

Background: NLRP3 is a key regulator of innate inflammation and is linked to inflammatory diseases.
Results: GPSM3 associates with NLRP3 and inhibits its function.
Conclusion: GPSM3 specifically inhibits NLRP3-dependent inflammasome activity by interacting with its leucine-rich repeat domain.
Significance: This association uncovers a putative new mechanism of NLRP3 control, linking a G protein modulator to NLRP3-dependent inflammatory diseases.
Keywords: Cellular Immune Response, Cytokine, G Protein, Inflammasome, Inflammation, GPSM, NLRP3, Nod-like Receptors
Abstract
Inflammasomes are multi-protein complexes that regulate maturation of the interleukin 1β-related cytokines IL-1β and IL-18 through activation of the cysteine proteinase caspase-1. NOD-like receptor family, pyrin domain containing 3 (NLRP3) protein is a key component of inflammasomes that assemble in response to a wide variety of endogenous and pathogen-derived danger signals. Activation of the NLRP3-inflammasome and subsequent secretion of IL-1β is highly regulated by at least three processes: transcriptional activation of both NLRP3 and pro-IL-1β genes, non-transcriptional priming of NLRP3, and final activation of NLRP3. NLRP3 is predominantly expressed in cells of the hematopoietic lineage. Using a yeast two-hybrid screen, we identified the hematopoietic-restricted protein, G protein signaling modulator-3 (GPSM3), as a NLRP3-interacting protein and a negative regulator of IL-1β production triggered by NLRP3-dependent inflammasome activators. In monocytes, GPSM3 associates with the C-terminal leucine-rich repeat domain of NLRP3. Bone marrow-derived macrophages lacking GPSM3 expression exhibit an increase in NLRP3-dependent IL-1β, but not TNF-α, secretion. Furthermore, GPSM3-null mice have enhanced serum and peritoneal IL-1β production following Alum-induced peritonitis. Our findings suggest that GPSM3 acts as a direct negative regulator of NLRP3 function.
Introduction
Phagocytic mononuclear cells such as monocytes and macrophages respond to host and environmental cues through numerous signal transduction pathways to coordinate inflammatory responses. The release of IL-1β and IL-18 from these cells plays a major role in inflammatory responses to challenge with infectious agents and in aseptic inflammatory conditions (1). These cytokines are produced as inactive cytoplasmic pro-cytokines and require specialized proteolytic processing and secretion mechanisms to be released as biologically active molecules. The proteolytic processing is carried out by a specific cysteine proteinase, caspase-1 (2). Caspase-1 is synthesized as an inactive pro-enzyme and activation of procaspase-1 is tightly regulated (3). Multiprotein complexes known as inflammasomes activate caspase-1 in response to numerous pro-inflammatory stimuli (4). While all inflammasomes activate caspase-1, inflammasomes are assembled from various core protein components each of which has a defined range of triggers.
Inflammasome activation of procaspase-1 is ultimately responsible for proteolytic processing of the precursors of IL-1β and IL-18 into mature, active cytokines (4). In addition to procaspase-1, inflammasomes are known to contain other cellular proteins, including the adapter proteins Apoptotic Speck protein containing a Card (or ASC)3 and Cardinal (also known as CARD8 or TUCAN), as well as a NOD-like receptor (NLR) protein family member (4). The mammalian genome encodes ∼23 NLR gene family members, each defined by a conserved tripartite protein structure of a variable number of leucine-rich repeat at the C terminus, a central nucleotide-binding oligomerization domain, and an N-terminal effector domain. Most NLR effector domains are either caspase activation and recruitment domains (CARD), leading to the designation NLRC, or pyrin domains, leading to the designation NLRP (5). The immunologic signaling pathways within which many of these NLR proteins participate have been elucidated both through genetic disease associations and through cell biological and biochemical analyses of their function (6).
Of all inflammasome-forming proteins, NLRP3 is activated by the broadest range of stimuli and has been implicated in the pathogenesis of a wide array of autoinflammatory conditions, sterile inflammatory conditions, and infectious diseases. Monosodium urate and calcium pyrophosphate crystals, underlying causes of the sterile inflammatory arthritides gout and pseudogout, cause the activation of caspase-1 and secretion of IL-1β; macrophages from NLRP3-deficient mice fail to secrete IL-1β in response to either stimuli (7). Additionally, crystals linked to various other pathologic inflammatory processes are known to signal through NLRP3, including asbestos, silica, and β-amyloid fibrils (8–10). Aluminum hydroxide, a vaccine adjuvant used in humans, also stimulates the release of NLRP3-inflammasome-processed cytokines. NLRP3-deficient mice have blunted immunologic responses to vaccinations accompanied by aluminum hydroxide, suggesting that NLRP3 plays an important role in the adaptive immune response in this setting as well (10, 11). In addition, several biochemical moieties produced by infectious disease agents or host inflammatory processes, named pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), are known to activate the NLRP3-dependent inflammasome; PAMPs and DAMPs include pore-forming toxins, pathogen-related RNA and DNA species, host cell-derived ATP and DNA, and hyaluronan generated from cellular damage (12). Activation of the NLRP3 inflammasome in response to these diverse stimuli is controlled by a series of transcriptional and post-transcriptional mechanisms that include up-regulation of NLRP3 and involvement of the chaperonin HSP90 and the deubiquitinase BRCC3 (13–15).
Some of us have recently described functional studies of G protein signaling modulator-3 (GPSM3), a newly identified signaling regulator with prominent expression in myeloid lineage cells and lower relative expression levels in other hematopoietic lineages as well as non-hematopoietic tissues (16). GPSM3 (a.k.a. AGS4 or G18; Ref. 17, 18) possesses two functional “GoLoco motifs” (18) for binding heterotrimeric G protein Gαi subunits and additionally binds to Gβ subunits during their synthetic pathway toward forming mature Gβγ dimers (19); these interactions with heterotrimeric G protein subunits are thought to underlie the effects of GPSM3 on chemokine receptor signaling that is critical to the development of inflammatory arthritis (16). More recently, we have described GPSM3 as also interacting directly with the adaptor protein 14-3-3 (20). To identify additional interacting partner(s) that might be involved in GPSM3-mediated signaling regulation, a yeast two-hybrid screen was performed using full-length GPSM3 as bait. This screen (19) identified one clone encoding a C-terminal fragment of NLRP3. Based their co-expression in myeloid lineage cells and the discovery of their potential association by yeast two-hybrid screening, we investigated the role of GPSM3 in NLRP3-mediated IL-1β production and further characterized this association using biochemical approaches. Our data indicate a modulatory function of GPSM3 on NLRP3-dependent IL-1β generation.
MATERIALS AND METHODS
Commercial Antibodies, Constructs, and Other Reagents
Horseradish peroxidase (HRP)-conjugated anti-hemagglutinin (HA) monoclonal antibody (clone 3F10) was obtained from Roche Diagnostics. Anti-Flag M2 antibody, and agarose-conjugated anti-Flag M2 antibody were purchased from Sigma. HRP-conjugated goat anti-mouse and goat anti-rabbit antibodies were from GE Healthcare (Piscataway, NJ). Anti-GFP Abfinity antibody was from Invitrogen. Mouse monoclonal anti-NLRP3 clone cryo-2 was from Adipogen (San Diego, CA) and sheep polyclonal anti-NLRP3 from R&D systems (Minneapolis, MN). Monoclonal anti-GPSM3 antibody was produced by the UNC Antibody Core Facility and has been previously described (20). All cDNAs used in this report were cloned in the pcDNA3.1 backbone vector (Invitrogen, Carlsbad, CA), with Flag- or HA-epitope tag sequences included in the forward PCR primer to produce N-terminal-tagged open-reading frames as described previously (20). Recombinant Staphylococcus aureus α hemolysin was generated as previously described (21). Aluminum hydroxide suspension (Imject Alum) was purchased from Thermo Scientific.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293) and THP-1 cell lines were each obtained from the American Type Culture Collection (ATCC) and maintained in DMEM or RPMI 1640 media (Invitrogen), respectively, supplemented with 10% fetal bovine serum (Cellgro, Manassas, VA) at 37 °C in a humidified atmosphere containing 5% CO2. Transient transfections of cell monolayers grown to 75–90% confluence were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Immunoprecipitation and Immunoblotting
Cells were lysed with ice-cold lysis buffer (20 mm HEPES, pH 7.5, 1 mm EDTA, 150 mm NaCl, 1% Nonidet P-40, and Complete protease inhibitor mixture tablets (Roche, Indianapolis, IN)) at 4 °C on a rocker platform for 30 min. Lysates were clarified by centrifugation at 16,000 × g for 15 min at 4 °C and quantified by the bicinchoninic acid (BCA) protein content assay (Pierce). For immunoprecipitation, lysates were incubated with specific antibody for 2 h at 4 °C followed by overnight incubation with protein-A/G agarose (Santa Cruz Biotechnology), or directly incubated with agarose-conjugated anti-Flag M2 antibody overnight. Pelleted antibody/bead complexes were then washed three times with lysis buffer and proteins eluted in Laemmli buffer. Eluted proteins or lysate samples were resolved on 4–12% precast SDS-polyacrylamide gels (Novex/Invitrogen), transferred to nitrocellulose, immunoblotted using primary and HRP-conjugated secondary antibodies, and visualized by chemiluminescence (ECL, GE Healthcare).
Bioluminescence Resonance Energy Transfer (BRET)
HEK293 cells were seeded in 12-well plates (3.5 × 105 cells/well) and transfected with fixed amount of RLuc-GPSM3 or RLuc-GPSM3 LSL mutant vector DNA (50 ng) and increasing amounts of GFP10-NLRP3 vector DNA (0–1500 ng) and corresponding (decreasing) amounts of pcDNA3 empty vector (1500 to 0 ng) to obtain a saturation curve. The others BRET assays were performed by transfecting cells with 50 ng of the RLuc fusion protein-expressing vector with 750 ng of GFP10 fusion protein expressing vector as indicated. 24 h post-transfection, cells were washed once, harvested, and resuspended in BRET buffer (phosphate-buffered saline with 1 mm CaCl2, 0.5 mm MgCl2, 0.1% glucose) and distributed in white 96-well microplates. BRET was initiated by adding coelenterazine-400a at a final concentration of 5 μm. Measurements of emitted light were collected on a Mithras LB-940 plate-reader (Berthold Technologies) using a BRET2 filter set.
Bimolecular Fluorescence Complementation (BiFC)
Fusion constructs were made similar to those previously described (19): namely, a fusion of the N-terminal fragment (aa 1–158) of yellow fluorescent protein (YN) to the N terminus of full-length GPSM3 (“YN-GPSM3”) and the C-terminal fragment (aa 159–238) of YFP (YC) to the C terminus of NLRP3 (“NLRP3-YC”). HEK293 cells were transfected with an equal amount of plasmids encoding the fusion proteins YN-GPSM3 and NLRP3-YC, and cells were incubated at 37 °C for 24 h. Total DNA quantity was normalized using empty pcDNA3.1 vector DNA. To measure fluorescence from formed complexes, transfected cells were washed, harvested, and resuspended in PBS. BiFC signal was acquired using a Mithras LB-940 plate-reader using an excitation/emission filter set of 485 and 510 nm. The level of expression of each fusion protein was quantified by Western blotting using a polyclonal antibody directed against the GFP.
Immunofluorescence Microscopy
HEK293 cells were seeded in a 12-well plate and transfected with 0.4 μg of each DNA: YN-GPSM3 and NLRP3-YC. The following day, cells were transferred to a poly-d-lysine (PDL)-coated coverslip in a 6-well plate and grown overnight. Cells were then fixed with 4% paraformaldehyde plus PBS for 10 min at room temperature. Coverslips were mounted using Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA) and examined by inverted epifluorescence microscopy (Olympus IX70) using a 40× objective.
Mice
Gpsm3(−/−) mice were generated as previously described (16). Nlrp3(−/−) mice were a kind gift from Dr. John Bertin (Millennium Pharmaceuticals) and were further backcrossed at UNC to achieve nine generations of backcrossing onto the C57BL/6 background. Nlrc4(−/−) mice were produced using C57BL/6 embryonic stem (ES) cells and were a kind gift from Dr. Vishva Dixit (Genentech) (23). Aim2(−/−) mice were generated using C57BL/6 ES cells by inGenious Targeting Laboratory (Ronkonkoma, NY).4
Bone Marrow-derived Macrophages (BMDMs) Isolation and Culture
BMDMs were prepared from 9–12-week-old mice. Briefly, femurs and tibiae were removed from CO2 euthanized mice. Both ends of the bones were cut and bone marrow cells were extracted by flushing with DMEM medium containing 10% FBS using a syringe with a 27-gauge needle. Cell suspension was filtered through a 75-μm cell strainer and then centrifuged and resuspended in RBC lysis buffer (150 mm NH4Cl, 10 mm KCO3, 0.1 mm EDTA, pH 7.4) for 10 min on ice. Cells were washed in complete medium and finally resuspended at a density of 4 × 106 cells/ml in macrophage-conditioned medium (DMEM containing 10% FBS and 20% L-929 culture supernatant as source of M-CSF). Cells were seeded in 10-cm tissue culture plate (4 × 106 cells/plate) in 10-ml final volume. Three days later, 10 ml of fresh macrophage-conditioned medium was added and replaced with 10 ml of fresh medium at day 6 and day 8. Cells were collected from culture dishes at day 8 by washing once with PBS and treated with 5 ml of Accutase (Sigma) for 5 min at 37 °C.
Ex Vivo Inflammasome Activation Assay
BMDMs were resuspended in serum-free RPMI 1640 and seeded in a 24-well plate at a density of 1 × 106 cells/ml/well. LPS (100 ng/ml) was added for 3 h. Subsequently, cells were stimulated with NLRP3's inflammasome activators as indicated: ATP (5 mm), α hemolysin (10 μg/ml), and aluminum hydroxide (130 μg/ml ImjectTM, Pierce) for 1 h. For AIM2 inflammasome activation, dsDNA (2 μg/ml, poly(dA:dT)/LyovecTM, Invivogen) was used and cells treated for 15 h. For NLRC4 inflammasome activation, LPS treated macrophages were then transduced with 250 ng/ml flagellin (Invivogen) using Profect-P2 (1.25 μl/ml, Targeting Systems) as described by Miao et al. (24) for 1 h. Culture supernatants were collected after centrifugation at 13,000 × g for 10 min, and cytokines (IL-1β and TNFα) were measured by enzyme-linked immunosorbent assay (ELISA) as described by Craven et al. (21).
Quantitative RT-PCR
THP-1 or bone marrow-derived macrophage (BMDM) cells were activated as previously described with LPS (100 ng/ml) and/or α-hemolysin (10 μg/ml). Cells were harvested and lysed in buffer RLT (Qiagen) + 1% 2-mercaptoethanol (Fisher) before immediately being placed on ice. RNA extraction was performed using the Qiagen RNeasy kit following the manufacturer's instructions, and isolated RNA was DNase-I treated and used to synthesize cDNA immediately (Thermo Verso cDNA synthesis kit). Relative gene expression was quantified using Veriquest Fast Probe PCR master mix (Affymetrix) and Taqman probes (Invitrogen) for GPSM3, NLPR3, and IL1B normalized to multiplexed β2-microglobin. Fold change in target gene expression was calculated using threshold values by the 2−ΔΔCt method, described by Livak and Schmittgen (25), and statistical significance was determined by one-way ANOVA with Bonferroni post-hoc (p < 0.05).
In Vivo Peritonitis Experiments
Gpsm3(−/−), Nlrp3(−/−), and wild type littermate control mice were first pretreated intraperitoneally (intraperitoneal) with LPS (20 mg/kg) and subsequently injected 2 h later with 350 μg Alum (Pierce), an NLRP3-dependent inflammasome activator (26). Peritoneal exudate and peripheral blood were both collected and peritoneal cavity rinses with 3 ml of PBS (intraperitoneal lavage) and both samples mixed together and cytokine content quantified as described previously for the ex vivo assay. Cardiac puncture was used to bleed mice. Serum was collected from coagulated blood that had been centrifuged.
RESULTS
GPSM3 Interacts with the C-terminal End of NLRP3
Yeast two-hybrid screening for potential GPSM3-associated proteins within a commercial, 106 clone human leukocyte cDNA library identified NLRP3, a critical component of the monocyte inflammasome complex, as a potential GPSM3-interacting protein. Fig. 1A shows the growth under auxotrophic selection of yeast transfected with the cDNA encoding full-length GPSM3 fused to the Gal4 DNA binding domain (bait) and the NLRP3-transcriptional activation domain fusion encoding the partial NLRP3 clone initially retrieved in the screen (prey). Sequence analysis of this prey clone revealed the last four leucine-rich repeats of NLRP3 (aa 831–979) in frame with the Gal4-transcriptional activation domain (Fig. 1B). To further characterize this interaction, cDNA encoding HA epitope-tagged NLRP3 and FLAG epitope-tagged GPSM3 were co-transfected in HEK293 cells which do not express either protein endogenously. Full-length NLRP3, as well as its C-terminal fragment (aa 831–979), were each efficiently co-immunoprecipitated with GPSM3 using anti-Flag antibodies (Fig. 1, C and D, respectively).
FIGURE 1.

Identification of NLRP3 as a GPSM3-interacting protein. A, Saccharomyces cerevisiae were co-transformed with indicated bait plasmids (either expressing the Gal4p DNA binding domain alone [pAS2-1] or as a fusion with full-length GPSM3 [pAS2-1/GPSM3]) and prey plasmids (either expressing the Gal4p activation domain alone [pGAD424] or as a fusion with the C terminus of NLRP3 [pACT2/NLRP3]). Transformed yeast were plated onto synthetic defined agar (Yc) lacking leucine (Leu−, to select for the prey plasmid) and tryptophan (Trp−, to select for the bait plasmid); growth on Yc Leu−Trp− medium demonstrates incorporation of both bait and prey plasmids (left panel). Growth on Yc Leu−Trp− medium also deficient in adenine (Ade−) and histidine (His−) indicates a positive protein-protein interaction (right panel). B, domain architecture of NLRP3 comprising the pyrin domain, nucleotide-binding domain (NBD), and eight leucine-rich repeats (LRRs) in comparison to the truncated domain structure for the NLRP3 fragment identified in the yeast two-hybrid screen. Additionally represented is the domain structure of GPSM3 comprising the three GoLoco motifs (GL1-GL3) and locations of point mutations used in this study. Amino acid numbering for each domain is based on data from UniProt identifier Q96P20 (NLRP3) and Q9Y4H4 (GPSM3). C and D, HEK293 cells were transiently co-transfected with plasmids expressing HA-tagged full-length NLRP3 (aa 1–979; panel C) or an NLRP3 C-terminal fragment (aa 831–979; panel D), along with Flag-tagged GPSM3, either of wild type sequence (WT), R81F + R152F double mutant (RF; abrogates GPSM3/Gαi·GDP interaction) or the LSL mutant in which the GPSM3 amino acids 58LSL were mutated to PLP to eliminate the GPSM3/Gβ interaction as previously described. Immunoprecipitation (IP) of GPSM3 was performed using agarose-conjugated anti-Flag M2 antibody and co-immunoprecipitating proteins were detected by immunoblotting (IB) with anti-HA epitope tag antibody. E, whole cell lysates from THP-1 cells were immunoprecipitated with anti-NLRP3 goat polyclonal antibody or with protein-A/-G agarose beads alone. GPSM3 was subsequently detected using an anti-GPSM3 mouse monoclonal antibody (clone 35.5.1) and NLRP3 was detected with anti-NLRP3 mouse monoclonal antibody. Results shown are representative of two independent experiments.
Our group has previously described different point mutations to GPSM3 that abolish specific interactions with its known heterotrimeric G protein binding partners: Gαi·GDP and monomeric Gβ subunits. For example, mutation to phenylalanine of two arginines at positions 81 and 152, within the critical triad sequences of the first and third GoLoco motifs, completely abolishes direct interaction with Gαi·GDP subunits (herein described as the RF mutant) (19) (Fig. 1B). Moreover, the point mutations 58LSL60-to-PLP, within a leucine-rich motif upstream of the first GoLoco motif, dramatically reduce the association of GPSM3 with monomeric Gβ subunits (herein described as the LSL mutant) (19) (Fig. 1B). While the RF mutation did not perturb co-immunoprecipitation between GPSM3 and NLRP3, the LSL mutation decreased the ability of GPSM3 to interact with full-length and truncated NLRP3 (831–979) suggesting a common interacting protein or shared binding site (Fig. 1, C and D, last lane), in a fashion similar to that observed for the GPSM3/Gβ interaction (19). To demonstrate that the GPSM3/NLRP3 association is not an artifact of protein overexpression, co-immunoprecipitation of endogenous NLRP3 and GPSM3 was performed from the human monocytic cell line, THP-1. A sheep polyclonal anti-NLRP3 antibody was used to immunoprecipitate endogenous NLRP3 and associated proteins. The immunoprecipitated complexes were analyzed for the presence of GPSM3 using immunoblot with a mouse monoclonal anti-GPSM3 antibody (19, 20). Immunoprecipitation with anti-NLRP3 antibodies led to capture of detectable endogenous GPSM3 as well as NLRP3, while mock immunoprecipitation failed to capture either protein (Fig. 1E).
Mapping the GPSM3 Interaction Site(s) within NLRP3
To further characterize the GPSM3/NLRP3 interaction, bioluminescence resonance energy transfer (BRET) was used to identify the minimal GPSM3 interaction domain within NLRP3. Full-length GPSM3, fused to the luminescence donor Renilla Luciferase (RLuc-GPSM3) was co-expressed with fusion proteins comprising various domains of NLRP3 fused to the acceptor fluorescent protein GFP10 (GFP10-NLRP3, Fig. 2A). Any interaction between RLuc-GPSM3 and the NLRP3 domain-GFP10 fusion proteins brings the luciferase donor and GFP acceptor proteins into close proximity, allowing energy transfer. Cellular co-transfection with a fixed quantity of RLuc-GPSM3 plasmid and a titration of increasing amounts of GFP10-NLRP3 (full-length) plasmid generated a saturable signal (Fig. 2B) with a maximum net BRET ratio of 0.3. Furthermore, when the RLuc-GPSM3 fusion contained the LSL mutation, the BRET signal with GFP10-NLRP3 was lost (Fig. 2B). Fig. 2C summarizes BRET experiment results between RLuc-GPSM3 and a variety of GFP10-NLRP3 truncation constructs (Fig. 2A). Expression of RLuc-GPSM3 with GFP10 fusion protein encoding solely the Pyrin domain of NLRP3 (Fig. 2C) gave no observable BRET signal. Truncation of individual leucine-rich repeat (LRR) domains, starting from the C terminus, revealed the importance of the last LRR (aa 911–932) to the GPSM3/NLRP3 interaction, as ∼50% of the signal was lost following its removal. However, a GFP10 fusion protein encoding both the pyrin and NBD domains (GFP10-NLRP3 536 stop) also produced ∼50% of the total netBRET signal (Fig. 2C).
FIGURE 2.

Characterization of the GPSM3/NLRP3 interaction using BRET. A, domain architecture of full-length and truncated GFP10-NLRP3 fusion constructs used to characterize the GPSM3/NLRP3 interaction. B, constant amount of wild type (or LSL mutant) RLuc-GPSM3 fusion expression vector was co-transfected into HEK293 cells with increasing amounts of GFP10-NLRP3 fusion expression vector; the resultant net BRET ratio was plotted as a function of the acceptor/donor ratio. C, C-terminal truncation mutants were used to identify the minimal determinants within NLRP3 required for the interaction with GPSM3. A stop codon was inserted by mutagenesis after each domain of the GFP10-NLRP3 fusion construct (as indicated) and subsequently tested in the BRET assay upon co-expression with RLuc-GPSM3. D, leucine-rich repeat domains were fused to GFP10 (called “GFP10-NLRP3 LRR”) and used in the same BRET experiment.
This BRET-based evidence of interaction with the N-terminal region of NLRP3 was surprising, given that the cDNA clone retrieved from the yeast two-hybrid screen contained only the last 4 LRRs of NLRP3 and interaction between GPSM3 and this C-terminal NLRP3 fragment was confirmed by reciprocal co-immunoprecipitation (Fig. 1). One possible explanation for also detecting a GPSM3 interaction with the NLRP3 N-terminal region is that the NBD domain is known to interact with the C-terminal LRR portion, forming an intramolecular hairpin-like structure (27). It is thus possible that, within the cellular context, the NBD domain of NLRP3 interacts with endogenous NLR proteins that might also interact with GPSM3. To determine whether GPSM3 could interact with the NLRP3 C terminus without bridging by the NLRP3-NBD domain, we made a GFP10 fusion construct expressing only the C-terminal LRRs of NLRP3 (GFP10-NLRP3-LRR). Additionally, a truncation of the GFP10-NLRP3-LRR fusion lacking the last LRR (aa 911–932) was generated. As expected, the construct containing all the C-terminal LRRs (i.e. NLRP3 without its pyrin and NBD domains) was sufficient to generate a robust BRET signal (Fig. 2D). Removal of the final LRR abolished the BRET signal, confirming that the last LRR is required for interaction with GPSM3. Removal of the residual C-terminal extension beyond the final LRR domain (i.e. “NLRP3 934 stop”) had no effect on the GPSM3/NLRP3 interaction as measured by BRET (Fig. 2D). The interaction between GPSM3 and the GFP10-NLRP3-LRR fusion protein was also confirmed by co-immunoprecipitation. As also observed in the BRET assay, the co-immunoprecipitation of GPSM3 and the GFP10-NLRP3-LRR fusion protein was disrupted by deletion of the last LRR domain, but not the residual C-terminal amino acids (Fig. 3).
FIGURE 3.
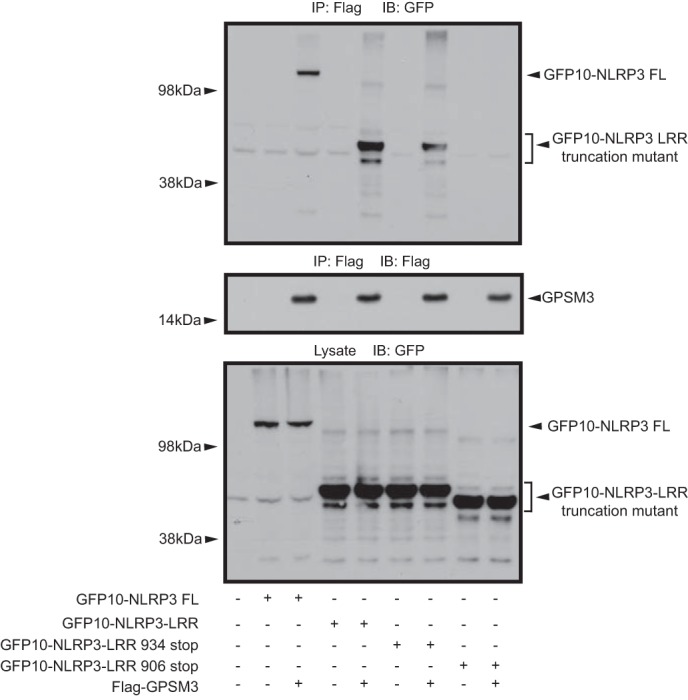
Co-immunoprecipitation of GFP10-NLRP3 protein constructs with GPSM3. Flag-tagged GPSM3 was co-transfected with the same GFP10-NLRP3 fusion proteins used to map the interaction by BRET (Fig. 2) and immunoprecipitation was performed using anti-Flag M2 agarose-conjugated antibody; co-immunoprecipitated proteins were detected by immunoblotting with an anti-GFP antibody.
Subcellular Localization of the GPSM3/NLRP3 Interaction
We employed a third protein-protein interaction assay, BiFC, to confirm the interaction between GPSM3 and NLRP3 as well as to study the localization of their complex, using previously described methods (19, 20). The N-terminal portion of the yellow fluorescent protein (YFP) was fused to full-length GPSM3 (“YN-GPSM3”) and the C-terminal part of YFP was fused to full-length NLRP3. When expressed alone or with the non-fused counterpart, no detectable fluorescence was measured. However, when both fusion proteins were expressed together (YN-GPSM3 + NLRP3-YC), a fluorescent signal was detectable in transfected cells (Fig. 4). As further confirmation of the co-immunoprecipitation and BRET results (Figs. 2 and 3), the LSL mutation in GPSM3 greatly reduced the BiFC signal (Fig. 4A). The YN-GPSM3/NLRP3-YC complex was seen to form punctate structures throughout transfected cells as observed by fluorescence microscopy (Fig. 4B). Ectopic expression of the GFP10-NLRP3 fusion, which is inherently fluorescent without binding the GPSM3 partner, was also observed to form similar punctate structures, suggesting that the GPSM3 interaction does not trigger this subcellular localization phenomenon nor perturb the localization of overexpressed NLRP3 (Fig. 4B). These large dots are likely oligomers of NLRP3, similar to those observed by others using immunofluorescence-based studies of either overexpressed NLRP3 or activation of native NLRP3 in NLRP3-expressing cells (28–30).
FIGURE 4.
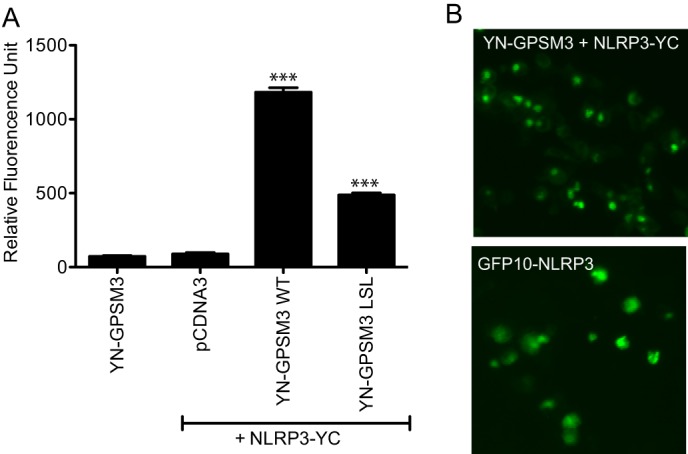
Analysis of the GPSM3/NLRP3 interaction using BiFC. A, HEK293 cells were transfected with plasmids expressing YN-GPSM3 (aa 1–158 of YFP fused to GPSM3) and NLRP3-YC (NLRP3 fused to aa 159–238 of YFP) and the fluorescence of reconstituted YFP fluorophore was measured at 24 h post-transfection. Cells were harvested and fluorescence quantified on a plate reader. Total DNA used for transfection was normalized using empty pcDNA3.1 vector DNA. Data are expressed as relative fluorescence units (RFU) using the means ± S.E. of at least three different experiments; *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by one-way ANOVA. B, epifluorescence microscopy images of HEK293 cells transfected with plasmids expressing the two YFP protein fusions YN-GPSM3 and NLRP3-YC (top panel) or the GFP10-NLRP3 alone (bottom panel).
Functional Effects of the GPSM3/NLRP3 Interaction on NLRP3-dependent IL-1β Secretion
To test the hypothesis that the GPSM3 interaction regulates NLRP3 function and inflammasome activation, bone marrow-derived macrophages (BMDMs) were generated from GPSM3-deficient mice (16) and IL-1β secretion quantified following inflammasome activation. Inflammasome-dependent IL-1β production requires initial priming using lipopolysaccharide (LPS) to induce pro-IL-β synthesis (31, 32). Following LPS stimulation, different inflammasome activators were added, and mature IL-1β secreted in the medium was quantified. Activators of the NLRP3-dependent inflammasome including ATP, α-hemolysin (HLA), and Alum induced robust IL-1β secretion with a significant increase in Gpsm3(−/−) mice after 4 h of stimulation compared with littermate control mice (Fig. 5, A–C, left panels). As expected, NLRP3-deficient mice showed no observable IL-1β secretion (Fig. 5, A–C, left panels). Inflammasome activation is known to trigger release of IL-1β but not that of cytokines not dependent on caspase-1 activation (26). TNF-α was also quantified in the culture supernatants from these stimulated macrophages and found to be secreted to the same extent by all three mouse genotypes (Fig. 5, A–C, right panels); these results suggest that GPSM3 regulation of cytokine secretion is specific for those dependent on NLRP3-mediated caspase-1 activation.
FIGURE 5.

GPSM3 deficiency leads to selective enhancement of the activation of the NLRP3-dependent inflammasome in macrophages ex vivo and in a mouse NLRP3-dependent peritonitis in vivo. BMDMs were generated from wild type (WT), Gpsm3(−/−) and Nlrp3(−/−) mice (panels A, B, and C), or with WT, Gpsm3(−/−), Nlrp3(−/−), Nlrc4(−/−), and Aim2(−/−) mice (panels D and E), and primed with LPS for indicated times at 50 ng/ml before treatment with specific NLR inducers. ATP at 1 mm, S. aureus HLA at 10 μg/ml and aluminum hydroxide and potassium salts (Alum) at 130 μg/ml were used to activate the NLRP3-inflammasome. Mouse macrophages were also separately transfected with dsDNA to activate AIM2 or flagellin peptide to activate NLRC4, as described under “Materials and Methods.” Cell culture supernatant was collected following treatment and secreted IL-1β and TNF-α quantified by ELISA as indicated. F, 12-weeks old, wild type littermate (WT), Gpsm3(−/−), and Nlrp3(−/−) mice were each initially primed by an intraperitoneal injection of LPS (20 mg/kg) and subsequently injected 2 h later with 350 μg of Alum (or saline vehicle, as indicated). Peritoneal exudate was collected and the peritoneal cavity rinsed with 3 ml of PBS (“intraperitoneal lavage”) and IL-1β content quantified by ELISA (right panel). Peripheral blood was also collected, and serum IL-1β was quantified by ELISA (left panel). Data are expressed in pg/ml using the means ± S.E. of three different experiments (n = 8–10); *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by one-way ANOVA.
The specificity of GPSM3 for the NLRP3 inflammasome signaling pathway was also tested using dsDNA stimulation, an AIM2-inflammasome activator (33), and a flagellin-derived peptide known to activate the NLRC4-inflammasome (34). BMDMs isolated from AIM2-, NLRC4-, and NLRP3-deficient mice were used as controls. At 18 h post-treatment, dsDNA was found to induce a similar release of IL-1β secretion in BMDMs from WT mice as compared with Gpsm3(−/−) mice. As predicted, BMDMs from Aim2(−/−) mice failed to secrete IL-1β upon dsDNA stimulation; interestingly, NLRP3-deficient BMDMs were found to have increased IL-1β release compared with BMDMs from WT control and Gpsm3(−/−) mice (Fig. 5C). Similarly, BMDMs from Nlrc4(−/−) mice showed a significant decrease in IL-1β secretion in response to flagellin, as expected (34) (Fig. 5D). Importantly, GPSM3-deficient and WT BMDMs responded equally to dsDNA and flagellin as an inflammasome stimulator (Fig. 5, D and E), suggesting that GPSM3 regulation of inflammasomes is limited to the NLRP3-inflammasome.
To explore the involvement of GPSM3 in NLRP3-dependent inflammasome activation in a more physiological context, we employed a model of Alum-induced peritonitis and quantified resultant IL-1β production from peritoneal lavage and blood serum. Gpsm3(−/−), Nlrp3(−/−), and wild type littermate control mice were first pretreated intraperitoneally (intraperitoneal) with LPS and subsequently injected two hours later with Alum, an NLRP3-dependent inflammasome activator (26). Peritoneal exudate and peripheral blood were both collected and cytokine content quantified. We observed that mature IL-1β secretion was higher in both the lavage and serum of Alum-treated Gpsm3(−/−) mice compared with WT controls, whereas blunted IL-1β secretion was observed from Nlrp3(−/−) mice (Fig. 5F).
To explore the potential mechanism by which GPSM3 regulates NLRP3-dependent IL-1β secretion, we undertook studies of Nlrp3, Il1b, and Gpsm3 expression in primary mouse macrophages and the THP1 human monocytic cell line. WT and Gpsm3(−/−) macrophages exhibited equivalent induction of Nlrp3 and Il1b mRNA after treatment with LPS and/or α-hemolysin (Fig. 6, A and B), further suggesting that GPSM3 alters IL-1β secretion through post-transcriptional regulation of NLRP3. In THP1 cells, priming with LPS increased pro-IL-1β-encoding mRNA and decreased GPSM3 mRNA expression (Fig. 6, C–E). GPSM3 protein levels were also found to be decreased after treatment with LPS (Fig. 6F), suggesting that priming by LPS relieves GPSM3-mediated inflammasome inhibition in part through GPSM3 protein down-regulation.
FIGURE 6.

Pro-IL-1β and GPSM3 are reciprocally regulated during LPS-induced inflammasome priming. BMDMs were generated from WT or Gpsm3(−/−) mice and primed with LPS for 3 h with 100 ng/ml and/or subsequently treated with HLA for 1 h, as indicated. Il1b and Nlrp3 mRNA levels in each cell line were assessed using quantitative RT-PCR as described under “Materials and Methods” (panels A and B). THP1 cells were treated with LPS (100 ng/ml) or vehicle for 3 h. GPSM3, IL1B, and NLRP3 mRNA levels were assessed by quantitative RT-PCR (panels C, D, and E). Data are expressed relative to the β2 M gene transcript; *, p < 0.05 by one-way ANOVA. GPSM3 and actin (loading control) protein levels were also assessed using immunoblot analysis with the indicated antibodies (panel F). Samples from three independent experiments are shown.
The GPSM3/NLRP3 Complex Includes HSPA8 and Is Independent of the GPSM3/14−3−3 Interaction
The chaperonin proteins HSP90, SGT1 (35–37), and HSP70 (38, 39) are known to be essential in regulating NLRP3-inflammasome activation (reviewed in Ref. 40). Cellular heat shock diminishes NLRP3-inflammasome mediated caspase-1 activation, suggesting that these chaperonins are part of an inactive, basal-state NLRP3-containing complex (41). The scaffolding protein RACK1 (“receptor for activated C kinases 1”), is required for the functioning of a HSP90-, SGT1-, and HSP70-containing chaperonin complex in rice which regulates innate immune signaling through nucleotide binding leucine-rich repeat (NB-LRR)-type R proteins (the rice orthologs of mammalian NLR proteins) and other innate immune signaling systems (37, 42, 43). Our yeast two-hybrid screen identified the heat shock protein HSPA8 (also known as HSC70 or HSP73) and RACK1 as additional GPSM3-interacting proteins, suggesting that GPSM3 may be involved in chaperonin-mediated regulation of NLRP3 function. We thus co-expressed Flag-tagged GPSM3 and HA-tagged NLRP3 in the presence or absence of HA-HSPA8 and HA-RACK1 for co-immunoprecipitation analyses. Immunoprecipitation of GPSM3 revealed formation of a complex comprising GPSM3, NLRP3, and HSPA8 but excluding RACK1 (Fig. 7A). Co-expression of HSPA8 increased complex formation between GPSM3 and NLRP3; surprisingly, co-expression of RACK1 also increased GPSM3-NLRP3 complex formation, and co-expression of RACK1 and HSPA8 was found to act synergistically on complex formation. The LSL mutant of GPSM3, previously found to have a greatly reduced interaction with NLRP3 (Fig. 1), also failed to interact with HSPA8 (Fig. 7, A and B, lane 7), suggesting a common binding site or partner. In addition, overexpression of RACK1 and HSPA8 was found to increase fluorescence signal between YN-GPSM3 and NLRP3-YC as observed using the BiFC assay (Fig. 7B). This result supports the idea of a synergistic effect of RACK1 and HSPA8 on GPSM3-NLRP3 complex formation.
FIGURE 7.

The heat shock protein HSPA8 is involved in the interaction between NLRP3 and GPSM3. A, to assess potential roles of HSPA8 and RACK1 in the interaction between NLRP3 and GPSM3, HEK293 cells were transiently co-transfected with plasmids encoding Flag-tagged GPSM3, HA-tagged NLRP3, HA-tagged HSPA8, and/or C-terminal HA-tagged RACK1. Immunoprecipitation (IP) of GPSM3 performed using agarose-conjugate anti-Flag M2 antibody revealed the presence of HSPA8 in the complex, but the absence of RACK1, when immunoblotted with anti-HA. B, HEK293 cells were transfected with plasmids expressing YN-GPSM3 and NLRP3-YC in the presence or absence of RACK1 and HSPA8 and the fluorescence of reconstituted YFP fluorophore was measured at 24 h post-transfection. Cells were harvested and fluorescence quantified on a plate reader. Total DNA used for transfection was normalized using empty pcDNA3.1 vector DNA. Data are expressed as relative fluorescence units (RFU) using the means ± S.E. of at least three different experiments; *, p < 0.05; **, p < 0.01; and ***, p < 0.001 by one-way ANOVA. C, to assess the role of the known GPSM3 interactor 14-3-3, HEK293 cells were similarly co-transfected with Flag-tagged wild type GPSM3 or GPSM3 R35A mutant (a loss-of-function mutant abolishing interaction with 14-3-3; Ref. 20) and HA-tagged HSPA8 and HA-tagged 14-3-3.
We recently described GPSM3 as interacting directly with the adaptor protein 14-3-3 (20). Given that numerous heat shock proteins are known to interact or form a complex with 14-3-3 proteins, including HSPA8 (44), we tested whether 14-3-3 proteins were involved in complex formation between GPSM3, HSPA8, and NLRP3. As previously described (20), mutation of a critical serine 35 within the 14-3-3-binding site of GPSM3 abolishes interaction with 14-3-3. However, the GPSM3 S35A mutant was seen to associate with HSPA8 and NLRP3 identically as wild-type GPSM3 (Fig. 7C), while completely abolishing the 14-3-3 interaction, as previously reported (20).
DISCUSSION
Activation of NLRP3 by various danger signals contributes to pathologic inflammatory responses that drive a number of disease states (12). Presumably the capacity to generate potent pathologic inflammatory responses has led to the evolution of a multistage activation process to rein in NLRP3 inflammasome activity to minimize inappropriate inflammation-driven pathologies. An unbiased screen for proteins that interact with the protein GPSM3 identified a potential interaction with NLRP3. Our present results support an endogenous and physiologic interaction between NLRP3 and GPSM3, and the notion that GPSM3 exerts a negative regulatory effect on the function of NLRP3. Interestingly, GPSM3 has been found to contribute to the induction of acute inflammation in a collagen antibody-induced arthritis mouse model of acute inflammatory arthritis. This pro-inflammatory effect is thought to be mediated by the influence of GPSM3 on monocyte apoptosis and chemotaxis in response to several G protein-coupled receptor activating chemokines (16). This proinflammatory role of GPSM3 does not contradict the negative regulation of the NLRP3 inflammasome that we now describe because induction of collagen antibody-induced arthritis has previously been shown as independent of NLRP3 inflammasome activation (45, 46). Our current data suggest that GPSM3 has pleiotropic effects on immune signaling pathways and that, in addition to modulation of G protein-coupled receptor signaling, it is also able to regulate NLRP3 inflammasome activation.
Using a combination of methodologies, we have shown that GPSM3 interacts with the distal C-terminal region of NLRP3 and absolutely requires the last leucine-rich repeat domain (LRR8). Because no structural information is yet available for either protein, we cannot exclude other determinants within NLRP3 that are also important for this association. The LRR domains of NLR proteins are generally believed to serve as ligand-binding domains for these innate immune sensors. Although the structure of the LRR domain of NLRP3 has not yet been elucidated, the LRR domains of NLR proteins are believed to adopt a horseshoe tertiary structure akin to other LRR-containing proteins. Crystal structures of the LRR portion of different TLR receptors have been elucidated in the presence of different molecular ligands including dsRNA (47), LPS (48), and lipoprotein (49), as well as the dsRNA-binding C terminus of the NLR protein NLRX1 (50). It is possible that GPSM3 interferes with the ability of NLRP3 to interact with activating ligands through interaction with the final LRR domain of NLRP3. A horseshoe tertiary structure for the LRR domains, if also present within NLRP3, might allow intramolecular interaction of the C-terminal LRR domains with the N-terminal NBD domain to facilitate the formation of a closed inactive state, similarly to that described by the NLRC4 crystal structure (51). The observed residual BRET signal between GPSM3 and NLRP3 constructs lacking the LRR domains raises the possibility of an endogenous NLR protein bridging truncated (LRR-lacking) NLRP3 mutants and GPSM3 (52). However, one could also speculate that, if GPSM3 is capable of simultaneously interacting with both the N terminus and the LRR domain of NLRP3, such an interaction could inhibit release of the NLRP3 N terminus and LRR domain, thereby locking the protein in an inactive form. Further experiments are clearly required to determine if either one of these mechanisms is involved in the inhibition of NLRP3 by GPSM3.
A two-step regulatory circuit for cellular IL-1β secretion, in which pro-IL-1β is induced by TLR stimulation and NLRP3 inflammasome-mediated caspase-1 activation is induced by a distinct second signaling event, has been a widely accepted model (53). The first step, or “signal 1,” of regulation of inflammasome-mediated IL-1β secretion is a cell surface receptor-induced NF-κB transcriptional activation event that can be initiated by various TLR-activating stimuli or by numerous cytokine receptors. TLR stimulation primes cells for IL-1β secretion by enhancing pro-IL-1β production and by increasing production of NLRP3, which is a very low abundance protein under basal conditions. Our data indicate that, as NLRP3 and IL1B are up-regulated, the negative regulator GPSM3 is transcriptionally down-regulated as well. A heterogenous group of stimuli then provide “signal 2,” leading to post-transcriptional activation of NLRP3 (reviewed in Ref. 53). The activation of NLRP3 by “signal 2” is now known to further require a post-transcriptional deubiquitination event that is catalyzed by the DUB class of deubiquitinases (22). These data suggest that, in its basal state, NLRP3 is ubiquitinated. A complex containing HSP90 and the ubiquitin ligase subunit SGT1 is required for NLRP3 signaling in mammalian cells, and these proteins may be involved in the baseline ubiquitination of NLRP3 that has been previously observed (40). We have now presented data demonstrating that one other chaperonin protein, HSPA8, is also found in complex with NLRP3 and GPSM3. These data suggest the possibility of a third mechanism by which GPSM3 inhibits NLRP3 activation: namely, that GPSM3 may exert a negative regulatory effect on NLRP3 by helping to bring the latter protein to a basal heat shock protein ubiquitination/chaperonin complex. Based on our present study and recent advances in the field, we propose a revised model of basal NLRP3 regulation (Fig. 8). The role of GPSM3 in promoting NLRP3 inactivation and whether this negative regulatory pathway can be exploited therapeutically in cases of pathologic NLRP3 activation remain areas for further study.
FIGURE 8.

Model of NLRP3 regulation by GPSM3. Formation of a network between GPSM3, chaperones, and NLRP3 maintains the steady-state level of NLRP3 by stabilizing it within this complex. Activation of the inflammasome requires two independent signals. Toll-like receptors (TLR) and tumor-necrosis factor receptors (TNFR) are known to trigger priming signals leading to up-regulation of NLRP3 and pro-IL1β through a NFκB-dependent pathway, as well as down-regulation of GPSM3 mRNA and protein levels. Non-transcriptional priming of NLRP3 allows inflammasome activation upon detection of the activation signal. The NLPR3 activation signal is triggered by endogenous DAMPs or exogenous PAMPs danger signals. Activation of NLRP3 can also be mediated by bacterial pore-forming toxins such as the HLA of S. aureus. Activation of NLRP3 leads to formation of a multiprotein complex containing oligomerized NLRP3, the adaptor ASC, and caspase-1. Once activated, caspase-1 cleaves pro-IL1β (and pro-IL-18), releasing the biological active form of IL-1β (and IL-18) and thereby triggering inflammation and antimicrobial activity.
This work was supported by National Institutes of Health Grants R01 AI088255 (to J. A. D.), U19 AI109965 (to J. A. D. and J. P. T.), and U54 GM104942 (to D. P. S.) via the WVCTSI Pilot Grants Program and by a Burroughs Wellcome Fund Career Award for Medical Scientists (to J. A. D.).
J. E. Wilson, A. S. Petrucelli, L. Chen, A. A. Koblansky, Y. Oyama, A. D. Truax, M. Schneider, A. B. Rogers, M. Mühlbauer, B. R. Barker, W.-C. Chou, W. J. Brickey, I. C. Allen, C. Jobin, D. Ramsden, B. K. Davis, and J. P. Ting, manuscript in preparation.
- ASC
- apoptotic speck protein containing a CARD
- CARD
- caspase activation and recruitment domain
- BRET
- bioluminescence resonance energy transfer
- BiFC
- bimolecular fluorescence complementation
- DAMP
- danger-associated molecular patterns
- PAMP
- pathogen-associated molecular patterns
- HLA
- α-hemolysin
- BMDM
- bone marrow-derived macrophages
- NLR
- NOD-like receptor
- GPSM
- G protein signaling modulator.
REFERENCES
- 1. Dinarello C. A. (1998) Interleukin-1β, interleukin-18, and the interleukin-1β converting enzyme. Ann. N.Y. Acad. Sci. 856, 1–11 [DOI] [PubMed] [Google Scholar]
- 2. Alnemri E. S., Livingston D. J., Nicholson D. W., Salvesen G., Thornberry N. A., Wong W. W., Yuan J. (1996) Human ICE/CED-3 protease nomenclature. Cell 87, 171. [DOI] [PubMed] [Google Scholar]
- 3. Ayala J. M., Yamin T. T., Egger L. A., Chin J., Kostura M. J., Miller D. K. (1994) IL-1β-converting enzyme is present in monocytic cells as an inactive 45-kDa precursor. J. Immunol. 153, 2592–2599 [PubMed] [Google Scholar]
- 4. Martinon F., Burns K., Tschopp J. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell 10, 417–426 [DOI] [PubMed] [Google Scholar]
- 5. Auffray C., Fogg D. K., Narni-Mancinelli E., Senechal B., Trouillet C., Saederup N., Leemput J., Bigot K., Campisi L., Abitbol M., Molina T., Charo I., Hume D. A., Cumano A., Lauvau G., Geissmann F. (2009) CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J. Exp. Med. 206, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dowds T. A., Masumoto J., Chen F. F., Ogura Y., Inohara N., Núñez G. (2003) Regulation of cryopyrin/Pypaf1 signaling by pyrin, the familial Mediterranean fever gene product. Biochem. Biophys. Res. Commun. 302, 575–580 [DOI] [PubMed] [Google Scholar]
- 7. Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 [DOI] [PubMed] [Google Scholar]
- 8. Halle A., Hornung V., Petzold G. C., Stewart C. R., Monks B. G., Reinheckel T., Fitzgerald K. A., Latz E., Moore K. J., Golenbock D. T. (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dostert C., Petrilli V. (2008) Asbestos triggers inflammation by activating the Nalp3 inflammasome. Med. Sci. 24, 916–918 [DOI] [PubMed] [Google Scholar]
- 10. Cassel S. L., Eisenbarth S. C., Iyer S. S., Sadler J. J., Colegio O. R., Tephly L. A., Carter A. B., Rothman P. B., Flavell R. A., Sutterwala F. S. (2008) The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. U.S.A. 105, 9035–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu Y., Hu X., Boumsell L., Ivashkiv L. B. (2008) IFN-γ and STAT1 arrest monocyte migration and modulate RAC/CDC42 pathways. J. Immunol. 180, 8057–8065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Menu P., Vince J. E. (2011) The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin. Exp. Immunol. 166, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauernfeind F. G., Horvath G., Stutz A., Alnemri E. S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B. G., Fitzgerald K. A., Hornung V., Latz E. (2009) Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayor A., Martinon F., De Smedt T., Pétrilli V., Tschopp J. (2007) A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 8, 497–503 [DOI] [PubMed] [Google Scholar]
- 15. Py B. F., Kim M. S., Vakifahmetoglu-Norberg H., Yuan J. (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 [DOI] [PubMed] [Google Scholar]
- 16. Giguère P. M., Billard M. J., Laroche G., Buckley B. K., Timoshchenko R. G., McGinnis M. W., Esserman D., Foreman O., Liu P., Siderovski D. P., Tarrant T. K. (2013) G-protein signaling modulator-3, a gene linked to autoimmune diseases, regulates monocyte function and its deficiency protects from inflammatory arthritis. Mol. Immunol. 54, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao X., Cismowski M. J., Sato M., Blumer J. B., Lanier S. M. (2004) Identification and characterization of AGS4: a protein containing three G-protein regulatory motifs that regulate the activation state of Giα. J. Biol. Chem. 279, 27567–27574 [DOI] [PubMed] [Google Scholar]
- 18. Kimple R. J., Willard F. S., Hains M. D., Jones M. B., Nweke G. K., Siderovski D. P. (2004) Guanine nucleotide dissociation inhibitor activity of the triple GoLoco motif protein G18: alanine-to-aspartate mutation restores function to an inactive second GoLoco motif. Biochem. J. 378, 801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giguère P. M., Laroche G., Oestreich E. A., Siderovski D. P. (2012) G-protein signaling modulator-3 regulates heterotrimeric G-protein dynamics through dual association with Gβ and Gαi protein subunits. J. Biol. Chem. 287, 4863–4874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giguère P. M., Laroche G., Oestreich E. A., Duncan J. A., Siderovski D. P. (2012) Regulation of the subcellular localization of the G-protein subunit regulator GPSM3 through direct association with 14–3-3 protein. J. Biol. Chem. 287, 31270–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Craven R. R., Gao X., Allen I. C., Gris D., Bubeck Wardenburg J., McElvania-Tekippe E., Ting J. P., Duncan J. A. (2009) Staphylococcus aureus α-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4, e7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopez-Castejon G., Luheshi N. M., Compan V., High S., Whitehead R. C., Flitsch S., Kirov A., Prudovsky I., Swanton E., Brough D. (2013) Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J. Biol. Chem. 288, 2721–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sutterwala F. S., Ogura Y., Szczepanik M., Lara-Tejero M., Lichtenberger G. S., Grant E. P., Bertin J., Coyle A. J., Galán J. E., Askenase P. W., Flavell R. A. (2006) Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 [DOI] [PubMed] [Google Scholar]
- 24. Miao E. A., Ernst R. K., Dors M., Mao D. P., Aderem A. (2008) Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U.S.A. 105, 2562–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 26. Franchi L., Núñez G. (2008) The Nlrp3 inflammasome is critical for aluminum hydroxide-mediated IL-1β secretion but dispensable for adjuvant activity. Eur. J. Immunol. 38, 2085–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tschopp J., Schroder K. (2010) NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215 [DOI] [PubMed] [Google Scholar]
- 28. Broz P., Newton K., Lamkanfi M., Mariathasan S., Dixit V. M., Monack D. M. (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broz P., von Moltke J., Jones J. W., Vance R. E., Monack D. M. (2010) Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shenoy A. R., Wellington D. A., Kumar P., Kassa H., Booth C. J., Cresswell P., MacMicking J. D. (2012) GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485 [DOI] [PubMed] [Google Scholar]
- 31. Allam R., Darisipudi M. N., Rupanagudi K. V., Lichtnekert J., Tschopp J., Anders H. J. (2011) Cutting edge: cyclic polypeptide and aminoglycoside antibiotics trigger IL-1β secretion by activating the NLRP3 inflammasome. J. Immunol. 186, 2714–2718 [DOI] [PubMed] [Google Scholar]
- 32. Guarda G., Braun M., Staehli F., Tardivel A., Mattmann C., Förster I., Farlik M., Decker T., Du Pasquier R. A., Romero P., Tschopp J. (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223 [DOI] [PubMed] [Google Scholar]
- 33. Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D. R., Latz E., Fitzgerald K. A. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miao E. A., Alpuche-Aranda C. M., Dors M., Clark A. E., Bader M. W., Miller S. I., Aderem A. (2006) Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 7, 569–575 [DOI] [PubMed] [Google Scholar]
- 35. Azevedo C., Sadanandom A., Kitagawa K., Freialdenhoven A., Shirasu K., Schulze-Lefert P. (2002) The RAR1 interactor SGT1, an essential component of R gene-triggered disease resistance. Science 295, 2073–2076 [DOI] [PubMed] [Google Scholar]
- 36. Austin M. J., Muskett P., Kahn K., Feys B. J., Jones J. D., Parker J. E. (2002) Regulatory role of SGT1 in early R gene-mediated plant defenses. Science 295, 2077–2080 [DOI] [PubMed] [Google Scholar]
- 37. Hubert D. A., Tornero P., Belkhadir Y., Krishna P., Takahashi A., Shirasu K., Dangl J. L. (2003) Cytosolic HSP90 associates with and modulates the Arabidopsis RPM1 disease resistance protein. EMBO J. 22, 5679–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Noël L. D., Cagna G., Stuttmann J., Wirthmüller L., Betsuyaku S., Witte C. P., Bhat R., Pochon N., Colby T., Parker J. E. (2007) Interaction between SGT1 and cytosolic/nuclear HSC70 chaperones regulates Arabidopsis immune responses. Plant Cell 19, 4061–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spiechowicz M., Zylicz A., Bieganowski P., Kuznicki J., Filipek A. (2007) Hsp70 is a new target of Sgt1–an interaction modulated by S100A6. Biochem. Biophys. Res. Commun. 357, 1148–1153 [DOI] [PubMed] [Google Scholar]
- 40. Kadota Y., Shirasu K., Guerois R. (2010) NLR sensors meet at the SGT1-HSP90 crossroad. Trends Biochem. Sci. 35, 199–207 [DOI] [PubMed] [Google Scholar]
- 41. Levin T. C., Wickliffe K. E., Leppla S. H., Moayeri M. (2008) Heat shock inhibits caspase-1 activity while also preventing its inflammasome-mediated activation by anthrax lethal toxin. Cell Microbiol. 10, 2434–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakashima A., Chen L., Thao N. P., Fujiwara M., Wong H. L., Kuwano M., Umemura K., Shirasu K., Kawasaki T., Shimamoto K. (2008) RACK1 functions in rice innate immunity by interacting with the Rac1 immune complex. The Plant cell 20, 2265–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoser R., Zurczak M., Lichocka M., Zuzga S., Dadlez M., Samuel M. A., Ellis B. E., Stuttmann J., Parker J. E., Hennig J., Krzymowska M. (2013) Nucleocytoplasmic partitioning of tobacco N receptor is modulated by SGT1. New Phytol. 200, 158–171 [DOI] [PubMed] [Google Scholar]
- 44. Ge F., Li W. L., Bi L. J., Tao S. C., Zhang Z. P., Zhang X. E. (2010) Identification of novel 14–3-3zeta interacting proteins by quantitative immunoprecipitation combined with knockdown (QUICK). J. Proteome Res. 9, 5848–5858 [DOI] [PubMed] [Google Scholar]
- 45. Yamazaki H., Takeoka M., Kitazawa M., Ehara T., Itano N., Kato H., Taniguchi S. (2012) ASC plays a role in the priming phase of the immune response to type II collagen in collagen-induced arthritis. Rheumatol. Int. 32, 1625–1632 [DOI] [PubMed] [Google Scholar]
- 46. Ippagunta S. K., Brand D. D., Luo J., Boyd K. L., Calabrese C., Stienstra R., Van de Veerdonk F. L., Netea M. G., Joosten L. A., Lamkanfi M., Kanneganti T. D. (2010) Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J. Biol. Chem. 285, 12454–12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu L., Botos I., Wang Y., Leonard J. N., Shiloach J., Segal D. M., Davies D. R. (2008) Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 49. Jin M. S., Kim S. E., Heo J. Y., Lee M. E., Kim H. M., Paik S. G., Lee H., Lee J. O. (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082 [DOI] [PubMed] [Google Scholar]
- 50. Hong M., Yoon S. I., Wilson I. A. (2012) Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 36, 337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu Z., Yan C., Liu P., Huang Z., Ma R., Zhang C., Wang R., Zhang Y., Martinon F., Miao D., Deng H., Wang J., Chang J., Chai J. (2013) Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175 [DOI] [PubMed] [Google Scholar]
- 52. Damiano J. S., Oliveira V., Welsh K., Reed J. C. (2004) Heterotypic interactions among NACHT domains: implications for regulation of innate immune responses. Biochem. J. 381, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Franchi L., Muñoz-Planillo R., Núñez G. (2012) Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]