNA replication and the division cycle in Escherichia coli. J. Mol. Biol. 1967;23:99–112. [Google Scholar]
- 22.Løbner-Olesen A., Skarstad K., Hansen F.G., von Meyenburg K., Boye E. The DnaA protein determines the initiation mass of Escherichia coli K- 12. Cell. 1989;57:881–889. doi: 10.1016/0092-8674(89)90802-7. [DOI] [PubMed] [Google Scholar]
- 23.Guelfo J.R., Rodriguez-Rojas A., Matic I., Blazquez J. A MATE-family efflux pump rescues the Escherichia coli 8-oxoguanine-repair-deficient mutator phenotype and protects against H(2)O(2) killing. PLoS. Genet. 2010;6:e1000931. doi: 10.1371/journal.pgen.1000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowosielska A., Marinus M.G. DNA mismatch repair-induced double-strand breaks. DNA Repair. 2008;7:48–56. doi: 10.1016/j.dnarep.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baxter J.C., Sutton M.D. Evidence for roles of the Escherichia coli Hda protein beyond regulatory inactivation of DnaA. Mol. Microbiol. 2012;85:648–668. doi: 10.1111/j.1365-2958.2012.08129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gon S., Camara J.E., Klungsoyr H.K., Crooke E., Skarstad K., Beckwith J. A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in Escherichia coli. EMBO J. 2006;25:1137–1147. doi: 10.1038/sj.emboj.7600990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujimitsu K., Su'etsugu M., Yamaguchi Y., Mazda K., Fu N., Kawakami H., Katayama T. Modes of overinitiation, dnaA gene expression, and inhibition of cell division in a novel cold-sensitive hda mutant of Escherichia coli. J. Bacteriol. 2008;190:5368–5381. doi: 10.1128/JB.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Løbner-Olesen A. Distribution of minichromosomes in individual Escherichia coli cells: implications for replication control. EMBO J. 1999;18:1712–1721. doi: 10.1093/emboj/18.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kline B.C., Kogoma T., Tam J.E., Shields M.S. Requirement of the Escherichia coli dnaA gene product for plasmid F maintenance. J. Bacteriol. 1986;168:440–443. doi: 10.1128/jb.168.1.440-443.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keasling J.D., Palsson B.O., Cooper S. Cell-cycle-specific F plasmid replication: regulation by cell size control of initiation. J. Bacteriol. 1991;173:2673–2680. doi: 10.1128/jb.173.8.2673-2680.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins J., Pritchard R.H. Relationship between chromosome replication and F’lac episome replication in Escherichia coli. J. Mol. Biol. 1973;78:143–155. doi: 10.1016/0022-2836(73)90434-8. [DOI] [PubMed] [Google Scholar]
- 32.Stouf M., Meile J.C., Cornet F. FtsK actively segregates sister chromosomes in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2013;110:11157–11162. doi: 10.1073/pnas.1304080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadoya R., Chattoraj D.K. Insensitivity of chromosome I and the cell cycle to blockage of replication and segregation of Vibrio cholerae chromosome II. MBio. 2012;3 doi: 10.1128/mBio.00067-12. doi:10.1128/mBio.00067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drlica K., Zhao X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997;61:377–392. doi: 10.1128/mmbr.61.3.377-392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Setsukinai K., Urano Y., Kakinuma K., Majima H.J., Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 2003;278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 36.Kellenberger-Gujer G., Podhajska A.J., Caro L. A cold sensitive dnaA mutant of E. coli which overinitiates chromosome replication at low temperature. Mol. Gen. Genet. 1978;162:9–16. doi: 10.1007/BF00333845. [DOI] [PubMed] [Google Scholar]
- 37.Atlung T., Løbner-Olesen A., Hansen F.G. Overproduction of DnaA protein stimulates initiation of chromosome and minichromosome replication in Escherichia coli. Mol. Gen. Genet. 1987;206:51–59. doi: 10.1007/BF00326535. [DOI] [PubMed] [Google Scholar]
- 38.Camara J.E., Skarstad K., Crooke E. Controlled initiation of chromosomal replication in Escherichia coli requires functional Hda protein. J. Bacteriol. 2003;185:3244–3248. doi: 10.1128/JB.185.10.3244-3248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh L.-S., Rouvière-Yaniv J., Drlica K. Bacterial DNA supercoiling and [ATP]/[ADP] ratio: changes associated with salt shock. J. Bacteriol. 1991;173:3914–3917. doi: 10.1128/jb.173.12.3914-3917.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran Q.H., Unden G. Changes in the proton potential and the cellular energetics of Escherichia coli during growth by aerobic and anaerobic respiration or by fermentation. Eur. J. Biochem. 1998;251:538–543. doi: 10.1046/j.1432-1327.1998.2510538.x. [DOI] [PubMed] [Google Scholar]
- 41.Kashket E.R. Stoichiometry of the H+-ATPase of Escherichia coli cells during anaerobic growth. FEBS Lett. 1983;154:343–346. doi: 10.1016/0014-5793(83)80179-3. [DOI] [PubMed] [Google Scholar]
- 42.Alhama J., Ruiz-Laguna J., Rodriguez-Ariza A., Toribio F., Lopez-Barea J., Pueyo C. Formation of 8-oxoguanine in cellular DNA of Escherichia coli strains defective in different antioxidant defences. Mutagenesis. 1998;13:589–594. doi: 10.1093/mutage/13.6.589. [DOI] [PubMed] [Google Scholar]
- 43.Gedik C.M., Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- 44.Rotman E., Kuzminov A. The mutT defect does not elevate chromosomal fragmentation in Escherichia coli because of the surprisingly low levels of MutM/MutY-recognized DNA modifications. J. Bacteriol. 2007;189:6976–6988. doi: 10.1128/JB.00776-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Connor T.R., Laval J. Physical association of the 2, 6-diamino-4-hydroxy-5N-formamidopyrimidine-DNA glycosylase of Escherichia coli and an activity nicking DNA at apurinic/apyrimidinic sites. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5222–5226. doi: 10.1073/pnas.86.14.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonura T., Town C.D., Smith K.C., Kaplan H.S. The influence of oxygen on the yield of DNA double-strand breaks in x-irradiated Escherichia coli K-12. Radiat. Res. 1975;63:567–577. [PubMed] [Google Scholar]
- 47.Kouzminova E.A., Kuzminov A. Fragmentation of replicating chromosomes triggered by uracil in DNA. J. Mol. Biol. 2006;355:20–33. doi: 10.1016/j.jmb.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 48.Rudolph C.J., Upton A.L., Lloyd R.G. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007;21:668–681. doi: 10.1101/gad.417607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Felczak M.M., Kaguni J.M. The rcbA gene product reduces spontaneous and induced chromosome breaks in Escherichia coli. J. Bacteriol. 2012;194:2152–2164. doi: 10.1128/JB.06390-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams S.D., David S.S. Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA. Nucleic Acids Res. 1998;26:5123–5133. doi: 10.1093/nar/26.22.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doetsch P.W., Cunningham R.P. The enzymology of apurinic/apyrimidinic endonucleases. Mutat. Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 52.Blaisdell J.O., Hatahet Z., Wallace S.S. A novel role for Escherichia coli endonuclease VIII in prevention of spontaneous G–>T transversions. J. Bacteriol. 1999;181:6396–6402. doi: 10.1128/jb.181.20.6396-6402.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang-Akiyama Q.M., Morinaga H., Kikuchi M., Yonekura S., Sugiyama H., Yamamoto K., Yonei S. KsgA, a 16S rRNA adenine methyltransferase, has a novel DNA glycosylase/AP lyase activity to prevent mutations in Escherichia coli. Nucleic Acids Res. 2009;37:2116–2125. doi: 10.1093/nar/gkp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foti J.J., Devadoss B., Winkler J.A., Collins J.J., Walker G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Swick M.C., Evangelista M.A., Bodine T.J., Easton-Marks J.R., Barth P., Shah M.J., Chung C.A., Stanley S., McLaughlin S.F., Lee C.C., et al. Novel conserved genotypes correspond to antibiotic resistance phenotypes of clinical isolates. PLoS ONE. 2013;8:e65961. doi: 10.1371/journal.pone.0065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Katayama T., Takata M., Sekimizu K. CedA is a novel Escherichia coli protein that activates the cell division inhibited by chromosomal DNA over-replication. Mol. Microbiol. 1997;26:687–697. doi: 10.1046/j.1365-2958.1997.5941967.x. [DOI] [PubMed] [Google Scholar]
- 57.Cambridge J., Blinkova A., Magnan D., Bates D., Walker J.R. A replication-inhibited unsegregated nucleoid at mid-cell blocks Z-ring formation and cell division independently of SOS and the SlmA nucleoid occlusion protein in Escherichia coli. J. Bacteriol. 2014;196:36–49. doi: 10.1128/JB.01230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jonas K., Liu J., Chien P., Laub M.T. Proteotoxic stress induces a cell-cycle arrest by stimulating lon to degrade the replication initiator DnaA. Cell. 2013;154:623–636. doi: 10.1016/j.cell.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riber L., Fujimitsu K., Katayama T., Lobner-Olesen A. Loss of Hda activity stimulates replication initiation from I-box, but not R4 mutant origins in Escherichia coli. Mol. Microbiol. 2009;71:107–122. doi: 10.1111/j.1365-2958.2008.06516.x. [DOI] [PubMed] [Google Scholar]
- 60.Katayama T., Akimitsu N., Mizushima T., Miki T., Sekimizu K. Overinitiation of chromosome replication in the Escherichia coli dnaAcos mutant depends on activation of oriC function by the dam gene product. Mol. Microbiol. 1997;25:661–670. doi: 10.1046/j.1365-2958.1997.5001872.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.