

Background: Guinea pig serum and liver contain an enzyme with l-asparaginase activity.
Results: H0W0T5_CAVPO (gpASNase1) displays a low micromolar Km with Asn. Structures of apo and ASP complex are presented.
Conclusion: gpASNase1is the likely identity of a guinea pig l-asparaginase endowed with anticancer properties.
Significance: The high sequence identity to the human enzymes and its lack of l-glutaminase activity make gpASNase1 a potential replacement for the bacterial enzymes.
Keywords: Anticancer Drug, Cancer, Cancer Therapy, Crystallography, Enzyme Kinetics, Asparaginase
Abstract
The initial observation that guinea pig serum kills lymphoma cells marks the serendipitous discovery of a new class of anti-cancer agents. The serum cell killing factor was shown to be an enzyme with l-asparaginase (ASNase) activity. As a direct result of this observation, several bacterial l-asparaginases were developed and are currently approved by the Food and Drug Administration for the treatment of the subset of hematological malignancies that are dependent on the extracellular pool of the amino acid asparagine. As drugs, these enzymes act to hydrolyze asparagine to aspartate, thereby starving the cancer cells of this amino acid. Prior to the work presented here, the precise identity of this guinea pig enzyme has not been reported in the peer-reviewed literature. We discovered that the guinea pig enzyme annotated as H0W0T5_CAVPO, which we refer to as gpASNase1, has the required low Km property consistent with that possessed by the cell-killing guinea pig serum enzyme. Elucidation of the ligand-free and aspartate complex gpASNase1 crystal structures allows a direct comparison with the bacterial enzymes and serves to explain the lack of l-glutaminase activity in the guinea pig enzyme. The structures were also used to generate a homology model for the human homolog hASNase1 and to help explain its vastly different kinetic properties compared with gpASNase1, despite a 70% sequence identity. Given that the bacterial enzymes frequently present immunogenic and other toxic side effects, this work suggests that gpASNase1 could be a promising alternative to these bacterial enzymes.
Introduction
In a seminal publication from 1953, John G. Kidd reported the unexpected observation that guinea pig serum has the ability to inhibit the growth of subcutaneous lymphomas transplanted into mice (1). It took a further 10 years before the serum ingredient responsible for this effect was revealed to be an enzyme with l-asparaginase activity (2). l-Asparaginases catalyze the hydrolysis of the amino acid l-asparagine to l-aspartate and ammonia. This discovery ushered in a new concept in cancer chemotherapy. Certain cancers, such as acute lymphoblastic leukemia (ALL),2 do not express the enzyme asparagine synthetase (3, 4). As a result, the Asn present in the blood becomes the sole source of this amino acid for ALL cells. Administration of l-asparaginase to ALL patients acts to deplete the blood of Asn, depriving the leukemic cells of this amino acid, ultimately leading to cell death.
For an l-asparaginase to be efficient at hydrolyzing the extracellular pool of Asn (∼50 μm Asn concentration in human blood (5)), its Km value must be in the low micromolar range. Although the observations with the guinea pig serum revealed the anti-cancer potential of l-asparaginases (in fact, the serum was used to treat an ALL patient, and a response was observed (6)), obtaining the particular guinea pig l-asparaginase for clinical use proved difficult, so other sources were explored. Escherichia coli has several l-asparaginases, but only one of them, the type II l-asparaginase, denoted here as EcII (gene name ansB), has the prerequisite low Asn Km property (Km of 11.5–15 μm (7, 8)). Interestingly, the type I E. coli l-asparaginase, denoted as EcI (gene name ansA) is structurally similar to EcII but has a Km in the millimolar range (9). EcII was approved by the Food and Drug Administration for the treatment of certain blood cancers such as ALL in 1978 under the brand name Elspar. Immunogenic and other complications with this treatment promoted the development of a pegylated version (approved by the Food and Drug Administration in 2006 under the name Oncospar). More recently, in 2011 the l-asparaginase from the bacterium Erwinia chrysanthemi (denoted here as Erw), which shows a different immunogenic profile compared with EcII, was also approved by the Food and Drug Administration (brand name Erwinaze). Asparec (pegcrisantaspase) is a pegylated version of Erw currently being developed by Jazz Pharmaceuticals together with Alize Pharma (10).
Although these newer bacterial l-asparaginase preparations seem to be superior to the original naked EcII, side effects persist. A reduced but still present immune intolerance remains because of the foreign origin of these drugs. Furthermore, the enzyme can still be cleared from the blood and an immune reaction elicited because of antibody formation against PEG (11–13). This suggests that a more human-like enzyme, which would be less immunogenic, could be a superior replacement (14–16).
Additionally, the bacterial l-asparaginases exhibit a l-glutaminase side-activity (i.e. the hydrolysis of l-glutamine to l-glutamate and ammonia). This side reaction could be behind the liver, pancreas, and blood coagulation side effects seen in patients treated with the bacterial l-asparaginases. It was recently reported that the l-glutaminase activity of l-asparaginases is not required for anticancer activity against asparagine synthetase-deficient cells (17). These factors prompted us seek out the guinea pig l-asparaginase that is behind the original observations made by Kidd.
A manuscript from 1966 reported that the guinea pig serum l-asparaginase has a molecular weight of ∼138 kDa, as measured by equilibrium sedimentation and gel filtration (18). To our knowledge, this information fails to identify the precise guinea pig serum l-asparaginase, because this value does not fit the monomeric size of any of the four guinea pig enzymes that have been associated with l-asparaginase activity (ranging in size from 34 to 61 kDa) or the expected size of their oligomeric form. The Km value of the serum guinea pig enzyme was reported to be 72 μm (19). This value is consistent with an enzyme that can effectively deplete the blood of Asn. Interestingly, it was reported that the guinea pig enzyme purified from serum lacked l-glutaminase activity (20). This observation suggests that, if indeed some of the side effects with the bacterial enzymes are due to this co-activity, the guinea pig enzyme will be devoid of this problem.
As mentioned above, a required property of the guinea pig serum l-asparaginase is a low micromolar Km value with the substrate Asn. Previously we reported that the guinea pig l-asparaginase annotated in UniProt as H0VQC8_CAVPO (referred to by us a gpASNase3 because of its homology to the E. coli type III enzyme) has a Km value of 2.24 mm with Asn (21). This property eliminates this enzyme as a candidate for being the guinea pig serum l-asparaginase.
Here we report that the guinea pig enzyme annotated as H0W0T5_CAVPO does indeed possess the required micromolar Km property with Asn. This is rather surprising because it is highly similar to its human homolog, called 60-kDa lysophospholipase and annotated as LPP60_HUMAN (69.8% amino acid identity, 88.6% homology), yet it was recently reported that this enzyme has a Km value of 11.5 mm with Asn (22). Both the human and guinea pig enzymes share more sequence homology with the EcI enzyme compared with the EcII enzyme, promoting us to refer to these enzymes as hASNase1 and gpASNase1, respectively. However, whereas hASNase1 does indeed share the high Km property with EcI, gpASNase1, as reported here, shares the low Km property with EcII.
In studying gpASNase1, one of our goals is to understand what makes this particular enzyme different from most of its mammalian homologs in having a low Km for Asn. To gain an insight into this question, we solved the crystal structure of gpASNase1 in its unliganded form (referred to as apo) and in complex with the product aspartate (referred to as Asp complex). Analysis of these gpASNase1 structures and their comparison with the known bacterial l-asparaginase structures provide a molecular understanding for the lack of l-glutaminase activity in mammalian l-asparaginases. Additionally, comparison to the hASNase1 homology model we generated provides a clue for the highly different Km values of otherwise very similar enzymes.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of Bacterial l-Asparaginases
A codon-optimized synthetic gene corresponding to the amino acid sequence of Erw (UniProt entry P06608) without the first 21-amino acid signaling peptide was synthetized by Genscript with NdeI and BamHI sites at the 5′ and 3′ ends, respectively. The open reading frame of EcII (UniProt entry P00805) without the first 22-amino acid signaling peptide was amplified via PCR using primers incorporated with NdeI and BamHI sites at the 5′ and 3′ ends, respectively. The PCR product was digested with NdeI and BamHI-HF, gel-purified, and ultimately ligated into a His6-SUMO-pET14b vector (where the His6 tag is followed by the yeast protein SUMO (small ubiquitin modifier) Smt3p tag of 101 residues) using Instant Sticky End DNA ligase (New England Biolabs). The ligation mixture was transformed into XL1 blue DH5α E. coli cells. Plasmids with correct insert genes (verified by sequencing) were then transformed into E. coli BL21(DE3) C41 cells for expression. The cells were grown at 37 °C in 2YT medium until reaching an optical density (at 600 nm) of 0.6–0.8. Expression of the targeted gene was induced using 0.3 mm isopropyl β-d-1-thiogalactopyranoside, and the temperature was reduced to 18 °C overnight. After lysis by sonication, the enzyme was purified on a nickel affinity column (GE Healthcare). The N-terminal His6-SUMO tag was then cleaved in solution by SUMO protease and loaded back through a nickel affinity column to separate the tag. The flow-through fraction containing purified enzyme in 25 mm Tris, pH 8.5, 100 mm NaCl was concentrated to 20–50 mg/ml, aliquoted, flash frozen, and stored at −80 °C.
Cloning, Expression, and Purification of Mammalian l-Asparaginases
Cavia porcellus (guinea pig) asparaginase UniProt entry H0W0T5 is annotated as an uncharacterized protein consisting of 565 amino acid residues (see Fig. 1A). For hASNase1, the sequence from UniProt entry Q86U10 was used. For our studies, we ordered synthetic genes (Genscript) codon-optimized for expression in E. coli. The synthetic genes for gpASNase1 and hASNase1 were transferred to a His6-TEV-pET14b expression vector using the NdeI and BamHI restriction sites at the 5′ and 3′ ends, respectively. The N-terminal hexahistidine tag followed by a TEV protease cleavage site has the following sequence: MGSSHHHHHHSSGGNENLYFQGH. The His6-TEV-gpASNase1(1–565)-pET14b and His6-TEV-hASNase1(1–573)-pET14b plasmids were transformed into BL21 (DE3) C41 E. coli cells for expression. Starter cultures of the plasmids were grown overnight and inoculated into 6 or 12 liters, respectively, of 2YT medium at a ratio of 1:100. The cells were grown at 37 °C to an optical density of 0.6–0.8, and overexpression was induced with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 0.1–0.15 mm. Growth continued at 18 °C overnight, at which point the cells were harvested by centrifugation, and the pellets were frozen at −20 °C. For purification of gpASNase1, the cell pellets were thawed, resuspended in lysis buffer (25 mm Tris-HCl, pH 7.5, 400 mm KCl, 10 mm MgCl2, 10 mm imidazole, 10% glycerol, 1% Triton X-100, 1 mm PMSF), and disrupted by sonication. The lysate was cleared by ultracentrifugation, and the supernatant was loaded onto an equilibrated 5-ml HisTrap HP nickel-Sepharose column (GE Healthcare), washed with buffer containing 25 mm Tris-HCl, pH 7.5, 200 mm KCl, 10 mm MgCl2, 30 mm imidazole followed by a wash with buffer containing 25 mm Tris-HCl, pH 7.5, 200 mm KCl, 10 mm MgCl2, 50 mm imidazole. The final wash step was performed using buffer containing 20 mm Tris-HCl, pH 7.5 before elution with buffer containing 20 mm Tris-HCl, pH 7.5, 200 mm imidazole yielding 237 mg of His6-TEV-gpASNase1(1–565). The protein was concentrated to 18 mg/ml, and ∼100 mg were loaded onto a Superdex 200 Hi Load 26/60 gel filtration column (GE Healthcare) pre-equilibrated with 20 mm Tris-HCl, pH 7.5, 100 mm KCl, 2 mm DTT. The protein was eluted in one peak, and the fractions were pooled (yielding ∼80 mg) and concentrated to 20 mg/ml and frozen at −80 °C.
FIGURE 1.

Comparison of mammalian gpASNase1 and hASNase1 with bacterial type I and II l-asparaginases. A, schematic of the gpASNase1, hASNase1, EcI, EcII, and Erw domain organization. The mammalian enzymes contain, in addition to the N-terminal l-asparaginase domain (yellow), a C-terminal domain of yet unknown function that is predicted to contain several ankyrin repeats. B, SDS-PAGE of purified EcII, Erw, gpASNase1, and hASNase1. First lane, marker; second lane, E. coli type II Δ22N (molecular mass = 34.9 kDa); third lane, Erwinia Δ21N (molecular mass = 35.3 kDa); fourth lane, gpASNase1 (molecular mass = 60.9 kDa); fifth lane, His-TEV hASNase1 (molecular mass = 63.5 kDa). C, structure-based sequence alignment of the l-asparaginase domains of gpASNase1, hASNase1, EcI, EcII, and Erw. Highlighted in red are active site residues, and in blue are active site carboxylic acid residues that originate from different regions but all interact with the substrate α-amino group. Arrowheads denote the functionally conserved tyrosine present in the C-terminal region of the mammalian enzymes (Tyr308 in gpASNase1) and in the N-terminal region of the bacterial enzymes (Tyr25 in EcII). EcI has a tyrosine in both the N- and C-terminal regions, and studies have yet to delineate which of these fulfills the analogous function. The secondary structure elements shown above the sequence (yellow arrows, β-strands; blue ribbons, helices) correspond to the structure of gpASNase1.
For His6-TEV-hASNase1(1–573)-pET14b, the same protocol was followed except that all buffers contained 20% glycerol. The lysis buffer also only contained 200 mm KCl, and the wash buffers used for nickel affinity chromatography contained 25 mm Tris-HCl, pH 7.5, 200 mm KCl, 10 mm MgCl2, and either 25, 50, or 75 mm imidazole. The protein was washed on the column overnight at 4 °C with wash buffer without imidazole. The next morning the column was washed with 20 mm Tris-HCl, pH 7.5, before elution with buffer containing 20 mm Tris-HCl, pH 7.5, and either 200 or 400 mm imidazole, yielding a total of 25 mg. The elution fractions were combined, diluted with 20 mm Tris, pH 7.5, and 2 mm DTT was added before the protein was loaded onto a Q Sepharose column. The protein was eluted over a gradient created by combining 20 mm Tris, pH 7.5, 2 mm DTT with buffer containing 20 mm Tris, pH 7.5, 2 mm DTT, 1 m KCl. The peak fractions were pooled (∼10 mg total), and the kinetic activity was immediately measured. After ∼8 h on ice, the protein began to rapidly lose activity. The activity of protein frozen at −80 °C overnight was also lower than that of fresh protein.
Kinetic Assay
The catalytic activity of the l-asparaginases was determined using a continuous spectroscopic enzyme-coupled assay (23, 24), which measures the production of l-aspartate through the 1:1 oxidation of reduced NADH. The conversion of NADH to NAD+ was measured spectrophotometrically as a decrease in absorbance at 340 nm at 37 °C. All measurements were taken in triplicate. Glutamic-oxalacetic transaminase (Sigma G2751) and malic dehydrogenase (Sigma M2634) were helper enzymes for the coupled enzymatic reactions; ∼5 and ∼1 unit were used, respectively. Our kinetic assay buffer was composed of 100 mm Tris-HCl, pH 7.5, 100 mm KCl, 200 μm α-ketoglutarate, and 200 μm NADH. The l-Asn substrate stock (Sigma-Aldrich A93003) was made fresh in 50 mm Tris-HCl, pH 7.5. Rates were fit to the Michaelis-Menten equation using SigmaPlot (Systat Software Inc). For hASNase1, the Km curve did not follow classic Michaelis-Menten kinetics. Its sigmoidal curve was fit using the Hill equation, yielding the Hill coefficient, n, which indicates cooperativity.
As mentioned above, for the bacterial enzymes, the His6-SUMO tag was removed. For the mammalian enzymes, we verified that the His6-TEV tag has no influence on the kinetics.
Crystallization of gpASNase1
A variety of crystallization screens (Qiagen) were applied to purified His6-TEV-gpASNase1(1–565) using 1 μl of 7.5–10 mg/ml of protein combined with 1 μl of reservoir solution in the sitting drop format in 96-well plates. Using this vapor diffusion method at 20 °C, several hits were obtained with buffers ranging from pH 6.0–8.5, various salts at 0.1–0.2 m, and different concentrations of PEG (400–20,000). Crystals grew after 2 weeks in condition 22 from the Protein Complex Suite (0.1 m HEPES, pH 7.0, 15% PEG 4000). For the Asp complex crystals, setup included co-crystallization with 10 mm l-aspartic acid sodium salt monohydrate (Sigma A6683) in the protein solution. For cryoprotection of the crystals, 30% ethylene glycol was added to the reservoir solution. 25 mm l-aspartic acid sodium salt monohydrate was additionally added to the cryoprotectant for the Asp-bound crystals.
Data Collection and Structure Solution of gpASase1 apo and Asp Complex
Diffraction data for gpASNase1 apo and Asp complex were collected at the Advanced Photon Source located at Argonne National Laboratory using the LS-CAT ID-G Beamline (see Table 2 for data collection and refinement statistics). The data were processed using XDS (25). The apo structure was solved by molecular replacement (Molrep (26) CCP4) using EcI as the starting model (PDB code 2HIM). Several programs were used for refinement, including Refmac5 (27), Phenix (28, 29), and the PDB_REDO web server (30). The models were built using Coot (31). Figures of the structures were made using MacPyMOL (PyMOL Molecular Graphics System, version 1.4; Schrödinger, LLC).
TABLE 2.
Data collection and refinement statistics
| Structure | apo | ASP complex |
|---|---|---|
| PDB codes | 4R8K | 4R8L |
| Data collection statistics | ||
| X-ray source and detector | LS-CAT ID-G | LS-CAT ID-G |
| MARCCD 300 | MARCCD 300 | |
| Wavelength (Å) | 0.97857 | 0.97857 |
| Temperature (K) | 100 | 100 |
| Resolution (Å)a | 2.20 (2.34-2.20) | 2.41 (2.55-2.41) |
| Number of reflections | ||
| Observed | 930,263 (136,419) | 262,581 (40,379) |
| Unique | 146,207 (23,169) | 55,164 (8,712) |
| Completeness (%) | 99.7 (98.5) | 92.6 (91.8) |
| Rsym (%) | 10.0 (61.0) | 10.4 (75.0) |
| Average I/σ(I) | 13.71 (2.78) | 12.24 (2.10) |
| Space group | P21 | I222 |
| Unit cell (Å): a, b, c | 98.8, 123.7, 121.0 | 123.2, 156.0, 158.8 |
| β (°) | 92.4 | |
| Wilson B-factors (Å2) | 32.5 | 49.2 |
| Refinement statistics | ||
| Refinement program | REFMAC5, Phenix 1.9, PDB_REDO server | REFMAC5, Phenix 1.9, PDB_REDO server |
| Rcryst (%) | 20.7 | 20.6 |
| Rfree (%) | 23.8 | 23.6 |
| Resolution range (Å) | 30.0–2.2 | 30.0–2.4 |
| Protein molecules per a.u. | 8 | 4 |
| Number of atoms | ||
| Protein | ||
| protA, protB | 2,716, 2,667 | 2,717, 2,724 |
| protC, protD | 2,724, 2,717 | 2,717, 2,724 |
| protE, protF, | 2,703, 2,717 | |
| protG, protH | 2,717, 2,710 | |
| Water molecules | 892 | 210 |
| HEPES molecules | 8 | |
| Asp molecules | 4 | |
| Root mean square deviation from ideal | ||
| Bond length (Å) | 0.010 | 0.010 |
| Bond angles (°) | 1.339 | 1.368 |
| Average B-factors (Å2) | ||
| Protein | ||
| protA, protB | 36.0, 40.7 | 46.6, 46.4 |
| protC, protD | 32.7, 33.9 | 42.7, 47.4 |
| protE, protF, | 41.5, 40.5 | |
| protG, protH | 36.9, 37.5 | |
| Water molecules | 32.4 | 32.1 |
| HEPES molecules | ||
| protA, protB | 33.7, 38.8 | |
| protC, protD | 35.6, 38.7 | |
| protE, protF, | 45.4, 32.5 | |
| protG, protH | 43.0, 33.1 | |
| Asp molecules | ||
| protA, protB | 52.7, 55.1 | |
| ProtC, protD | 51.8, 34.0 | |
| Ramachandran plot (%) | ||
| Most favored regions | 98.6 | 97.2 |
| Additionally allowed regions | 1.1 | 2.8 |
| Outlier regions | 0.2 | 0.0 |
a Last shell in parentheses.
Homology Modeling of hASNase1
Two homology models of hASNase1 were generated using the automated modeling mode of Swiss-Model (32, 33). Protomers A and D of the Asp-bound complex structure of gpASNase1 (PDB code 4R8L) were used as templates for the protomer containing the C-terminal and N-terminal regions of the active site, respectively, given that these protomers of gpASNase1 had the best electron density in these areas.
RESULTS
Expression, Purification, and Kinetic Characterization of Mammalian and Bacterial l-Asparaginases
EcI (cytoplasmic, high Km), EcII (periplasmic, low Km), and the Erwinia homolog Erw (periplasmic, low Km) all contain a similar l-asparaginase domain of ∼340 residues. In contrast, the mammalian homologs, including gpASNase1 and hASNase1, have an additional C-terminal domain of ∼200 residues that is predicted to contain several ankyrin repeats (Fig. 1A). For this study, we expressed and purified the bacterial EcII and Erw as well as the mammalian gpASNase1 and hASNase1 enzymes (Fig. 1B). A structure-based sequence alignment of these l-asparaginases, including EcI, is presented in Fig. 1C.
The bacterial l-asparaginases, because of their clinical importance, are well characterized, and the kinetic properties that we measure for these enzymes (Table 1) are consistent with previous reports (8). The kinetic properties of the human homolog, hASNase1, have been recently reported, albeit for the C-terminal truncated version (22). Our determined steady state parameters for full-length hASNase1 reveal a higher kcat (14.4 versus 6.7 s−1) and lower Km (3.0 mm versus 11.5 mm) relative to the C-terminal truncated version (Table 1). These differences could be due to the lower stability of truncated hASNase1, which is why we elected to study the full-length version. The Km value we determined for hASNase1 is still ∼60–200 times higher than that measured for the clinically used EcII and Erw enzymes. Therefore, this human l-asparaginase is not a suitable replacement for the bacterial enzymes.
TABLE 1.
Steady state kinetic parameters for examined bacterial and mammalian l-asparaginases
| Enzyme name | No. of residues | kcat | Km | kcat/Km | Hill |
|---|---|---|---|---|---|
| s−1 | μm | s−1 μm−1 | |||
| EcIIa,b | 326 | 48.9 ± 0.6 | 14.9 ± 1.3 | 4.4 | NAc |
| Erwb,d | 327 | 207.5 ± 3.6 | 47.5 ± 3.5 | 4.4 | NA |
| gpASNase1e | 565 | 38.6 ± 1.4 | 57.7 ± 6.4 | 0.8 | NA |
| hASNase1f | 573 | 14.4 ± 0.4 | 2,960 ± 131g | 0.005 | 2.5 ± 0.2 |
a E. coli type II Δ22N; also known as l-asparaginase II; gene name ansB.
b The N-terminal truncation results in an enzyme lacking the periplasmic signal peptide.
c NA, not applicable.
d Erwinia Δ21N.
e UniProt entry H0W0T5_CAVPO, gene name ASPG.
f UniProt entry LPP60_HUMAN; also known as 60-kDa lysophospholipase, gene name ASPG.
g For enzymes such as hASNase1 that do not follow Michaelis-Menten kinetics, this term is called [S]0.5 or K0.5.
We next examined the kinetic properties of gpASNase1, which as mentioned before, has 69.8% sequence identify with hASNase1 when comparing the full-length sequences, and an even higher identity of 83.4% when limiting the comparison to the more relevant l-asparaginase domain. Notably, all predicted active site residues between hASNase1 and gpASNase1 are conserved (Fig. 1C), suggesting that these homologs will have similar properties. However, gpASNase1 has a ∼2-fold faster rate (38.6 s−1) and ∼60-fold lower Km (57.7 μm) as seen in Table 1. Noteworthy, the Km of gpASNase1 is very similar to that of Erw (47.5 μm). Because it is this property that would most strongly govern the ability of the enzyme to deplete the blood Asn level, this low Km guinea pig enzyme could likely act as a suitable replacement for the bacterial enzymes in the clinic.
Quaternary Structure of the l-Asparaginase Domain of gpASNase1
Despite using the full-length gpASNase1 enzyme in the crystallization setups, when we solved the structure, we could only detect density for the l-asparaginase domain (Fig. 1A). Apparently proteolysis occurring during the 2 weeks required for crystal growth released the catalytic domain, and it is only this that could form well ordered crystals. Using the protease specificity prediction server, PROSPER, there were two predicted cleavage sites that corresponded to the truncated gpASNase1 we observed in the crystal structure (∼8–362). Although our results are consistent with the first cleavage site (residue 364), it is also possible that the residues were disordered up to the second cleavage site (residue 372), and no electron density was observed (34). Consistent with this interpretation is the fact that upon addition of EDTA to inhibit metalloproteases, no crystals were obtained.
The apo l-asparaginase domain of gpASNase1 (residues ∼8–361) was solved at 2.20 Å resolution and refined to Rwork/Rfree of 20.7%/23.8% (Table 2). To obtain the Asp complex, gpASNase1 was co-crystallized with 10 mm Asp. The Asp complex structure was solved at 2.41 Å resolution and refined to Rwork/Rfree of 20.6%/23.6% (Table 2). Because ultimately it is the l-asparaginase domain that is the focus of this report, unless noted otherwise, we retain the nomenclature gpASNase1 and hASNase1 when referring to this domain and add the prefix full-length when discussing that version.
The related bacterial l-asparaginases have all been shown to be tetrameric—more specifically, a dimer of dimers. In our hands, full-length gpASNase1 elutes as a tetramer from a gel filtration column. Consistent with this, an analytical ultracentrifugation sedimentation equilibrium experiment indicates a tetrameric state (data not shown). gpASNase1 also forms tetramers in the crystals, indicating that the C-terminal ankyrin repeats do not influence the oligomeric state. Like the bacterial enzymes, gpASNase1 is a dimer of dimers (Fig. 2, A and B). The tight dimer interface has an area of ∼1,900 Å2, whereas that forming the weak dimer interface has ∼1,100 Å2 (calculated using PISA (35)).
FIGURE 2.
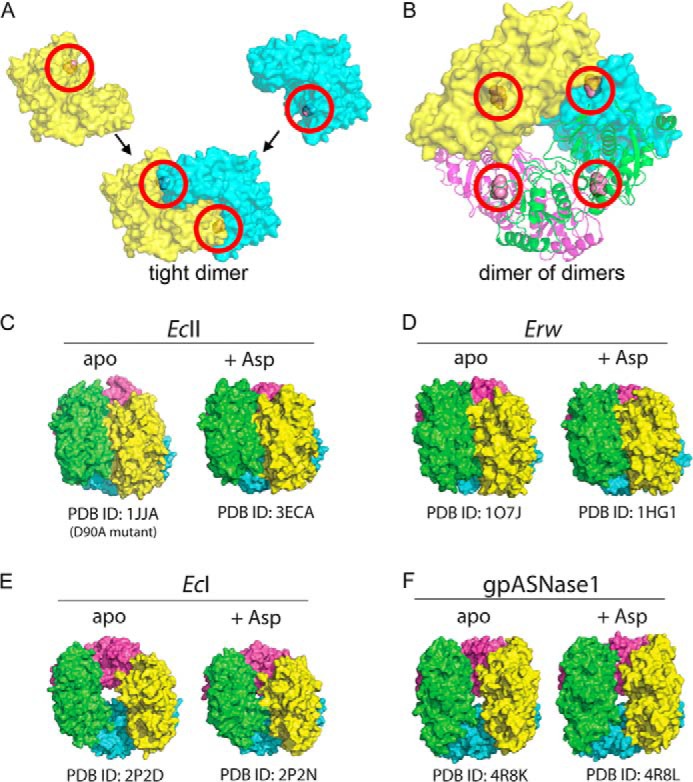
Quaternary structure of gpASNase1 and comparison to bacterial enzymes. l-Asparaginases of this family build a tetramer that is composed of a dimer of dimers; two protomers build a tight dimer, and two tight dimers assemble into the tetramer. In this figure, each protomer in the tetramer is colored differently. A, the two protomers (depicted as yellow and cyan surface representation) that build the gpASNase1 tight dimer assemble a complete active site (circled in red); the Asp bound at the active site is shown as a spheres object. B, two tight dimers come together to form the tetramer. One tight dimer is shown as a surface representation, and the other tight dimer is shown as a ribbon representation. Active sites are circled in red. C, the quaternary arrangement in EcII is unchanged upon binding of the product of the reaction Asp. D, likewise, the quaternary arrangement in Erw is unchanged upon Asp binding. E, in contrast, the quaternary arrangement in EcI transitions from a donut shape to a more compact structure upon binding Asp. F, gpASNase1 is similar to the high Km EcI by having a donut shape in the absence of ligand. Interestingly, it maintains this structure in the presence of Asp. In not changing structure upon Asp binding, gpASNase1 is similar to the low Km l-asparaginases EcII and Erw.
The low Km bacterial l-asparaginases, EcII and Erw, display the same compact tetrameric arrangement regardless of the absence or presence of ligand (Fig. 2, C and D). In contrast, the high Km EcI enzyme adopts a donut shape in the absence of ligand that changes to a more compact form in the presence of ligand (Fig. 2E). Interestingly, gpASNase1 also adopts a donut shape in the absence of ligand, but unlike EcI, it retains the open donut shape in the presence of ligand (Fig. 2F). In other words, just as observed for the low Km bacterial enzymes, binding of ligand to gpASNase1 does not induce a profound quaternary conformational change (for detailed substrate-induced conformational changes see below). This could be due to the fact that EcI is an allosteric enzyme requiring a second molecule of Asn to bind to a site distant from the active site (9). gpASNase1 follows classic Michaelis-Menten kinetics, and no cooperativity was observed. It is tempting to speculate that l-asparaginases that do not require a conformational change for ligand binding are characterized by a low Km (e.g. EcII, Erw, gpASNase1), whereas those that do require a conformational change for ligand binding, such as the EcI, are characterized by high Km.
Topology of a gpASNase1 Protomer
The four protomers that make up the gpASNase1 tetramer adopt a relatively identical structure, with an root mean square deviation of 0.13–0.27 Å (note, the asymmetric unit of the Asp complex contained four protomers that built a complete tetramer). A ribbon diagram of a gpASNase1 protomer from the Asp complex is shown in Fig. 3. Protomers from the EcI and EcII enzymes are also shown for comparison. This family of l-asparaginases is composed of an N-terminal and a C-terminal domain. The active site is located in the N-terminal domain, indicated by the pink space-filling object representing the bound product of the reaction, Asp (EcI has an additional allosteric binding site, indicated by the orange space-filling object).
FIGURE 3.
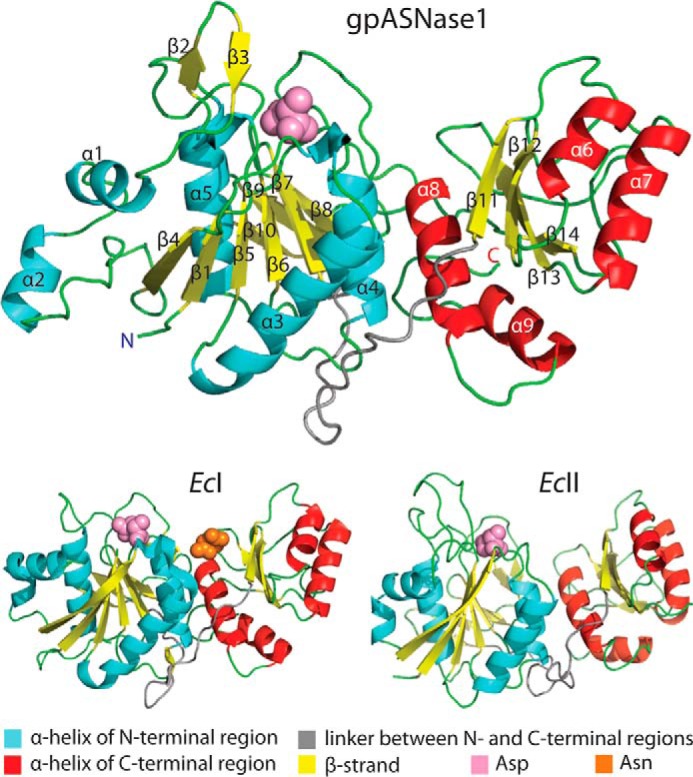
Protomer structure comparison. Cartoon diagram of gpASNase1 (top), EcI (bottom left), and EcII (bottom right). The two-domain fold of the gpASNase1 l-asparaginase domain is very similar to that of the E. coli enzymes. Helices in the N- and C-terminal domains are colored cyan and red, respectively. Strands are colored yellow, loops are in green, and the linker between the N- and C-terminal domains is in gray. Although gpASNase1 has a higher sequence identity with EcI (43.5%) versus EcII (26.4%), its kinetic properties are more similar to that of EcII. Labels of secondary structure elements correspond to those in the sequence alignment, Fig. 1C. The active site bound Asp molecule present in all structures is depicted as pink spheres. The allosteric Asn molecule present in the EcI structure is depicted as orange spheres.
Substrate-induced Conformational Change in gpASNase1
Although binding of Asp does not cause a change in the quaternary arrangement, it does induce a major tertiary structural change that is localized to two regions. These two regions, one located at the N terminus (residues Gly21–Gly34), and one at the C terminus (residues Thr305–Asn316), approach each other at the interface between the two protomers that build the tight dimer (Fig. 4A). The labile N-terminal region, which is near the Asp binding site (Asp depicted in pink), is built from two anti-parallel β-strands connected by a short loop. This region tilts toward the neighboring protomer as a result of Asp binding.
FIGURE 4.

Comparison of gpASNase1 with and without bound product Asp. A, overlay of the apo and Asp complex gpASNase1 structures. The overall quaternary structure is unchanged upon Asp binding (left panel). However, two regions, one N-terminal and one C-terminal, undergo a pronounced change in conformation in response to Asp binding (middle and right panels). B, surface representation of gpASNase1 in the apo (left panel, protomers in yellow and light blue) and Asp-bound state (right panel, protomers in orange and cyan). The Asp molecule from the complex structure is modeled onto the apo structure to demonstrate how in the absence of Asp the active site is exposed. In contrast, the two regions close like lids over the bound reaction product. C, close-up view of the active site of the apo and Asp-bound gpASNase1 structures. Asp has been modeled into the Fo − Fc difference density map (green) contoured at 3 σ. Most of the active site residues are in the same position. However, Tyr308′ (located in the C-terminal region on the neighboring protomer) dramatically changes orientation upon Asp binding. The ligand-bound conformation brings Tyr308′ in close proximity to the conserved catalytic residue Thr19. Asp84 also rotates to coordinate the Asp α-amino group. D, schematic representation of the interactions between Asp (red) and active site residues (black). The dashed lines indicate potential interactions, with distances in Å.
Even more dramatic is the change to the C-terminal region, where a loop that contains Tyr308′ becomes more ordered, and Tyr308′ flips from an inside pointing conformation to one pointing at the adjacent protomer (the prime is used to denote a residue from an adjacent protomer). The stabilization and change in conformation of the C-terminal region acts as a lid closing on the bound Asp, largely confining it to the active site (Fig. 4B).
A close-up of the Asp bound at the active site, including an overlay with the apo-structure, is shown in Fig. 4C (see Fig. 4D for a schematic of gpASNase1-Asp interactions). Most active site residues maintain the same position between the apo and Asp complex structure. The most significant changes are in the conformation of Asp84 and the previously mentioned Tyr308′. Asp84 preserves its main chain position between the two states but rotates its side chain to interact with the amino group of Asp. The change in position of Tyr308′ is more dramatic, adopting a totally different position in the Asp complex structure. This new position brings Tyr308′ in close proximity to Thr19. The homologous threonine in the EcII l-asparaginase has previously been implicated in the reaction mechanism, forming a covalent intermediate with the substrate Asn (36). The potential role of this tyrosine in the hydrolysis reaction is discussed below.
DISCUSSION
Structural Comparison of gpASNase1 to EcII and Erw
By its nature of having a micromolar Km with Asn, gpASNase1 is more similar to EcII and Erw than to hASNase1 (Table 1). To understand the factors that make gpASNase1 have this similarity to the low Km bacterial enzymes, we overlaid our gpASNase1-Asp complex structure with the EcII-Asp complex structure (PDB code 3ECA) (37) and the Erw succinate complex structure (PDB code 1HG0) (38) (Fig. 5A). The rationale for choosing this particular Erw structure is the lower number of gaps in the model, especially in the N-terminal region, which is important for the reaction.
FIGURE 5.
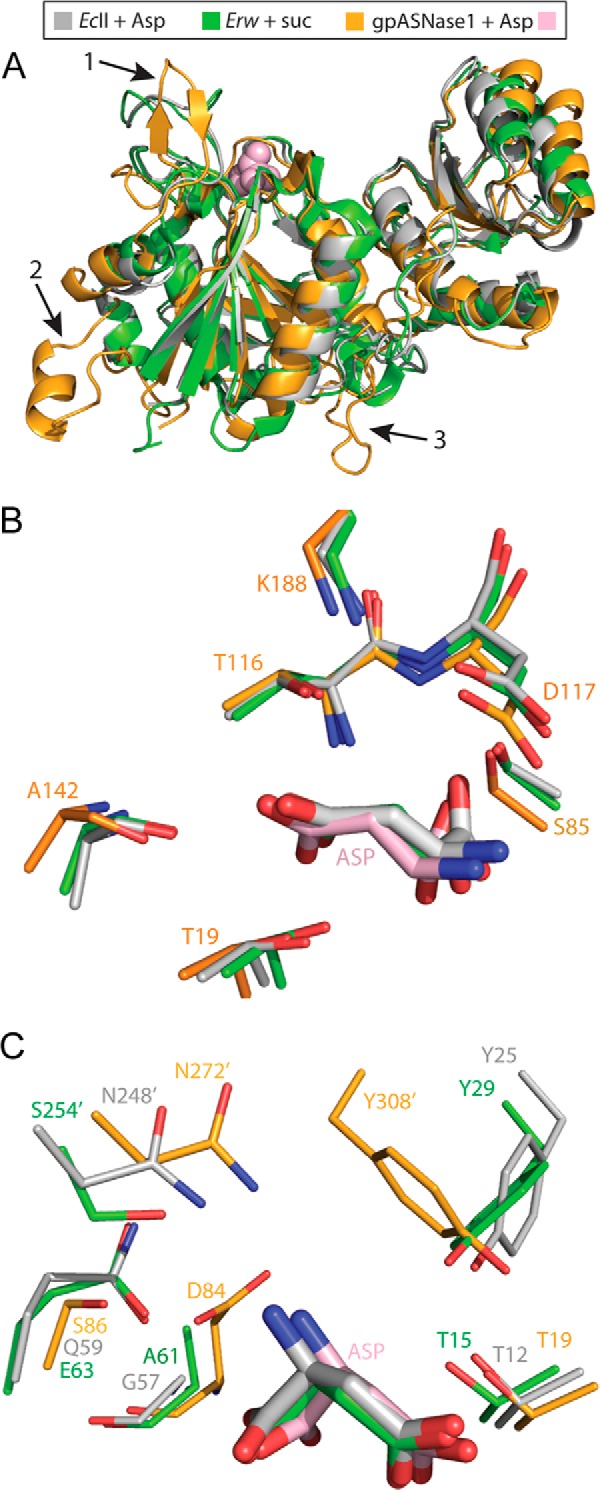
Structural comparison of the low Kml-asparaginases gpASNase1, EcII, and Erw. A, overlay of a gpASNase1 protomer (orange) with that of EcII (gray) and Erw (green). The Asp bound in the gpASNase1 active site is depicted as pink spheres. Three arrows point to the regions that differ most between the enzymes; see text for details. B, close-up of the active site, with the residues conserved between the three enzymes shown as sticks (thicker sticks represent the bound ligand). Numbering corresponds to gpASNase1. C, close-up of the active site, showing those residues not conserved among the three (apart from the catalytic threonine, present in all, for orientation). The most notable difference is the origin of the threonine-activating tyrosine residue. The bacterial enzymes contain this tyrosine in the N terminus (Tyr25 and Tyr29), whereas gpASNase1 supplies the tyrosine from the C terminus, coming from a neighboring protomer (Tyr308′).
Although the quaternary arrangement of the four protomers that build the dimer of dimers differs (Fig. 2), the protomers of gpASNase1, EcII, and Erw exhibit a very similar two-domain structure (root mean square deviation of gpASNase1 to EcII 1.25 Å over 172 common Cα atoms; to Erw 1.37 Å over 171 Cα atoms) (Fig. 5A). We do observe three notable differences. First is in the N-terminal region, indicated by arrow 1 (Gly21–Gly34), which is more open in gpASNase1. This is an important difference because this region is near the active site. Second, an insert present in gpASNase1 and other mammalian l-asparaginases (Thr41–Leu73) but absent in the bacterial enzymes is indicated by arrow 2 (see also Fig. 1C). In the structure, the insert is at the surface far from the active site, and its function is not clear. A third notable difference between the bacterial l-asparaginases and gpASNase1 is the linker region that connects the N-terminal domain to the C-terminal domain (Trp216–Asp237), as indicated by arrow 3.
Inspection of the active site reveals both conservation and divergence between gpASNase1 and the bacterial enzymes. Conserved are the two catalytic threonines (Thr19 and Thr116, all numbering per gpASNase1), the serine that interacts with the substrate carboxylic moiety (Ser85), and the aspartic acid (Asp117) that binds the substrate amino moiety, which is being positioned by a lysine residue (Lys188) (Fig. 5B).
Despite the conservation both in sequence and positioning of the above active site residues, several notable differences are observed (Fig. 5C). First, whereas in the bacterial enzymes a tyrosine (Tyr25 in EcII and Tyr29 in Erw) from the N-terminal region approaches the catalytic threonine, in gpASNase1 this same functionality is supplied from the C-terminally located Tyr308′. As a reminder, the prime denotes a residue from a neighboring protomer.
An additional active site difference is the position of Ser86. In gpASNase1, Ser86 is in proximity but too distant for a direct interaction (4.6 Å) to the substrate amino group. In contrast, the analogous residues, Gln59 (EcII) or Glu63 (Erw), are sufficiently close to make this interaction (3.2–3.3 Å).
This comparison demonstrates that gpASNase1 conserves many of the essential active site residues, and those that are missing (e.g. the N-terminal tyrosine in the bacterial enzymes) are supplied by the same residue originating from a different part of the structure. The other differences appear minor, consistent with the similar kinetic properties of gpASNase1 and the bacterial enzymes.
Rationalizing the Very Different Kinetic Properties of gpASNase1 and hASNase1
The high structural similarity of gpASNase1 to EcII and Erw begs the question, why is the human homolog, which is highly identical to gpASNase1, endowed with a much higher Km value for Asn? To address this question, we generated a homology model for hASNase1 that is based on our gpASNase1 structure. As expected for protein sequences that are >80% identical and almost 95% similar, the root mean square deviation between the experimentally derived gpASNase1 structure and the homology hASNase1 structure is a minuscule 0.06 Å. Despite this very small global root mean square deviation, one region has pronounced differences that are due to a two-residue insertion present in hASNase1 (Figs. 1C and 6). Notably, this insertion is just prior to the tyrosine residue seen in gpASNase1 that approaches the active site from a neighboring protomer, being positioned in proximity to the catalytic threonine Thr19.
FIGURE 6.

Structural comparison of gpASNase1 and the homology model of hASNase1. The >80% sequence identity of the l-asparaginase domains of gpASNase1 and hASNase1 allowed us to generate a highly predictive homology model for hASNase1. A, overlay of a gpASNase1 protomer (orange) and a hASNase1 protomer (beige). The most notable difference is at the C-terminal region where Tyr308′ is located (circled in black). B, focus on the dimer interface. Colors as indicated in the figure. Asp is shown as pink spheres. C, focus on the N- and C-terminal regions, as defined in Fig. 4A. Notice that most significant differences between gpASNase1 and the homology model of hASNase1 occur in these active site regions.
Previous work on EcII employing the T89V mutant (Thr116 in gpASNase1) revealed that a covalent intermediate is formed between EcII Thr12 (Thr19 in gpASNase1) and the substrate Asn (36). This work suggested that the hydrolysis of Asn commences by nucleophilic attack by Thr12. Presumably this covalent intermediate could not break down because of the T89V mutation. If this proposed mechanism is correct, then a likely role for Tyr25 in EcII (Tyr308′ in gpASNase1) is to activate Thr12. By analogy, in gpASNase1 the role of activating Thr19 falls on Tyr308′. The fact that this loop is one of the most pronounced differences between gpASNase1 and hASNase1 suggests that the insertion present in hASNase1 acts to modify the dynamic properties of this region and that this leads to a much increased Km value. Supporting this interpretation is the different conformation we observed for this region between the gpASNase1 apo and Asp complex structures (Fig. 4A). This demonstrates a substrate-induced conformational change, which acts to bring Tyr308′ to its activating conformation. The different sequence and the insertion present in hASNase1 likely modify the properties of this important region.
An alternative explanation for the vastly different kinetic properties between gpASNase1 and hASNase1 has to do with the allosteric behavior of the latter. Unlike gpASNase1, for which we could not detect any allosteric properties, for hASNase1 we measured a Hill coefficient of 2.5 (Table 1). Interestingly, the high Km EcI enzyme has also been shown to be allosterically regulated by its substrate Asn, with an almost identical Hill coefficient of 2.6 (9). However, the putative Asn allosteric site as observed in EcI is not conserved in hASNase1. Nevertheless, it is possible that the high Km property of hASNase1 is due to its allosteric regulation. How this functions in hASNase1 must await the determination of its structure.
Structural Basis for Lack of l-Glutaminase Activity in gpASNase1
The low Km property of gpASNase1 makes it a promising alternative to the clinically used EcII and Erw enzymes. As important as its low Km property is its lack of l-glutaminase activity, because this side reaction has been implicated in the toxicity observed with the bacterial enzymes (39–41). We therefore asked, what is the reason for the lack of gpASNase1 l-glutaminase activity?
To clarify this important point, we first wanted to understand what allows EcII and Erw to hydrolyze both Asn and Gln. Because Erw has a higher intrinsic l-glutaminase activity compared with EcII (10% versus 2% relative to the ASNase activity (42)), we focused on the Erwinia enzyme in addressing this question. Fig. 7A shows an overlay of the Erw-Asp complex in pink (PDB code 2JK0) (43) and the Erw-Glu complex in green (PDB code 1HFW) (38), zoomed on the substrate binding site. The hydrolysis occurs at the amino acid side chain, so that part of the substrate must occupy a similar position to allow nucleophilic attack by the catalytic threonine. Therefore, it is the position of the main chain amino group that must adjust to a larger degree to accommodate the one-carbon longer Glu versus Asp. In Erw, the amino group is bound by two carboxylic acids, Glu63 and Asp96. The comparison reveals that Glu63 adjusts its position to a larger degree to allow the substrate amino group to bind deeper in the active site (Fig. 7A).
FIGURE 7.
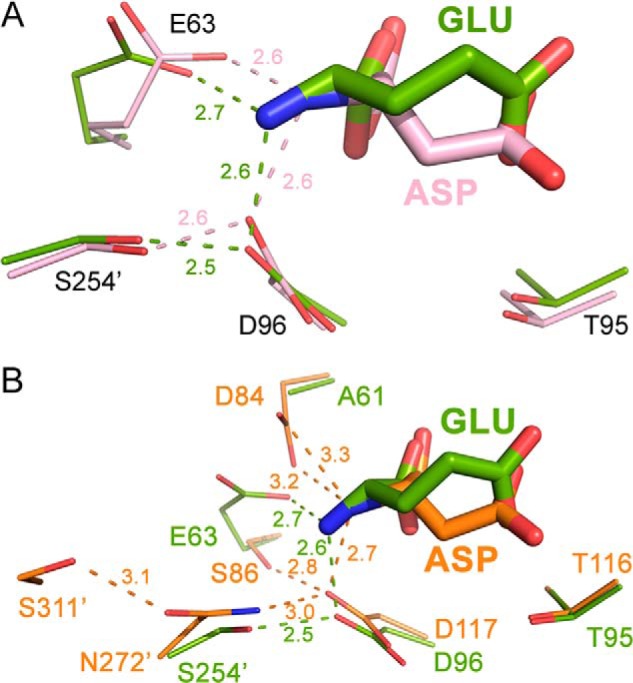
Rationalizing the lack of l-glutaminase activity in gpASNase1. A, overlay of the Erw-Glu complex (green; PDB code 1HFW) and the Erw-Asp complex (pink; PDB code 2JK0) with a focus on select active site residues (thicker sticks represent the bound ligand). Binding of the α-amino group in the longer Glu is accommodated by change in conformation of Glu63 with some minor adjustment of Asp96. B, overlay of the same Erw-Glu complex with the gpASNase1-Asp complex (orange). The residues in gpASNase1 that bind the substrate α-amino group are predicted to be less flexible compared with the residues in Erw that fulfill the same function. The inability to bind the Gln α-amino group deeper in the active site prevents productive binding in gpASNase1.
We performed the analogous comparison to the Erw-Glu structure using this time the gpASNase1-Asp complex structure (Fig. 7B, orange). This revealed that the active site residues that bind the substrate amino group differ between Erw and gpASNase1. Although Asp117 of gpASNase1 plays the role of Asp96 of Erw, gpASNase1 lacks Glu63, having Ser86 instead in that position. To compensate for the lack of Glu63, Asp84 in gpASNase1 adopts its function (being Ala61 in Erw). To accommodate the longer Glu in gpASNase1, these residues would need to modify their positions. However, it seems that the gpASNase1 active site residue Asp119 is tightly held in place by Asn272′, which in turn is held in place by Ser311′. Moreover, the replacement in gpASNase1 of the Erw Glu63 is Asp84, and by its nature of having one less methylene group, the aspartic acid would possess reduced flexibility. We propose that together, the less labile position of Asp117 and the intrinsically less flexible Asp84 do not allow the longer glutamic acid to bind productively in the gpASNase1 active site, explaining its lack of l-glutaminase activity.
Is gpASNase1 the Guinea Pig Serum l-Asparaginase
Arguments can be made for and against the low Km l-asparaginase discussed here being the guinea pig serum enzyme responsible for the cell killing property. Arguments against gpASNase1 being the serum enzyme include its size (∼240 kDa as tetramer), whereas the serum enzyme has been reported to be ∼138 kDa (18). The only way gpASNase1 can be consistent with this data is that in the serum, the gpASNase1 tetramer dissociates to dimers (∼120 kDa), or alternatively, the ankyrin repeats are cleaved off when the enzyme is secreted (leaving still a tetramer, but now of ∼140 kDa). This leads to an additional point against gpASNase1 being the serum enzyme, namely that it lacks a clear signal peptide for secretion in its sequence. This leaves the why and how of its secretion into the blood as open questions. It is unlikely to be due to the ankyrin repeats, because other mammalian enzymes also contain this C-terminal domain yet are not secreted.
Conversely, arguments in favor of gpASNase1 being the serum l-asparaginase include its low Km property, as would be required for an enzyme that can deplete the blood Asn level. Additionally, the Km value that we measure (50 μm) is similar to that reported for the serum-purified enzyme (70 μm) (19). Moreover, of the four guinea pig genes that have been associated with l-asparaginase activity, we have previously ruled out the gene product of one of them (gpASNase3 (21)), whereas the gene products of two additional genes are by homology expected to have a very high Km for Asn. This leaves gpASNase1 as the sole candidate for being the serum enzyme, unless there is a yet to be discovered fifth guinea pig enzyme with l-asparaginase activity.
However, probably the most convincing argument for gpASNase1 being indeed the guinea pig serum l-asparaginase comes from a series of patents filed in the late 1990s by the Japanese company Hayashibara (44). In these patent filings, the researchers describe how, after partial purification of the guinea pig serum, the sequences of short peptides were isolated from fractions having l-asparaginase activity. Those peptide sequences were ultimately used to clone an enzyme that is identical to gpASNase1. This would suggest that gpASNase1 is the guinea pig serum l-asparaginase. The caveat is that the cloning used mRNA from guinea pig liver cells. Interestingly, it was previously reported that the liver enzyme, like the serum enzyme, has cell killing properties (30). So ambiguity remains as to whether the liver l-asparaginase is the same as the one secreted into the serum.
Conclusions
In summary, we report that the guinea pig enzyme annotated as H0W0T5_CAVPO is a low Km l-asparaginase, lacking detectable l-glutaminase activity. Structures of its unliganded apo state and as a complex with Asp, representing the first structures of a mammalian l-asparaginase from this enzyme family, reveal the regions that become ordered upon presence of ligand, and the binding mode of the substrate. These structures also suggest reasons for its lack of l-glutaminase activity and explain why the human homolog hASNase1 has such dramatically different kinetic properties. In total, this work reveals that gpASNase1 would be a good contender for replacing the clinically used bacterial l-asparaginases; this enzyme displays the same low Km l-asparaginase activity, is mostly devoid of the toxicity-causing l-glutaminase activity, and is likely to be much less immunogenic.
Acknowledgment
We thank the staff at LS-CAT for assistance in data collection.
This work was supported, in whole or in part, by National Institutes of Health Grants R21 CA155424 and T32 HL0507692 (to A. M. S.).
The atomic coordinates and structure factors (codes 4R8K and 4R8L) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ALL
- acute lymphoblastic leukemia
- ASNase
- l-asparaginase
- gpASNase1
- guinea pig l-asparaginase (UniProt entry H0W0T5, H0W0T5_CAVPO)
- hASNase1
- human l-asparaginase (UniProt entry Q86U10, LPP60_HUMAN)
- EcI
- E. coli type I l-asparaginase (UniProt entry P0A962, ASPG1_ECOLI)
- EcII
- E. coli type II l-asparaginase (UniProt entry P00805, ASPG2_ECOLI)
- Erw
- E. chrysanthemi l-asparaginase (Uniprot entry P06608, ASPG_DICCH)
- PDB
- Protein Data Bank.
REFERENCES
- 1. Kidd J. G. (1953) Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum: I. course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J. Exp. Med. 98, 565–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Broome J. D. (1963) Evidence that the l-asparaginase of guinea pig serum is responsible for its antilymphoma effects: I. properties of the l-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J. Exp. Med. 118, 99–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haley E. E., Fischer G. A., Welch A. D. (1961) The requirement for l-asparagine of mouse leukemia cells L5178Y in culture. Cancer Res. 21, 532–536 [PubMed] [Google Scholar]
- 4. Neuman R. E., McCoy T. A. (1956) Dual requirement of Walker carcinosarcoma 256 in vitro for asparagine and glutamine. Science 124, 124–125 [DOI] [PubMed] [Google Scholar]
- 5. Cooney D. A., Capizzi R. L., Handschumacher R. E. (1970) Evaluation of l-asparagine metabolism in animals and man. Cancer Res. 30, 929–935 [PubMed] [Google Scholar]
- 6. Dolowy W. C., Henson D., Cornet J., Sellin H. (1966) Toxic and antineoplastic effects of l-asparaginase. Study of mice with lymphoma and normal monkeys and report on a child with leukemia. Cancer 19, 1813–1819 [DOI] [PubMed] [Google Scholar]
- 7. Ho P. P., Milikin E. B., Bobbitt J. L., Grinnan E. L., Burck P. J., Frank B. H., Boeck L. D., Squires R. W. (1970) Crystalline l-asparaginase from Escherichia coli B: I. purification and chemical characterization. J. Biol. Chem. 245, 3708–3715 [PubMed] [Google Scholar]
- 8. Derst C., Henseling J., Röhm K. H. (2000) Engineering the substrate specificity of Escherichia coli asparaginase: II. selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Sci. 9, 2009–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yun M. K., Nourse A., White S. W., Rock C. O., Heath R. J. (2007) Crystal structure and allosteric regulation of the cytoplasmic Escherichia coli l-asparaginase I. J. Mol. Biol. 369, 794–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salzer W. L., Asselin B. L., Plourde P. V., Corn T., Hunger S. P. (2014) Development of asparaginase Erwinia chrysanthemi for the treatment of acute lymphoblastic leukemia. Ann. N.Y. Acad. Sci. 10.1111/nyas.12496 [DOI] [PubMed] [Google Scholar]
- 11. Armstrong J. K., Hempel G., Koling S., Chan L. S., Fisher T., Meiselman H. J., Garratty G. (2007) Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 110, 103–111 [DOI] [PubMed] [Google Scholar]
- 12. Avramis V. I., Panosyan E. H. (2005) Pharmacokinetic/pharmacodynamic relationships of asparaginase formulations: the past, the present and recommendations for the future. Clin. Pharmacokinet. 44, 367–393 [DOI] [PubMed] [Google Scholar]
- 13. Müller H. J., Löning L., Horn A., Schwabe D., Gunkel M., Schrappe M., von Schütz V., Henze G., Casimiro da Palma J., Ritter J., Pinheiro J. P., Winkelhorst M., Boos J. (2000) Pegylated asparaginase (Oncaspar) in children with ALL: drug monitoring in reinduction according to the ALL/NHL-BFM 95 protocols. Br. J. Haematol. 110, 379–384 [DOI] [PubMed] [Google Scholar]
- 14. Boos J., Werber G., Ahlke E., Schulze-Westhoff P., Nowak-Gottl U., Würthwein G., Verspohl E. J., Ritter J., Jurgens H. (1996) Monitoring of asparaginase activity and asparagine levels in children on different asparaginase preparations. Eur. J. Cancer 32A, 1544–1550 [DOI] [PubMed] [Google Scholar]
- 15. Dellinger C. T., Miale T. D. (1976) Comparison of anaphylactic reactions to asparaginase derived from Escherichia coli and from Erwinia cultures. Cancer 38, 1843–1846 [DOI] [PubMed] [Google Scholar]
- 16. Müller H. J., Beier R., da Palma J. C., Lanvers C., Ahlke E., von Schütz V., Gunkel M., Horn A., Schrappe M., Henze G., Kranz K., Boos J. (2002) PEG-asparaginase (Oncaspar) 2500 U/m2 BSA in reinduction and relapse treatment in the ALL/NHL-BFM protocols. Cancer Chemother. Pharmacol. 49, 149–154 [DOI] [PubMed] [Google Scholar]
- 17. Chan W. K., Lorenzi P. L., Anishkin A., Purwaha P., Rogers D. M., Sukharev S., Rempe S. B., Weinstein J. N. (2014) The glutaminase activity of l-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 123, 3596–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yellin T. O., Wriston J. C., Jr. (1966) Purification and properties of guinea pig serum asparaginase. Biochemistry 5, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 19. Ohnuma T., Bergel F., Bray R. C. (1967) Enzymes in cancer. Asparaginase from chicken liver. Biochem. J. 103, 238–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wriston J. C., Jr. (1970) Asparaginase. Methods Enzymol. 98, 732–742 [DOI] [PubMed] [Google Scholar]
- 21. Schalk A. M., Lavie A. (2014) Structural and kinetic characterization of guinea pig l-asparaginase type III. Biochemistry 53, 2318–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karamitros C. S., Konrad M. (2014) Human 60-kDa lysophospholipase contains an N-terminal l-asparaginase domain that is allosterically regulated by l-asparagine. J. Biol. Chem. 289, 12962–12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fernandez C. A., Cai X., Elozory A., Liu C., Panetta J. C., Jeha S., Molinelli A. R., Relling M. V. (2013) High-throughput asparaginase activity assay in serum of children with leukemia. Int. J. Clin. Exp. Med. 6, 478–487 [PMC free article] [PubMed] [Google Scholar]
- 24. Hejazi M., Piotukh K., Mattow J., Deutzmann R., Volkmer-Engert R., Lockau W. (2002) Isoaspartyl dipeptidase activity of plant-type asparaginases. Biochem. J. 364, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Joosten R. P., Salzemann J., Bloch V., Stockinger H., Berglund A. C., Blanchet C., Bongcam-Rudloff E., Combet C., Da Costa A. L., Deleage G., Diarena M., Fabbretti R., Fettahi G., Flegel V., Gisel A., Kasam V., Kervinen T., Korpelainen E., Mattila K., Pagni M., Reichstadt M., Breton V., Tickle I. J., Vriend G. (2009) PDB_REDO: automated re-refinement of X-ray structure models in the PDB. J. Appl. Crystallogr. 42, 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 32. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 34. Song J., Tan H., Perry A. J., Akutsu T., Webb G. I., Whisstock J. C., Pike R. N. (2012) PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 36. Palm G. J., Lubkowski J., Derst C., Schleper S., Röhm K. H., Wlodawer A. (1996) A covalently bound catalytic intermediate in Escherichia coli asparaginase: crystal structure of a Thr-89-Val mutant. FEBS Lett. 390, 211–216 [DOI] [PubMed] [Google Scholar]
- 37. Swain A. L., Jaskólski M., Housset D., Rao J. K., Wlodawer A. (1993) Crystal structure of Escherichia coli l-asparaginase, an enzyme used in cancer therapy. Proc. Natl. Acad. Sci. U.S.A. 90, 1474–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aghaiypour K., Wlodawer A., Lubkowski J. (2001) Structural basis for the activity and substrate specificity of Erwinia chrysanthemi l-asparaginase. Biochemistry 40, 5655–5664 [DOI] [PubMed] [Google Scholar]
- 39. Distasio J. A., Salazar A. M., Nadji M., Durden D. L. (1982) Glutaminase-free asparaginase from vibrio succinogenes: an antilymphoma enzyme lacking hepatotoxicity. Int. J. Cancer 30, 343–347 [DOI] [PubMed] [Google Scholar]
- 40. Durden D. L., Salazar A. M., Distasio J. A. (1983) Kinetic analysis of hepatotoxicity associated with antineoplastic asparaginases. Cancer Res. 43, 1602–1605 [PubMed] [Google Scholar]
- 41. Ollenschläger G., Roth E., Linkesch W., Jansen S., Simmel A., Mödder B. (1988) Asparaginase-induced derangements of glutamine metabolism: the pathogenetic basis for some drug-related side-effects. Eur. J. Clin. Invest. 18, 512–516 [DOI] [PubMed] [Google Scholar]
- 42. Narta U. K., Kanwar S. S., Azmi W. (2007) Pharmacological and clinical evaluation of l-asparaginase in the treatment of leukemia. Crit. Rev. Oncol. Hematol. 61, 208–221 [DOI] [PubMed] [Google Scholar]
- 43. Papageorgiou A. C., Posypanova G. A., Andersson C. S., Sokolov N. N., Krasotkina J. (2008) Structural and functional insights into Erwinia carotovora l-asparaginase. FEBS J. 275, 4306–4316 [DOI] [PubMed] [Google Scholar]
- 44. Ario T., Madoka T., Kozo Y., Masashi K. (March 25, 2003) U. S. Patent 6,537,547 B1