

Background: The phycobilisome is assembled from many subunits, but the entire structure has not been determined.
Results: Coupled cross-linking/MS revealed neighboring residues within the interfaces between subunits.
Conclusion: The rods completely cover the core cylinders and are not in a staggered assembly form.
Significance: Energy transfer from rods to cores overcomes a jump of over 30 Å without loss of efficiency.
Keywords: Cyanobacteria, Energetics, Mass Spectrometry (MS), Molecular Modeling, Photosynthesis
Abstract
The phycobilisome (PBS) is an extremely large light-harvesting complex, common in cyanobacteria and red algae, composed of rods and core substructures. These substructures are assembled from chromophore-bearing phycocyanin and allophycocyanin subunits, nonpigmented linker proteins and in some cases additional subunits. To date, despite the determination of crystal structures of isolated PBS components, critical questions regarding the interaction and energy flow between rods and core are still unresolved. Additionally, the arrangement of minor PBS components located inside the core cylinders is unknown. Different models of the general architecture of the PBS have been proposed, based on low resolution images from electron microscopy or high resolution crystal structures of isolated components. This work presents a model of the assembly of the rods onto the core arrangement and for the positions of inner core components, based on cross-linking and mass spectrometry analysis of isolated, functional intact Thermosynechococcus vulcanus PBS, as well as functional cross-linked adducts. The experimental results were utilized to predict potential docking interactions of different protein pairs. Combining modeling and cross-linking results, we identify specific interactions within the PBS subcomponents that enable us to suggest possible functional interactions between the chromophores of the rods and the core and improve our understanding of the assembly, structure, and function of PBS.
Introduction
One of the most critical steps in all light-harvesting complex (LHC)2 systems is the transfer of absorbed energy by one LHC to adjacent LHC and from there to the reaction center (RC). Such transfer exists in the LHII/LHI/RC system of purple non-sulfur bacteria (1), Lhca1–4/RC (2) of plant and algal PSI, and the LHCII/CP29-CP24/CP47-CP43/RC of plants and algal photosystem II (PSII) (3, 4). It has been proposed that the structural gap that typically exists between the LHC chromophores and the RC chromophores (5) protects the LHC system from the potential damaging effect of the redox reactions occurring in the RCs. The organization of LHCs in all of the systems listed above into smaller units enables organisms to control the flux of excitation energy onto the RC, as well as change the absorption cross-section as a function of the light quality and quantity. In most LHCs, the chromophores are bound to the protein matrix in a dense fashion, with energy transfer between chromophores occurring over short distances (<10 Å). Phycobilisomes (PBS) serve as the major photosynthetic light-harvesting complex in cyanobacteria and red algae, expanding the range of absorbed wavelengths and increasing the overall light energy conversion efficiency (6–9). The PBS is assembled sequentially from phycobiliprotein (PBP) monomeric units, each one composed of two homologous α and β subunits (10–12). Subunits bind 1–3 bilin chromophores of different types, covalently attached to conserved cysteine residues. The monomers assemble further into trimers (via interactions between α and β subunits from different monomers), two of which further assemble into hexamers (mostly by interactions between α subunits) (13), and finally, the hexamers assemble further into the two main substructures: the core and rods (through interaction between the β subunits). Each PBS can contain between two and five core cylinders surrounded by six to eight rods. Higher energy photons are absorbed by the rod components: phycoerythrin (λmax =560 nm), phycoerythrocyanin (λmax = 570 nm), and phycocyanin (PC, λmax = 620 nm), and the absorbed energy is efficiently transferred to the core, which contains allophycocyanin (APC, λmax = 652 nm). The central cavities formed inside the tube-like structures contain additional proteins, typically called linker proteins (LPs) (14). The LPs have been proposed to have a structural role in mediating the interactions (“linking”) between PBPs and also have the ability to modify the spectral properties of the pigmented components (15). The LPs that occupy the rods and those that occupy the core have significant sequence homology leading to structural similarities (12) and probably carry similar structural-functional roles. Unlike other LHCs, the PBS chromophores are not densely packed, with 20–50 Å separation between nearest neighboring chromophores, as a function of the level of assembly. As already mentioned above, a large physical gap also exists between the PBS and the RCs (both PSII and PSI). A similar physical gap exists within the PBS complex itself: energy absorbed within the rods has to be efficiently transferred to the core (containing APC and the terminal emitter minor components ApcE and ApcD (8)). Each substructure contains 10s to 100s of chromophores that absorb at roughly the same wavelength. Intercomplex energy jumps are typically bathochromic by 20–50 nm, a large enough energy gap to ensure directionality. On the basis of the crystal structures of some isolated PBPs, assembly into higher level complexes also aligns the chromophores into orientations that are identical or related by a 180° rotation (15, 16). However, there is no direct experimental proof that such alignment exist in these assemblies in vivo. One could assume that alignment could benefit energy transfer within the rods and within the core cylinders; however, this is not necessarily correct because in addition to relative alignment, geometry and distance also play important roles in determining the kinetics and efficiency of energy transfer. According to the present model of PBS, the energy jump between rods and core are between components that do not have similar geometries. The rod (PC) to core (APC) jump of ∼30 Å occurs between protein discs (the terminal rod PC trimer and the core cylinders) at nearly right angles to one another, yet appears to have the same almost perfect energy transfer efficiency as between the co-linear rings in the rods (15) or within the cores. The same co-linear energy transfer is found within the entire complex of the unique cyanobacterium Acaryochloris marina, whose PBS is composed of single rods containing both PC and APC (17), yet the measured energy transfer kinetics appear to be quite similar. It would thus appear that there must be some organizational component that allows the PBS to overcome the relatively large energy transfer distances, without loss of efficiency.
The most accepted structural model of the entire PBS shows the rods radiating of the central core substructure (6, 8, 18). This proposed structure is based on electron microscopy analysis of single PBS complexes and leaves two open questions: (i) What is the method of attachment of the PBS subcomponents, the rods and core? This question is functionally important, because it determines how chromophores from rods and core are positioned relative to each other and thus determines the most probable pathways of energy transfer. (ii) In what way are the intra-cylindrical LPs arranged? Answering this question could clarify the enigma of the source of the unidirectional energy flow and the fine-tuning of light energy transfer inside PBS to the final energy emitter. Despite the great number of studies dealing with the whole PBS structure, a satisfying model of the PBS and the rod-core interface has not yet been achieved. Mismatches exist between the known dimensions of the subcomplexes and the prevalent model. In the current model, the fashion by which 6 rods assemble onto a tricylindrical core is architecturally difficult if one considers the dimensions of the available outer surface of the core and the diameters of the rods (6). One possibility to overcome this mismatch in dimensions is that the rods do not attach flush onto the core cylinders but rather are assembled in a staggered arrangement with only a fraction (approximately half) of each rod contacting the core (8). It is also possible that the PBS are tightly packed within the interthylakoid membrane stromal space (19) and that rods could associate with more than one PBS core, making all chromophores equally efficient in energy transfer. This organization would, however, place the position of the rod aperture (where the LPs are located) in-between complexes. We have recently shown that by stabilizing the connection between rods and cores by chemical cross-linking, efficient energy transfer can be obtained in different binding modes (20). Thus it is certainly possible that the PBS does not indeed require a single, unique structural mode for proper function.
To date, many PBPs structures have been determined by x-ray crystallography, including phycoerythrin, phycoerythrocyanin, PC, APC, the small core linker (ApcC), and parts of the rod linker (CpcC), as well as isolated domains of the core-membrane linker (ApcE). In some crystal lattices, higher assemblies of hexamers and rods can be observed. Two structures of LPs associated with PC or phycoerythrin hexamers in which the LPs themselves are not visible because of crystal symmetry have also been determined (15, 21); however, not a single high resolution structure of the assembly of rods onto core cylinders has been determined. While striving to determine the structure of the entire PBS (20), we also embarked on developing a method to try to analyze the interfaces between rods and core and between PBPs and LPs and thus shed light on the relative chromophore orientations in the rod and core subcomplexes. We began with the assumption that to identify the rod-core interface we must first isolate intact and functional PBSs in high phosphate buffer. Here, we present the results of the integration of chemical cross-linking under native conditions and MS, combined with existing high resolution crystallographic structures of isolated subunits and molecular modeling to reach conclusions about the architecture of the complex. Using limiting length cross-linking reagents and appropriate conditions, we were able to cross-link residues that are neighbors in the native complex and identify the cross-linked peptides by MS. Combining these results with modeling, we developed models that incorporate the matrix of identified cross-links within structures of the subunits. On the basis of these results, we were able to propose structural arrangement of PBS subcomponents inside the core and between the rods and core elements.
EXPERIMENTAL PROCEDURES
Growth of Cells and Isolation of PBSs
Thermosynechococcus vulcanus cells were grown as 2 liters of BG11 medium in a 10 liters of cylinder at 55 °C under fluorescent lights. The cells were collected by centrifugation and kept at −20 °C until use. The cells were resuspended in 50 mm phosphate buffer, washed, and resuspended in 0.9 m phosphate buffer at pH between 7.5 and 8.0 (Buffer A). Later, the cells were passed twice through a French Press at 1,500 p.s.i. followed by 10 min of centrifugation at 3,500 × g to discard unbroken cells. The blue supernatant was then incubated in buffer containing 3% Triton X-100 for 60 min at room temperature and centrifuged for 45 min at 20,000 × g. The supernatant containing the soluble PBS separated from the thylakoid membranes was then loaded on a step sucrose gradient (0.5, 0.75, 1, and 1.3 m) in buffer A, and ultracentrifuged for 16 h at 175,000 × g. The blue band obtained at the interface between 1 and 1.3 m sucrose was collected and found to contain the intact PBS by room temperature absorbance and fluorescence measurements.
Cross-linking and Complex Isolation
Functional PBSs were cross-linked with 5 mm glutaraldehyde (GA) for 5 min at room temperature or by 1 mm bis-[sulfosuccinimidyl] suberate (BS3) for 3 h on ice in buffer A. Cross-linking was quenched by the addition of 100 mm Tris buffer. The cross-linked PBSs were concentrated by ultrafiltration using a Centricon 100 K (Amicon). The concentrated complexes were separated by HPLC-anion exchange chromatography followed by room temperature absorbance and fluorescence analysis.
MS Analysis
All MS/MS procedures were carried out at the Smoler Proteomic Center at the Technion.
In Gel Proteolysis and Mass Spectrometry Analysis
Protein in-gel digestion was performed in 10% acetonitrile and 10 mm ammonium bicarbonate with modified trypsin (Promega) for 16 h at 37 °C. The resulting peptides were then resolved by reverse phase chromatography on 0.075 × 200-mm fused silica capillaries (J&W) packed with Reprosil reversed phase material (Dr Maisch GmbH, Ammerbuch-Entringen, Germany). The peptides were eluted with linear 65-min gradients of 5 to 45% and 15 min at 95% acetonitrile with 0.1% formic acid in water at flow rates of 0.25 μl/min. Mass spectrometry was performed by an ion trap mass spectrometer (Orbitrap, Thermo) in a positive mode using repetitively full MS scan followed by collision-induced dissociation of the seven most dominant ion selected from the first MS scan. The mass spectrometry data were clustered and analyzed using the Sequest software (J. Eng and J. Yates, University of Washington and Finnigan, San Jose) searching against the cyanobacteria database.
Identification of cross-linked peptides was done using Massmatrix (20). In silico tryptic digestion was simulated for protein fragmentation. The following modifications were configured for peptides search: fixed − iodoacetamide/carbamidomethyl of cysteine, variable − deamidation of asparagine/glutamine, oxidation of methionine, Tris quenched GA (185.14 Da), and a 64.03-Da mass modification for GA (omitting two water molecules) cross-linking on Lys or Arg and modification of 138.06 Da for BS3. To find the statistically significant peptide pairs, the search configuration was set to produce a false discovery rate of significant peptide pairs of less than 2.5% (precursor ion tolerance = 0.02 Da, product ion tolerance = 0.5 Da, maximum number of PTM/peptide = 2, minimum peptide length = 5, maximum peptide length = 45, minimum PP score = 2.5, minimum PPtag score = 1.2, maximum number of matches/spectrum = 2, maximum number of combinations/match = 2, maximum number of cross-links/peptide = 2). Significance was determined by Massmatrix PP and PPtag score. The PP score gives the probability that a peptide match is a random occurrence, and the PPtag gives the probability that a peptide match has a random pattern of amino acid residue tags. All protein hits had a decoy percentage of less than 2.5%, and all chosen peptides are statistically significant both in PP and PPtag scores (p < 0.01). Peptides with more than one matching spectrum were discarded. We used the SMS2 score as an additional criterion for peptide selection, as described by Kalisman et al. (22). A typical MS/MS product ion spectra obtained for a cross-linked peptide between CpcA and CpcB is shown in Fig. 1.
FIGURE 1.
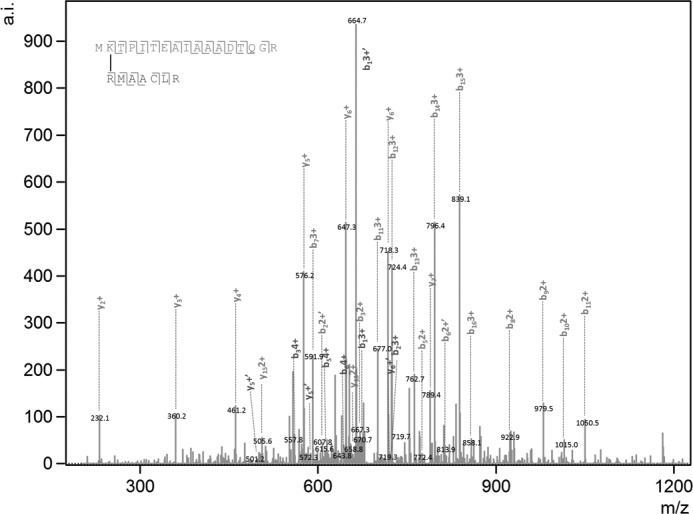
Typical product ion spectra used by MassMatrix. Shown are the product ion (MS/MS) spectra obtained for cross-linked peptides between peptide MKTPITEAIAAADTQGR (CpcA-Lys2) and RMAACLR (CpcB-Arg78) upon treatment with glutaraldehyde.
Subcomponent Models
High resolution crystal structures of PBS subunits have been previously determined for most of the components of the PBS from different cyanobacterial strains. These include APC, PC, ApcC, and short sections of ApcE (Protein Data Bank (PDB) codes 3DBJ, 3O18, 1B33, 3OSJ, and 3OHW, respectively). The high level of sequence homology between PBS subunits (12) from different organisms allowed us to use these structures directly for comparison or to build homology-based models (using the Swiss-Model Automated Protein Modeling Server (23)) with a high degree of confidence, for those proteins for which crystal structures have not yet been obtained for T. vulcanus. REP domains (pfam00427) were modeled based on structures 3OSJ and 3OHW in the PDB. Using the structural alignment tool implemented in PyMOL (24), we superimposed the four REP models described above and used the identical structural units as a REP consensus structure.
Model Filtration
We used 29 Å as a cutoff for the distance between all the identified cross-links in each of the models. Distances were measured between the relevant Cαs using a Python script. The models that were past the distance cutoff were energetically minimized by FireDock and ranked again according to the binding energy of the complex.
Statistical Analysis
We calculated all possible cross-links between Lys and Arg residues for each of the selected models to find the probability of obtaining these results randomly. The chance of randomly picking the identified cross-links in the selected models is found to be APC-ApcC (P(3) = 2.28*10−4), PC-APC (P(3) = 7.32*10−4) and APCHexamer-ApcC-ApcE(REP) (P(5) = 2.38*10−8) The difference between the means of all possible cross-linking distances to the experimental cross-linking distances was calculated using a t test. Although the null hypothesis is that there is no difference between the two populations (in case which results were collected by random), a significant p value was found (p ≪ 0.01), and the null hypothesis was rejected.
Two-dimensional Gel Electrophoresis
Two-dimensional gel electrophoresis was used for improved separation and analysis of the isolated complexes. For the first dimension we used Immobiline DryStrip NL (pH 3–7, 13 cm) (GE). The strips were rehydrated in rehydration solution (40 mm DDT, 4% CHAPS, 8 m urea, 2 m thiourea, 2% IPG, 1 μl of bromphenol blue) in the presence of the concentrated isolated PBS solution (A600 > 3). Isoelectric focusing was done with an Ettan IPGphor3 (GE) and maintained at 20 °C. The program was 150 V for 4 h; 10 h gradient from 150 to 1,000 V; 4 h gradient from 1,000 to 2,000 V; 2 h at 2,000 V; 3 h gradient from 2,000 to 3,000 V; and finally 1 h at 3,000 V (total, 26,850 Vh). The strips were equilibrated for 15 min in 0.05 m Tris-HCl (pH 8.8), 6 m urea, 30% (v/v) glycerol, 2% (w/v) SDS, 20 mm DTT and then for 15 min in the same buffer but containing 125 mm iodoacetamide in place of DTT. The equilibrated strips were applied to a vertical 7% SDS page used on S.E. 600 Ruby (Amersham Biosciences) and were subsequently scanned with an Ettan DIGE Imager (GE) fluorescence scanner. The scanner was equipped with Cy3 (λex = 550 nm, λem = 570 nm) and Cy5 (λex = 650 nm, λem = 670 nm) filters. Spots appearing only after cross-linking were taken out using a capillary and sent to mass spectrometry.
Fluorescence and Absorbance Measurements
Fluorescence measurements were carried out at room temperature with a Cary Eclipsed spectrofluorometer, using a 3-ml quartz cuvette (1 cm × 1 cm). Absorbance measurements were carried out on a Cary Eclipsed spectrophotometer, using 3-ml quartz cuvette (1 cm × 1 cm).
RESULTS
Mild Cross-linking of the PBS and Isolation of PBS Interface Adducts
Functional T. vulcanus PBSs (Tv-PBS) complexes were isolated in a high concentration of phosphate buffer (high phosphate buffer) followed by sucrose gradient centrifugation (20). Tv-PBS energy transfer to APC was monitored by excitation of PC at 540 nm, with emission from APC at 660–680 nm and minimal fluorescence at 645–655 nm (which would indicate the presence of disconnected rods). Only functionally intact samples were chosen for cross-linking. Isolated PBSs were then incubated with either GA (∼7 Å spacer length) or BS3 (11.4 Å spacer linker), as described under “Experimental Procedures.” GA (25, 26) and BS3 (22, 27) have previously been used as a cross-linking reagent in MS/MS analysis of protein interfaces. Following cross-linking, the stabilized PBS was transferred to a low ionic strength Tris-HCl buffer to induce complex disassembly. We isolated cross-linked adducts by three sequential protocols. In the first protocol, no additional isolation procedures were applied (hence called P1), and all of PBS proteins are present and could be separated by SDS-PAGE. New bands resulting from cross-linking were analyzed by MS. These cross-linked adducts could potentially include all of the PBS components. The P1 proteins were also further purified by HPLC-anion exchange chromatography to separate different populations of cross-linked PBPs (Fig. 2A). In this second protocol (hence called P2), each fraction was analyzed for PC to APC energy transfer by fluorescence. Only a single fraction, exhibiting energy transfer in low ionic strength Tris-HCl buffer was identified (Fig. 2B). This fraction contains the rod-core interface components and was analyzed by MS. In our third protocol (hence called P3), the P2 sample exhibiting energy transfer was further separated into its component cross-linked subunits using two-dimensional gel electrophoresis as described in detail under “Experimental Procedures.” After cross-linking, five different adducts were identified on these gels (Fig. 3, compare A with B and C). The positions of the spots were identified by the fluorescence of the phycocyanobilin (PCB) chromophores that, for T. vulcanus PBPs, preserve their fluorescent characteristics although treated with SDS (15). Note that after BS3 cross-linking, no PC fluorescence was detected, indicating energy transfer from PC to APC (Fig. 3, B versus C). Spots exhibiting detectable PC or APC fluorescence were extracted for further analysis.
FIGURE 2.
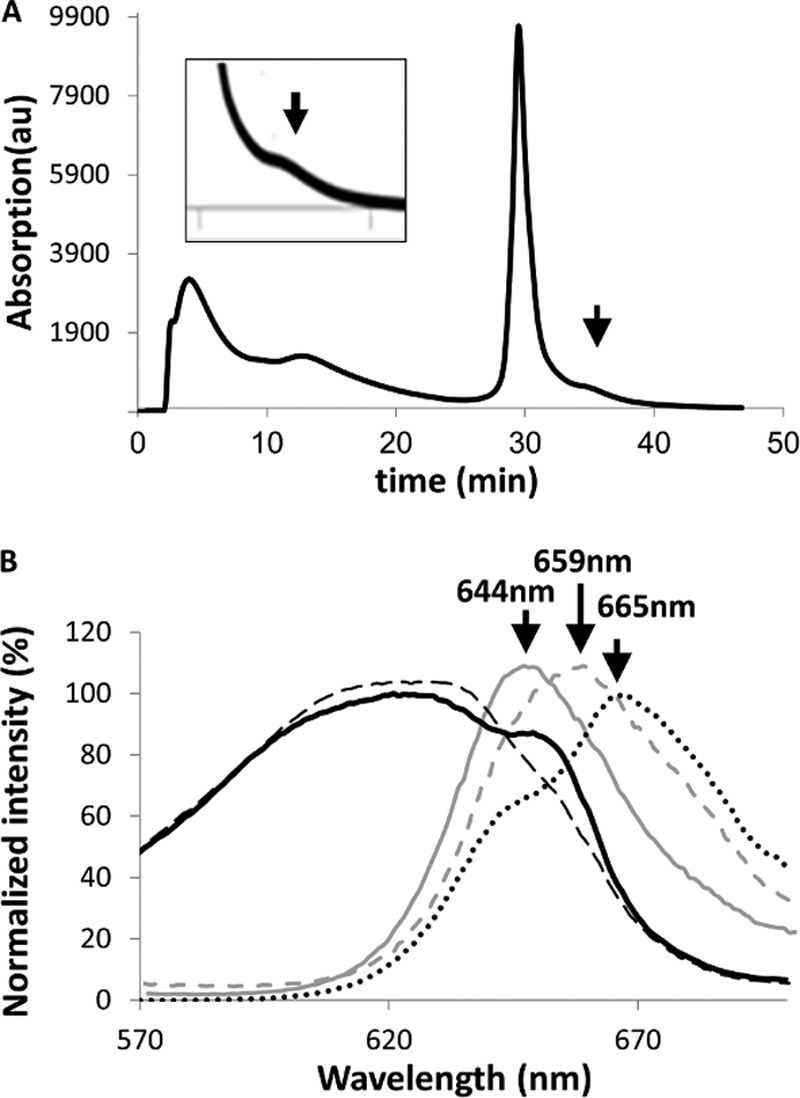
Isolation of functional PC-APC complexes. A, separation of cross-linked PC-APC products by HPLC-anion exchange chromatography (protocol 2). The arrow indicates the elution position of a single fraction that exhibited energy transfer to APC (λex = 540 nm). B, absorption and fluorescence of the PBS before and after cross-linking and isolation. PBS absorption in 0.9 m phosphate buffer (black dashed line), fluorescence emission of isolated non-cross-linked PBS in low ionic strength buffer (solid gray line), and in high phosphate buffer (gray dashed line). The absorption (black solid line) and fluorescence emission (black dotted line) spectra of the isolated cross-linked PC-APC fractions in low ionic strength buffer are also shown. In all fluorescence measurements, λex = 540 nm. The cross-linked adduct presents a PBS-like absorption curve with the contributions of PC at 620 nm and APC at 620 and 650 nm. PC transfers energy to APC is indicated by the strong emission at >660 nm in the cross-linked sample.
FIGURE 3.
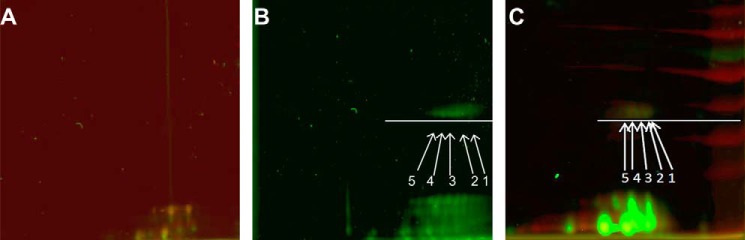
Separation of subfractions of the cross-linked PC-APC by two-dimensional gel electrophoresis (protocol 3). A, analysis of non-cross-linked PBSs shows the absence of cross-linked products with lower mobility in the second dimension. B, spots 1–5 appear following cross-linking with GA. C, spots 1–5 appear following cross-linking with BS3. Spots produce fluorescence when scanned using the Cy3 and Cy5 laser/filter sets. Red, PC; green, APC.
The overall quantity of functionally active P2 and P3 adducts were small because it was necessary to prevent the production of more extreme adducts, which would complicate our assessment of the most prevalent interfaces. The protocols also avoid to the greatest degree possible the formation of adducts between entire PBSs in solution. The P2 samples exhibited clear absorption from both PC and APC components (Fig. 2B), whereas the fluorescence shows equally clear energy transfer from bulk APC to the terminal energy emitters, ApcD and ApcE.
Analysis of the Cross-linked Complexes by MS
Following isolation of P1, P2, or P3 samples, proteins were denatured using SDS, digested with trypsin and submitted to MS analysis. Both functional termini of GA react predominantly with the primary amines (Lys, Arg, and N termini) (25). The cross-linking reagent BS3 reacts predominantly with the primary amines of Lys residues (22). Reactivity toward other residue types is known to be significantly weaker and therefore was not considered in this study. Using Massmatrix (28), the predicted spectra of all of the thousands of possible cross-linked peptides that may result from linking nearest neighbors of all of the PBS components were compared with the experimental fragmentation spectra of each sample. To perform molecular modeling of potential interaction interfaces, we postulated that we would require a minimum of three adducts for each putative interaction. We used two criteria to identify adducts that appeared in the fragment database at significant rates: The MassMatrix pp scores (−log10(probability)) and the MS score criteria previously described by Kalisman et al. (22). Calculation of false discovery rate was obtained using a decoy data set (29). We performed the experiments with a minimal (< 2.5%) rate of false positive results. A negative control search for cross-linked peptides between CpcD (the LP found at the distal end of the rods), and any core component (APC or linkers) was made using the P1 and P2 MS data sets. As expected, not a single peptide was found between these components. This result also strengthens our belief that no significant interactions occur between PBS complexes during the duration of the cross-linking reaction. Using the three protocols, 32 unique peptide adducts were identified (Table 1 and Fig. 4).
TABLE 1.
Cross-linked dipeptides identified by MS
| Subunits | Source protocol no. | Cross-linked residues (positions) |
p value | Distance on selected model (Cα-Cα) | |
|---|---|---|---|---|---|
| Residue 1 | Residue 2 | ||||
| Å | |||||
| PC-APCa | P1, P2, P3 | CpcA-Lys83 (82–86) | ApcA-Arg36 (36–38) | 1 × 10−5.2 | 29.0 |
| P1, P2, P3 | CpcB-Arg37 (36–47) | ApcA-Arg36 (33–43) | 1 × 10−7.5 | 24.5 | |
| P1, P2, P3 | CpcB-Lys36 (33–37) | ApcB-Lys26 (17–28) | 1 × 10−5 | 28.7 | |
| P3 | CpcA-Lys32 (BS3) (31–33) | ApcB-Lys28 (18–28) | 1 × 10−2.5 | 29.5 | |
| APC-ApcC | P1, P2 | ApcA-Lys27 (26–36) | ApcC-Lys45 (39–49) | 1 × 10−2.5 | 23.6 |
| P1, P2 | ApcA-Lys27 (26–36) | ApcC-Arg42 (39–45) | 1 × 10−3.3 | 27.3 | |
| P1, P2 | ApcB-Arg77 (77–83) | ApcC-Lys45 (43–49) | 1 × 10−4 | 22.0 | |
| APC-ApcE | P2 | ApcA-Lys52 (50–61) | ApcE-Lys818 (811–822) | 1 × 10−4 | 26.9 |
| P2 | ApcA-Lys6 (1–16) | ApcE-Arg1008 (1002–1010) | 1 × 10−2.9 | 22.7 | |
| ApcE-ApcC | P2 | ApcC-Lys5 (1–15) | ApcE-Lys339 (339–349) | 1 × 10−2.5 | 24.0 |
| P2 | ApcC-Arg2 (2–5) | ApcE-Arg1010 (1009–1021) | 1 × 10−3.6 | 18.6 | |
| P2 | ApcC-Lys45 (39–49) | ApcE-Arg961 (961–964) | 1 × 10−2.5 | 28.4 | |
| ApcF-APC | P2 | ApcA-Lys27 (26–35) | ApcF-Arg21 (18–26) | 1 × 10−2.8 | 32.4b |
| P2 | ApcA-Arg49 (39–52) | ApcF-Arg2 (2–10) | 1 × 10−3.8 | 23.0 | |
| ApcF-ApcE | P2 | ApcF-Arg37 (29–39) | ApcE-Arg1053 (1049–1062) | 1 × 10−2.5 | 36.0b |
| P2 | ApcF-Arg17 (11–21) | ApcE-Arg490 (487–495) | 1 × 10−3 | NAc | |
| ApcD-ApcE | P2 | ApcD-Arg141 (124–144) | ApcE-Arg729 (729–734) | 1 × 10−3.3 | NA |
| P2 | ApcD-Arg48 (47–58) | ApcE-Arg433 (428–434) | 1 × 10−2.5 | NA | |
| P2 | ApcD-Arg2 (1–20) | ApcE-Arg729 (729–734) | 1 × 10−3 | NA | |
| ApcD-ApcC | P1, P2 | ApcD-Arg93 (89–100) | ApcC-Arg2 (2–5) | 1 × 10−4.8 | 24.1 |
| CpcG2-PC | P1, P2 | CpcG2-Arg104 (95–111) | PcB-Arg78 (78–91) | 1 × 10−2.9 | NA |
| CpcG2-ApcF | P1, P2 | CpcG2-Arg189 (185–196) | ApcF-Arg21 (18–28) | 1 × 10−3.6 | NA |
| P1, P2 | CpcG2-Arg182 (177–184) | Apc-Phe26 (22–28) | 1 × 10−8.4 | NA | |
| P1, P2 | CpcG2-Arg164 (164–166) | ApcF-Arg21 (18–28) | 1 × 10−6 | NA | |
| CpcA-CpcB | P1 | CpcA-Arg33 (33–43) | CpcB-Arg37 (33–43) | 1 × 10−2.5 | 16.5 |
| P1 | CpcA-Lys2 (1–17) | CpcB-Arg78 (78–84) | 1 × 10−7.7 | 15.7 | |
| P1 | CpcA-Arg33 (31–33) | CpcB-Arg78 (78–91) | 1 × 10−2.9 | 29.0 | |
| P1 | CpcA-Lys32 (31–33) | CpcB-Lys7 (1–15) | 1 × 10−7.4 | 19.7 | |
| P1 | CpcA-Lys32 (31–42) | CpcB-Arg37 (37–43) | 1 × 10−2.6 | 13.8 | |
| P1 | CpcA-Lys32 (31–42) | CpcB-Arg43 (37–43) | 1 × 10−2.6 | 20.8 | |
| P1 | CpcA-Arg33 (33–42) | CpcB-Lys36 (33–37) | 1 × 10−3.1 | 17.1 | |
| P1 | CpcA-Arg33 (33–42) | CpcB-Lys7 (1–15) | 1 × 10−3 | 19.0 | |
a All peptide pairs were identified using MassMatrix according to the criteria detailed under “Experimental Procedures.”
b Distances longer then the 29 Å cutoff were positioned manually.
c NA, unmodeled flexible loop. Distance measurements are not reliable.
FIGURE 4.
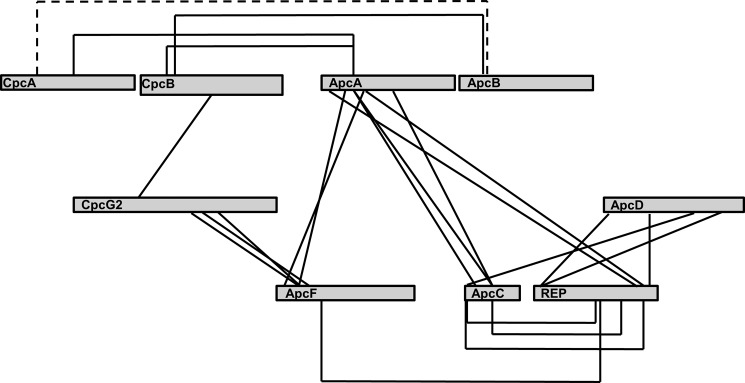
Schematic map of the identified cross-linked residues. Gray rectangles represent the different PBS subunits. Solid lines show identified cross-links using GA. The dashed line shows the BS3 cross-link.
Calibration and Validation of the Method
The combined distance between two α-carbons of cross-linked Lys and/or Arg was estimated to be 22.7 ± 1.0 Å: the sum of the length of GA and the lengths of lysine and arginine side chains (7.1 and 8.1 Å, respectively). We used the high resolution crystal structure of T. vulcanus PC (Tv-PC; PDB code 3O18) to map out the distances of the cross-linked peptides between α and β PC subunits that we identified from P1 data sets. In this calibration of the method, we used all possible levels of assemblies of PC: trimers, hexamers, or rods (according to these levels of assembly in the 3O18 structure). The distances between cross-linked residues (between α-carbons) were measured for each of the possible assemblies. For each resulting set of cross-linked pairs, the minimal distances were measured. We measured all of the possible distances for all levels of assembly and identified, for each set of cross-linked peptides, the most likely occurring pairs. This is based on the assumption that the cross-linking reaction under the conditions used occurs between nearby residues. A cutoff value for models filtration should be the highest possible distance between two cross-linked residues. Following scaling of the probable intra-PC distances, the average distance between pairs of α-carbons was found to be 19 ± 10 Å. Therefore, we used 29 Å distance as the maximal cutoff distance for peptide pair filtration. This value is similar to values used in previous studies. Leitner et al. (30) used 30 Å as the maximal distance using the DSS cross-linking reagent, and Kalisman et al. (22) estimated 28–33 Å as the maximal distance using the BS3 cross-linking reagent.
Analysis of PBS Subcomplex Interactions
The APC (αβ)3 Trimer-ApcC complex
In the 1B33 crystal structure (from Mastigocladus laminosus), the mode of binding of the ApcC (LC) linker within APC trimers was visualized (31) because of the unique lack of 3-fold symmetry in the asymmetric unit. To check the veracity of our method of using cross-linking/MS data as constraints in molecular modeling, we used the interaction between T. vulcanus ApcC and APC as a positive control, using the P1 sample. A computationally based docking procedure was carried out between a homology-based model of Tv-ApcC and the Tv-APC (αβ)3 trimer (PDB code 3DBJ), using the GRAMM-X docking server (32). The following criteria were applied to filter the docking models to those that might agree with the following cross-linking/MS results: (i) the maximal separation between residues of identified cross-links must be no more than 29 Å; (ii) steric clashes between any of the protein/co-factor atoms are prohibited; and (iii) ApcC was assumed to reside only inside the APC trimeric ring, so all solutions in the periphery of the timer were discarded. Following filtering, we used FireDock (33) to energetically minimize the model, followed by calculation of relative binding energies of the proposed complexes by PISA (34). Distances between all potential cross-linked fragments were calculated for the 300 best GRAMM-X models to find a possible docking model candidate. After filtration, only four models (1.3%) remained (Table 2). These models were ranked by binding energy calculation, and in this fashion the best model was chosen. The model that passed all of the filtration tests and had the lowest energy is nearly identical to the 1B33 structure (Fig. 5A). The RMSD of all equivalent α-carbons between the full T. vulcanus APC trimer-ApcC complex and the1B33 structure was found to be 0.955 Å. Two of the other three models position the ApcC subunit in the same site, with different orientations, whereas the last model crosses the APC trimer aperture and is significantly different.
TABLE 2.
Top rated models by distances and binding energy
| Model | Binding energy | Cα-Cα average distance |
|---|---|---|
| kcal/mol | Å | |
| PC-APCa | −7.0 | 27.4 |
| PC-APC | 2.1 | 28 |
| APC-ApcCa | −3.2 | 21.6 |
| APC-ApcC | −1.1 | 21 |
| APC-ApcC | −1.1 | 27 |
| APC-ApcC | −1.3 | 26 |
| APC[ApcC]-REPa | −0.5 | 24.1 |
a Selected models based on PISA-calculated energy considerations.
FIGURE 5.

Filtration of protein-pair models based on the modeling limitations. Experimentally derived peptide couples from proteins obtained by cross-linking were mapped onto structural models of the different proteins. Possible interactions were obtained using the 29 Å distance maximum as described under “Experimental Procedures.” The top ranked models are indicated with stars. The process was carried out for the proteins pairs. A, APC-ApcC. B, PC-APC monomers. C, APC-ApcC (the result of modeling in A with the REP domain of ApcE.
Analysis of the PC-APC Interface
Cross-linking/MS data from the P2 and P3 samples were used to analyze to the interface between the rods and core components. Crystallographic models of PC and APC monomers were docked using GRAMM-X, and distances were measured on the resulting models. In the initial step of modeling, we used only monomeric units (of PC and APC) to prevent redundancy. However, because of the 3-fold symmetry of the trimer, and the presence of three identical copies of each monomer, we also tried modeling the cross-linked pairs in other fashions, where not all cross-links occur between a single PC monomer to a single APC monomer. Such attempts were not consistent with our distance constraints, as described above.
Following the model building step, trimers or hexamers could be superimposed onto the modeled monomer-based complex. Only the first two criteria mentioned above were used, i.e. PC and APC were allowed to interact on any surface. After filtration, only two models of 300 (<1%) remained. These two models are almost identical and represent the most likely mode of interaction between PC and APC hexamers (Fig. 5B and Table 2). Model 1 had a lower energy and no violations, and therefore we continued our structural analysis on this model. In addition, we performed the same analysis of P2 cross-linking/MS data obtained using the BS3 cross-linker and found that these data supports only model 1.
Visualization of the predicted model 1 interface enabled us to examine the chemical attributes of the resulting model. The interaction between the two proteins is formed exclusively between ApcA and CpcB subunits. The surface electrostatic potential complimentarity between these two proteins is quite significant (Fig. 6A, lower panel). These putative interactions (Fig. 6A, upper panel) are formed mainly as follows: (i) the first patch is a negative protrusion formed by the conserved residues ApcA-Ser145/Asp148 and a less conserved residue ApcA-Glu147 (as indicated by the ConSurf (35) conservation analysis server), which associates with a positive pocket containing conserved residues CpcB-Lys36/Arg37; and (ii) a second interaction is formed by ApcA-Lys137 and CpcB-Asp167, both conserved residues. This interface will be discussed more fully below.
FIGURE 6.

Schematic view of the PBS subcomponents interactions, as were revealed by cross-linking/MS and structural analysis. A tricylindrical core scheme is presented (core cylinders are in gold, brown, and blue), interacting with a single rod cylinder (light blue). A, close-up view on the proposed interaction of CpcB and ApcA monomers. Top panel, the negative protrusion formed by residues ApcA Glu147 and Asp148 interacts with the positive pocket formed by CpcB Lys36 and Arg37 (spheres represent carbons, oxygen, and nitrogen atoms colored in green, red, and blue, respectively). Bottom panel, electrostatic and structural fitness for the interaction area (surface representation; positive and negative surfaces are colored blue and red, respectively). B, top panel, close-up view on the proposed interaction of ApcC and REP4. The interaction is formed mainly by REP4 Glu68, GLu73, and Gln126 with ApcC Arg15, Arg17, and Arg36 (spheres). Bottom panel, electrostatic and structural fitness for the interaction area (surface representation). C, model of the full APC hexamer. The complex consists the APC (αβ) monomers (brown), minor core components ApcE[PBP domain]/ApcF (magenta), ApcD containing monomer (green), REP4 domain (blue cartoon), and ApcC (red cartoon). The hexamer has been opened up by ∼30° for the observer's convenience (right panel). In the left panel, the hexamer has been rotated by 90° to visualize the hexameric back. The distal trimer contains ApcC. A gray disc represents the possible localization of PC rod adjacent to ApcF. In B and C, the protein subunits have been manually separated by ∼5 Å along the vertical axis to assist in identification of the interacting residues.
Analysis of the Interaction between the ApcE REP Domains and the APC/ApcC Trimer
It has been suggested that each core cylinder is terminated by an APC trimer containing the ApcC linker. We analyzed both P1 and P2 sample cross-linked adducts containing peptides from APC and ApcE. These were applied to potential docking models between the T. vulcanus (APC (trimer)-ApcC) complex obtained above, with a homology-based model of a partial structure of T. vulcanus ApcE. The cross-linking/MS data (Table 1) contained six REP-containing cross-linked peptides: one from REP1, one from REP3, and four from REP4. Four cross-linked peptides mapping to stretches of ApcE of unknown structure were not used for modeling. The same procedure described above was applied, including the caveat that the REP domain is located within the core cylinders. Only a single model remained after filtration, with an average distance of 25.9 Å for all cross-links (Fig. 5C). The REP domains were found to interact with their surrounding inter-APC hexamer residues and ApcC linkers dominantly by polar interactions (Fig. 6B), including with the surrounding co-factors, as was revealed by PISA and visual analysis with PyMOL (Fig. 6C, right panel).
Interactions of the Minor Core Components, ApcD and ApcE/ApcF
Although each core cylinder contains four APC trimers, each with 3-fold symmetry, the presence of ApcC, ApcD, ApcE, and ApcF breaks this symmetry. We wanted to see whether we could model a core cylinder more precisely by positioning these minor components, relative to the central REP domains. Three cross-links between ApcD and the ApcE REP domains (PDB codes 3OSJ and 3OHW) were identified. These cross-linking/MS results, together with the APC-REP model described in the section above, dramatically lowered the possible positions of minor core components inside the cylinder (Fig. 6C). Using the cross-linking/MS results, we could position the ApcD subunit as a component of an APC/ApcC trimer. Localization of ApcF was determined to be in the APC trimer lacking ApcC, based on the following considerations: (i) limitation of length for the single identified cross-link with REP indicated three possible settings inside the hexamer and (ii) no cross-links were identified between ApcD and ApcF or between ApcC and ApcF. We thus propose that ApcD and ApcE[PBP domain]/ApcF are not present in the same trimer (Fig. 6C). In addition, in the P1 and P2 data, cross-links were found between peptides from the rod-core linker CpcG2 and ApcF (Table 1). These links are all within putatively unstructured sections of CpcG2, and thus modeling is not possible. However, these results indicate that ApcF may interact directly with a CpcG2 containing rod (Fig. 6C, left panel). Because we assume that the ApcE[PBP domain] points toward the membrane (to provide efficient energy transfer to PSII), these results enable us to precisely position the ApcE[PBP domain]/ApcF monomer orientation with respect to the membrane.
DISCUSSION
Since their discovery and initial characterization in the 1960s and 1970s (7, 36–40), many models of the fashion of assembly of the PBS have been proposed (6, 8, 10, 11). The PBS has been structurally and functionally characterized at high resolution and precision at the level of isolated components (6, 41–44); however, analysis of the entire complex in isolated form is complicated by its inherent instability and need for high phosphate buffer for stabilization. These problems notwithstanding, many studies have been performed, and the overall picture that emerges shows that the PBS is highly efficient in energy transfer to both PSII and PSI (45, 46) and is under tight control to avoid overexcitation at high light fluencies (47, 48). We have recently shown that for isolated PBS from T. vulcanus, stabilized by cross-linking with GA, functional energy transfer can occur in different rod-core architectures (20). In this previous study, the resolution of the cryo-TEM analysis was not high enough to provide detailed information on the possibility that only certain residues of PC and APC interact. The present study, in which cross-linking was performed under the mildest condition that still afforded the isolation of an active PC-APC couple, has now provided at least one such interaction interface. Because the energy transfer step between the rods and cores is clearly a critical step for ensuring efficient energy transfer to the photosystems, we wanted to see whether we could identify at the molecular level precise interaction interfaces. Use of coupled cross-linking/MS has enabled such dissection of heterogeneous and large complexes (22); however, at the outset we realized that the PBS would present a unique problem, because six to eight apparently identical rods assemble onto three to five quite similar core cylinders. This problem of multiplicity is further complicated by the internal 3-fold symmetry found in all PBS elements. We also could not separate by this method peptides from PC612 and PC620 that we isolated from T. vulcanus (44), because they have the same sequence (and no adducts were found from the peptide that includes the N-methylated asparagine B72 residue). We thus could not assess the possible role of PC612 in assembly or function. Notwithstanding, we attempted to model the most prevalent and likely interface by this method. Isolation of the minimal fraction that continues to perform PC to APC energy transfer (P2 and P3 samples) was critical, and we thus optimized the cross-linking to prevent the production of larger and more complicated adducts. The negative and positive controls described above indicate that cross-linking was indeed performed on intact functional PBS complexes, and inter-PBS cross-links have been avoided. The different association interfaces could be the result of the existence of different types of CpcG LPs, however, not all PBS containing organisms have the same number of CpcG encoding genes. For instance, it appears that Synechocystis sp. PCC 6803 PBS complexes assemble into the classical tricylindrical complex with only a single form of CpcG (49). On the other hand, because the rods all contain the same PC in the same arrangement, and the face forming the interface is most likely made up of the β-subunits, it is not inconceivable that in all cases the same exact residues on the end of the rod must align with the exact same residues on the circumference of the core cylinders. We applied the cross-linking/MS limitation to the docking of single monomers of PC and APC; however, we did not limit the potential interaction interfaces to single monomer. Because each monomer represents one-sixth of a hexamer, we could then superimpose hexamers onto the monomers and obtain an expanded model of the rod-core interface (Fig. 7). The angle formed between the two superimposed hexamers is not precisely perpendicular, which could be a result of the fact that we are modeling a single interface that represents multiple interfaces (depending on the existence of symmetry on the rod-core association).
FIGURE 7.

The proposed model of PC-APC interaction. A, the selected PC-APC interaction model is represented by a green PC trimer attached to a complete APC cylinder. The black rectangle denotes the area enlarged in B. B, enlargement of the interaction interface. The hexamers of PC and APC were superimposed on the matching monomers to reveal the super structure of rod-core. At this level of enlargement, only the edges of the ApcA subunits are visible at the bottom, and these edges contact the PC trimer (on top). Chromophores (PCB) is the superimposed hexamers are represented as sticks. Lines connect residues found to cross-link.
As has been previously suggested, one major difference between the different PBS models is whether the rods associate in a fashion that the entire rod sits flush in a perpendicular manner onto a core cylinder (6, 7, 11) or whether a staggered assembly exists (required if the rods fan out from the core), such that only a fraction (no more than 50%) of the rod actually associates with the core cylinder, with the remaining part of the rod not associated with the core (8, 10, 50). On the basis of the cross-linking/MS analysis of the T. vulcanus PBS, we suggest that the former model is correct. In Fig. 7, we have assembled a rod hexamer onto half of a core cylinder, based on the PC-APC interactions. It is quite clear that the PC circumference is flush with the edge of the cylinder, and thus if the second APC hexamer was added to complete the cylinder, the entire rod would fit over the cylinder. This arrangement also provides complete coverage of the rod chromophores over the cylinder chromophores.
In the model presented here, the distance between the centers of the dipoles of the two closest chromophores from the terminal rod hexamer and the core cylinder hexamer is 34 Å. This distance would certainly appear to preclude exciton coupling as participating in the mechanism of rod to core energy transfer. At this distance, energy transfer is typically associated with the FRET model (51). The distance of 34 Å between the closest chromophores of PC and APC hexamers in the model we present here is in agreement with previous studies of energy transfer in PCs higher assemblies and the distances between pairs of chromophores, which energy most likely travels through (8, 52, 53). Although one might assume that because of the relatively long distance between the terminal rod emitter and the initial core acceptor, energy transfer would be concomitantly slow. However, measurements of rod to core energy transfer rates have been shown to be on the same order as the inter-rod rates. It has been suggested that the β84 PCB of PC monomers is the lowest energy chromophore; thus the β84 PCB chromophores situated at the ends of the rods function as the rod terminal energy donor to the core (8). In the interface between PC and APC modeled in this study, the dipole moment of the terminal β84 PCB is rotated ∼70° in comparison to the closest APC chromophore, an α84 PCB. Calculations in the literature (43, 54) show that this angle is close to the 60° angle found between chromophores in trimers, hexamers, and larger assemblies. This angle may promote fast and or efficient energy transfer, although the separation here is indeed quite large. On the other hand, the α84 PCB of APC may be excitonically coupled to the adjacent β84 PCB (55, 56); thus the actual overlap between donor and acceptor is between the single PC PCB and the APC dimeric PCB. This interaction has not been calculated in the past but may hold the key to fast and efficient energy transfer from the rod to core. Measurements carried out on PBSs isolated from red algae to study the rod-core energy transfer time indicated that this transfer involves small number of components with kinetics on the scale of picoseconds and a more dominant component with a kinetics of tens of picoseconds (57). Thus, although the separation between the terminal rod chromophores to the initially excited core chromophores is significantly greater than the distances between chromophores within the rods or cores, the effect on energy transfer efficiency or kinetics is small. This observation reiterates the importance of the protein surroundings of the chromophores that functionally tighten the rod-core interaction. The unique protein environment of the PBS chromophores has already been shown to impart significant coupling and energy transfer between quite distant chromophore pairs (55, 58), as well as impart on the entire complex the directionality of energy transfer that is absolutely required for proper light harvesting functionality. The rod-core protein interface must also strengthen the coupling between the closest chromophores, affording faster energy transfer, as well as preventing alternative energy quenching modes.
Close electrostatic and structural examination of the proteins contact area reveals a good fit between the monomers. The interaction interface of PC-APC was found to be between conserved residues of CpcB and ApcA. During normal photosynthetic activity, the stroma becomes somewhat more basic because of proton uptake into the lumen. Even a minor change to the protein environment caused by proton depletion (changing the stroma to pH 8.0) might affect the stability of the PBS, and the presence of multiple interacting residues that are strongly charged could assist in overcoming the environmental change. The configuration of two adjacent positive residues in CpcB (Lys36 and Arg37) that according to our model interact with two adjacent negative residues on ApcA (Glu147 and Asp148) might have such a stabilizing role.
In addition to proposing the fashion by which the rods assemble onto the core, additional structural modeling on the core was performed using the cross-linking/MS data. The PBSs have been proposed to be connected to the photosynthetic membrane by the ApcE core component (59–61). The N-terminal 240-amino acid region of ApcE pfam00502 domain (P502) is homologous to ApcA and contains a single chromophore. Together with ApcF, these two subunits form a unique monomer which is red-shifted compared with APC (λmax = 670 nm). Repeats of varying 15–90-amino acid spacers (ARM) and pfam00427 rod-linker like domains (REP) are positioned C terminally to P502 (14, 60, 62) and are believed to be involved in core interactions. The first ARM domain is 20 amino acids long and therefore has to be structurally close to the P502 domain and probably in the same cylinder. The second and fourth ARM domains are 86 and 48 amino acids long, probably connect between hexamers, and may interact with the thylakoid membrane and PSII (14). The third ARM domain is 16 amino acids long and connects between REP3 and REP4, which may be positioned inside two structurally close hexamers. Considering the significant homology that the rod and core LPs share, REP4 can be inserted into the rod or core hexamers. As previously suggested, the core subcomplex contains two copies of ApcE (14), together contributing eight REP domains to the structure. A single tricylindrical core contains APC hexamers, populated with two copies of REP1–3. Therefore, the additional REP domain from every ApcE might contribute to the assembly of the fourth and fifth APC cylinders in a pentacylindrical core or the starting point for rod assembly in a tricylindrical core.
The APC-ApcC-ApcE (REP) model obtained in this study (Fig. 8) fits extremely well with the experimental and docking results of Gao et al. (63). In this study, the pfam00427 domain of the rod-linker protein was crystallized. The interaction area with PC trimer was characterized using mutations and a pulldown assay. In the work presented here, we identified the interactions of ApcE (REP) with the complementary trimer inside a hexamer. Interaction differences of REPs with their surroundings were calculated using PISA (Table 3). These differences might explain the fashion by which LPs provide structural elements that ensure that absorbed energy is funneled toward the final energy emitter of the PBS.
FIGURE 8.

The complete model of a core hexamer. The selected model of APC hexamer containing ApcC and REP domain. A, REP (blue cartoon) interacts with ApcC (red cartoon) and is in close proximity to some of the chromophores (blue sticks). B–D, the same hexamer as in A contains the minor core components ApcD (magenta cartoon) and ApcF-ApcE(1–240) (brown cartoon) with the P502-loop (orange) in three 90° vantage points.
TABLE 3.
PISA-based calculated energy of the interface of the four REP domains with their intra-APC hexamer surrounding
| REP domain | Core subcomponent | Total ΔGa |
|---|---|---|
| kcal/mol | ||
| REP1 | ApcC | −0.77 ± 0.33 |
| ApcA | 0.53 ± 0.56 | |
| ApcB | −0.96 ± 0.07 | |
| REP2 | ApcC | 3.03 ± 0.42 |
| ApcA | 3.83 ± 0.48 | |
| ApcB | −1.72 ± 0.36 | |
| REP3 | ApcC | −3.43 ± 0.76 |
| ApcA | −0.98 ± 0.49 | |
| ApcB | −0.06 ± 0.00 | |
| REP4 | ApcC | −3.00 ± 0.34 |
| ApcA | −0.39 ± 0.54 | |
| Chromophore: ApcB | −0.12 ± 0.21 |
a The interaction energies of the minimized models were calculated using PISA. The ΔG values are the summary of all interactions within the specified components.
The positions of the minor core components ApcD and ApcE [P502]-ApcF could also be modeled. The cross-linking/MS-based model of the core agrees very well with previous evidence by EM. Arteni et al. (41) showed additional densities in one trimer of the four trimers that compose the basal core cylinder. Those densities appear on both of the basal cylinders in an anti-parallel fashion. The researcher proposed that it might be the ARM2 domain of ApcE. If this is indeed correct, the N-terminal domain of ApcE is necessarily in close proximity and in a fixed position (41). A similar model was also proposed by Kirilovsky and co-workers (64) when examining the orange carotenoid protein photoprotective component interaction with the PBS.
Based on the structural homology of CpcG2 pfam00427 domain to other LPs (REPs), we propose that the pfam00427 domain of CpcG2 in Tv may function within the center of the PC hexamer, whereas its flexible tail interacts specifically with the ApcF subunit in the PBS core. Previous study on the role of CpcGs proteins carried out by Kondo et al. (49) suggested that CpcG2s have a crucial role in rod-core interaction. These findings indicate that CpcG2 interacts with a red-shifted absorption core component. Kondo et al. also have suggested that rods that include CpcG2 might interact with the thylakoid membrane and PSI. The first of these suggestions agrees with our results, which indicate that the C terminus tail of CpcG2 participates in a close interaction with ApcF. If the ApcF-ApcE [P502] heteromonomer is positioned toward the membrane (Fig. 6), the CpcG2 C-terminal tail might interact with both the membrane and ApcF. Our results support this kind of configuration, and in this orientation, this rod type could interact with PSI and/or PSII, as recently suggested (46).
Acknowledgments
We thank the staff of the Smoler Center for Proteomics for assistance in developing the methods for coupled cross-linking/MS of the PBS proteins.
This work was supported by United States-Israel Binational Science Fund Grant 2009406 and Israel Science Foundation Grant 1576/12. We also acknowledge the support from the Nancy and Stephen Grand Technion Energy Program and the Technion Russell Berrie Nanotechnology Institute.
- LHC
- light-harvesting complex
- PBP
- phycobiliprotein
- PBS
- phycobilisome
- PCB
- phycocyanobilin
- PDB
- Protein Data Bank
- PSI
- photosystem I
- PSII
- photosystem II
- RC
- reaction center
- Tv
- Thermosynechococcus vulcanus
- APC
- allophycocyanin
- PC
- phycocyanin
- LP
- linker protein
- GA
- glutaraldehyde
- BS3
- bis-[sulfosuccinimidyl] suberate.
REFERENCES
- 1. Cogdell R. J., Gardiner A. T., Roszak A. W., Law C. J., Southall J., Isaacs N. W. (2004) Rings, ellipses and horseshoes: how purple bacteria harvest solar energy. Photosynth. Res. 81, 207–214 [DOI] [PubMed] [Google Scholar]
- 2. Croce R., van Amerongen H. (2013) Light-harvesting in photosystem I. Photosynth. Res. 116, 153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cogdell R. J., Gardiner A. T., Hashimoto H., Brotosudarmo T. H. (2008) A comparative look at the first few milliseconds of the light reactions of photosynthesis. Photochem. Photobiol. Sci. 7, 1150–1158 [DOI] [PubMed] [Google Scholar]
- 4. van Amerongen H., Croce R. (2013) Light harvesting in photosystem II. Photosynth. Res. 116, 251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noy D. (2008) Natural photosystems from an engineer's perspective: length, time, and energy scales of charge and energy transfer. Photosynth. Res. 95, 23–35 [DOI] [PubMed] [Google Scholar]
- 6. Adir N. (2005) Elucidation of the molecular structures of components of the phycobilisome: reconstructing a giant. Photosynth. Res. 85, 15–32 [DOI] [PubMed] [Google Scholar]
- 7. Glazer A. N. (1989) Light guides: directional energy transfer in a photosynthetic antenna. J. Biol. Chem. 264, 1–4 [PubMed] [Google Scholar]
- 8. MacColl R. (1998) Cyanobacterial phycobilisomes. J. Struct. Biol. 124, 311–334 [DOI] [PubMed] [Google Scholar]
- 9. Watanabe M., Ikeuchi M. (2013) Phycobilisome: architecture of a light-harvesting supercomplex. Photosynth. Res. 116, 265–276 [DOI] [PubMed] [Google Scholar]
- 10. Anderson L. K., Toole C. M. (1998) A model for early events in the assembly pathway of cyanobacterial phycobilisomes. Mol. Microbiol. 30, 467–474 [DOI] [PubMed] [Google Scholar]
- 11. Adir N., Dines M., Klartag M., McGregor A., Melamed-Frank M. (2006) Assembly and disassembly of phycobilisomes. In Microbiology Monographs: Inclusions in Prokaryotes (Shively J. M., ed) pp. 47–77, Springer, Berlin/Heidelberg [Google Scholar]
- 12. Apt K. E., Collier J. L., Grossman A. R. (1995) Evolution of the phycobiliproteins. J. Mol. Biol. 248, 79–96 [DOI] [PubMed] [Google Scholar]
- 13. Marx A., Adir N. (2013) Allophycocyanin and phycocyanin crystal structures reveal facets of phycobilisome assembly. Biochim. Biophys. Acta 1827, 311–318 [DOI] [PubMed] [Google Scholar]
- 14. Liu L. N., Chen X. L., Zhang Y. Z., Zhou B. C. (2005) Characterization, structure and function of linker polypeptides in phycobilisomes of cyanobacteria and red algae: an overview. Biochim. Biophys. Acta 1708, 133–142 [DOI] [PubMed] [Google Scholar]
- 15. David L., Marx A., Adir N. (2011) High-resolution crystal structures of trimeric and rod phycocyanin. J. Mol. Biol. 405, 201–213 [DOI] [PubMed] [Google Scholar]
- 16. Jiang T., Zhang J., Liang D. (1999) Structure and function of chromophores in R-Phycoerythrin at 1.9 A resolution. Proteins 34, 224–231 [PubMed] [Google Scholar]
- 17. Chen M., Floetenmeyer M., Bibby T. S. (2009) Supramolecular organization of phycobiliproteins in the chlorophyll D-containing cyanobacterium Acaryochloris marina. FEBS Lett. 583, 2535–2539 [DOI] [PubMed] [Google Scholar]
- 18. Adir N. (2008) Structure of the phycobilisome antennae in cyanobacteria and red algae. In Photosynthetic Protein Complexes: A Structural Approach (Fromme P., ed) pp. 243–274, Wiley, Weinheim [Google Scholar]
- 19. Collins A. M., Liberton M., Jones H. D., Garcia O. F., Pakrasi H. B., Timlin J. A. (2012) Photosynthetic pigment localization and thylakoid membrane morphology are altered in Synechocystis 6803 phycobilisome mutants. Plant Physiol. 158, 1600–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. David L., Prado M., Arteni A. A., Elmlund D. A., Blankenship R. E., Adir N. (2014) Structural studies show energy transfer within stabilized phycobilisomes independent of the mode of rod-core assembly. Biochim. Biophys. Acta 1837, 385–395 [DOI] [PubMed] [Google Scholar]
- 21. Ficner R., Lobeck K., Schmidt G., Huber R. (1992) Isolation, crystallization, crystal structure analysis and refinement of B-phycoerythrin from the red alga Porphyridium sordidum at 2.2 A resolution. J. Mol. Biol. 228, 935–950 [DOI] [PubMed] [Google Scholar]
- 22. Kalisman N., Adams C. M., Levitt M. (2012) Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U.S.A. 109, 2884–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 25. Migneault I., Dartiguenave C., Bertrand M. J., Waldron K. C. (2004) Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. BioTechniques 37, 790–796, 798–802 [DOI] [PubMed] [Google Scholar]
- 26. Fournier C. T., Cherny J. J., Truncali K., Robbins-Pianka A., Lin M. S., Krizanc D., Weir M. P. (2012) Amino termini of many yeast proteins map to downstream start codons. J. Proteome Res. 11, 5712–5719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang H., Liu H., Niedzwiedzki D. M., Prado M., Jiang J., Gross M. L., Blankenship R. E. (2014) Molecular mechanism of photoactivation and structural location of the cyanobacterial orange carotenoid protein. Biochemistry 53, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu H., Freitas M. A. (2009) Automated diagnosis of LC-MS/MS performance. Bioinformatics 25, 1341–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi H., Nesvizhskii A. I. (2008) False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J. Proteome Res. 7, 47–50 [DOI] [PubMed] [Google Scholar]
- 30. Leitner A., Reischl R., Walzthoeni T., Herzog F., Bohn S., Forster F., Aebersold R. (2012) Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteomics 11, 10.1074/mcp.M111.014126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reuter W., Wiegand G., Huber R., Than M. E. (1999) Structural analysis at 2.2 A of orthorhombic crystals presents the asymmetry of the allophycocyanin-linker complex, AP.LC7.8, from phycobilisomes of Mastigocladus laminosus. Proc. Natl. Acad. Sci. U.S.A. 96, 1363–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tovchigrechko A., Vakser I. A. (2006) GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 34, W310–W314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mashiach E., Schneidman-Duhovny D., Andrusier N., Nussinov R., Wolfson H. J. (2008) FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 36, W229–W232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 35. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gantt E., Conti S. F. (1966) Granules associated with the chloroplast lamellae of Porphyridium cruentum. J. Cell Biol. 29, 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Edwards M. R., Gantt E. (1971) Phycobilisomes of the thermophilic blue-green alga Synechococcus lividus. J. Cell Biol. 50, 896–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tandeau de Marsac N. (2003) Phycobiliproteins and phycobilisomes: the early observations. Photosynth. Res. 76, 193–205 [DOI] [PubMed] [Google Scholar]
- 39. Bryant D. A., Guiglielmi G., Tandeau de Marsac N., Castets A., Cohen-Bazire G. (1979) The structure of cyanobacterial phycobilisomes: a model. Arch. Microbiol. 123, 113–127 [Google Scholar]
- 40. Glazer A. N., Lundell D. J., Yamanaka G., Williams R. C. (1983) The structure of a “simple” phycobilisome. Ann. Microbiol. (Paris) 134B, 159–180 [DOI] [PubMed] [Google Scholar]
- 41. Arteni A. A., Ajlani G., Boekema E. J. (2009) Structural organisation of phycobilisomes from Synechocystis sp. strain PCC6803 and their interaction with the membrane. Biochim. Biophys. Acta 1787, 272–279 [DOI] [PubMed] [Google Scholar]
- 42. Arteni A. A., Liu L. N., Aartsma T. J., Zhang Y. Z., Zhou B. C., Boekema E. J. (2008) Structure and organization of phycobilisomes on membranes of the red alga Porphyridium cruentum. Photosynth. Res. 95, 169–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holzwarth A. R., Bittersmann E., Reuter W., Wehrmeyer W. (1990) Studies on chromophore coupling in isolated phycobiliproteins. III. Picosecond excited state kinetics and time-resolved fluorescence spectra of different allophycocyanins from Mastigocladus laminosus. Biophys. J. 57, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adir N., Lerner N. (2003) The crystal structure of a novel unmethylated form of C-phycocyanin, a possible connector between cores and rods in pycobilisomes. J. Biol. Chem. 278, 25926–25932 [DOI] [PubMed] [Google Scholar]
- 45. Dong C., Tang A., Zhao J., Mullineaux C. W., Shen G., Bryant D. A. (2009) ApcD is necessary for efficient energy transfer from phycobilisomes to photosystem I and helps to prevent photoinhibition in the cyanobacterium Synechococcus sp. PCC 7002. Biochim. Biophys. Acta 1787, 1122–1128 [DOI] [PubMed] [Google Scholar]
- 46. Liu H., Zhang H., Niedzwiedzki D. M., Prado M., He G., Gross M. L., Blankenship R. E. (2013) Phycobilisomes supply excitations to both photosystems in a megacomplex in cyanobacteria. Science 342, 1104–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tian L., Gwizdala M., van Stokkum I. H., Koehorst R. B., Kirilovsky D., van Amerongen H. (2012) Picosecond kinetics of light harvesting and photoprotective quenching in wild-type and mutant phycobilisomes isolated from the cyanobacterium Synechocystis PCC 6803. Biophys. J. 102, 1692–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tian L., van Stokkum I. H., Koehorst R. B., Jongerius A., Kirilovsky D., van Amerongen H. (2011) Site, rate, and mechanism of photoprotective quenching in cyanobacteria. J. Am. Chem. Soc. 133, 18304–18311 [DOI] [PubMed] [Google Scholar]
- 49. Kondo K., Geng X. X., Katayama M., Ikeuchi M. (2005) Distinct roles of CpcG1 and CpcG2 in phycobilisome assembly in the cyanobacterium Synechocystis sp. PCC 6803. Photosynth. Res. 84, 269–273 [DOI] [PubMed] [Google Scholar]
- 50. Grossman A. R., Schaefer M. R., Chiang G. G., Collier J. L. (1993) The phycobilisome, a light-harvesting complex responsive to environmental conditions. Microbiol. Rev. 57, 725–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ren Y., Chi B., Melhem O., Wei K., Feng L., Li Y., Han X., Li D., Zhang Y., Wan J., Xu X., Yang M. (2013) Understanding the electronic energy transfer pathways in the trimeric and hexameric aggregation state of cyanobacteria phycocyanin within the framework of Forster theory. J. Comput. Chem. 34, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 52. Matamala A. R., Almonacid D. E., Figueroa M. F., Martinez-Oyanedel J., Bunster M. C. (2007) A semiempirical approach to the intra-phycocyanin and inter-phycocyanin fluorescence resonance energy-transfer pathways in phycobilisomes. J. Comp. Chem. 28, 1200–1207 [DOI] [PubMed] [Google Scholar]
- 53. Figueroa M., Martínez-Oyanedel J., Matamala A. R., Dagnino-Leone J., Mella C., Fritz R., Sepúlveda-Ugarte J., Bunster M. (2012) In silico model of an antenna of a phycobilisome and energy transfer rates determination by theoretical Forster approach. Protein Sci. 21, 1921–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sauer K., Scheer H. (1988) Excitation transfer in C-phycocyanin: Forster transfer rate and exciton calculations based on new crystal-structure data for C-phycocyanins from Agmenellum quadruplicatum and Mastigocladus laminosus. Biochim. Biophys. Acta 936, 157–170 [Google Scholar]
- 55. McGregor A., Klartag M., David L., Adir N. (2008) Allophycocyanin trimer stability and functionality are primarily due to polar enhanced hydrophobicity of the phycocyanobilin binding pocket. J. Mol. Biol. 384, 406–421 [DOI] [PubMed] [Google Scholar]
- 56. Womick J. M., Miller S. A., Moran A. M. (2010) Toward the origin of exciton electronic structure in phycobiliproteins. J. Chem. Phys. 133, 024507 [DOI] [PubMed] [Google Scholar]
- 57. Suter G. W., Holzwarth A. R. (1987) A kinetic model for the energy transfer in phycobilisomes. Biophys. J. 52, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Curutchet C., Kongsted J., Muñoz-Losa A., Hossein-Nejad H., Scholes G. D., Mennucci B. (2011) Photosynthetic light-harvesting is tuned by the heterogeneous polarizable environment of the protein. J. Am. Chem. Soc. 133, 3078–3084 [DOI] [PubMed] [Google Scholar]
- 59. Capuano V., Braux A. S., Tandeau de Marsac N., Houmard J. (1991) The “anchor polypeptide” of cyanobacterial phycobilisomes. Molecular characterization of the Synechococcus sp. PCC 6301 apce gene. J. Biol. Chem. 266, 7239–7247 [PubMed] [Google Scholar]
- 60. Gao X., Wei T. D., Zhang N., Xie B. B., Su H. N., Zhang X. Y., Chen X. L., Zhou B. C., Wang Z. X., Wu J. W., Zhang Y. Z. (2012) Molecular insights into the terminal energy acceptor in cyanobacterial phycobilisome. Mol. Microbiol. 85, 907–915 [DOI] [PubMed] [Google Scholar]
- 61. Mullineaux C. W. (2008) Phycobilisome-reaction centre interaction in cyanobacteria. Photosynth. Res. 95, 175–182 [DOI] [PubMed] [Google Scholar]
- 62. Krogmann D. W., Pérez-Gómez B., Gutiérrez-Cirlos E. B., Chagolla-López A., González de la Vara L., Gómez-Lojero C. (2007) The presence of multidomain linkers determines the bundle-shape structure of the phycobilisome of the cyanobacterium Gloeobacter violaceus PCC 7421. Photosynth. Res. 93, 27–43 [DOI] [PubMed] [Google Scholar]
- 63. Gao X., Zhang N., Wei T. D., Su H. N., Xie B. B., Dong C. C., Zhang X. Y., Chen X. L., Zhou B. C., Wang Z. X., Wu J. W., Zhang Y. Z. (2011) Crystal structure of the N-terminal domain of linker L(R) and the assembly of cyanobacterial phycobilisome rods. Mol. Microbiol. 82, 698–705 [DOI] [PubMed] [Google Scholar]
- 64. Jallet D., Gwizdala M., Kirilovsky D. (2012) ApcD, ApcF and ApcE are not required for the orange carotenoid protein related phycobilisome fluorescence quenching in the cyanobacterium Synechocystis PCC 6803. Biochim. Biophys. Acta 1817, 1418–1427 [DOI] [PubMed] [Google Scholar]