

Background: Galectin-3, a β-galactoside-binding protein, is expressed by many types of tumor cells.
Results: Galectin-3 secreted from a tumor works as a soluble inhibitory ligand of the NK cell receptor NKp30 to inhibit NK cell immune responses against tumors.
Conclusion: Galectin-3 regulates the antitumor immunity of human NK cells.
Significance: This novel mechanism may provide a new therapeutic target for tumor treatment.
Keywords: Galectin, Immunosuppression, Natural Killer Cells (NK Cells), Tumor, Tumor Therapy
Abstract
Human Galectin-3 (Gal-3), a β-galactoside-binding protein expressed by tumor cells, has been reported to act as an immune regulator in antitumor T cells. However, its effect on natural killer (NK) cells is elusive. Using a recombinant human NK cell-activating receptor, NKp30 fusion protein (NKp30-Fc), we found that soluble NKp30-Fc could immunoprecipitate Galectin-3. The direct interaction between NKp30 and Galectin-3 was further confirmed using surface plasmon resonance experiments. Because Galectin-3 was mainly released from tumor cells in a soluble form in our study, the binding assay was performed to show that soluble Galectin-3 specifically bound to NK cells and NKp30 on the surface of the NK cells. Functionally, when soluble Galectin-3 was added to the NK-tumor cell coculture system, the NKp30-mediated, but not NKG2D-mediated, cytolysis and CD107a expression in the NK cells were inhibited, and these phenotypes could be restored by preincubation of soluble Galectin-3 with NKp30-Fc fusion protein or the addition of anti-Gal-3 antibody alone. Moreover, genetic down-regulation of Galectin-3 (shGal-3) resulted in tumor cells being more sensitive to NK cell lysis, and, reversely, Galectin-3-overexpressing HeLa cells (exGal-3) became less sensitive to NK cell killing. The results of these in vitro experiments were supported by studies in shGal-3-HeLa or exGal-3-HeLa xenograft non-obese diabetic/severe combined immunodeficiency mice after NK cell adoptive immunotherapy, indicating that Galectin-3 strongly antagonizes human NK cell attack against tumors in vivo. These findings indicate that Galectin-3 may function as an immune regulator to inhibit NK cell function against tumors, therefore providing a new therapeutic target for tumor treatment.
Introduction
Galectin-3 (Gal-3),4 one of the 15 members of the β-galactoside-binding lectin family, contains either one or two carbohydrate recognition domains that have a high affinity for β-galactosides (1). The overall homology of human intergalectins is ∼20%, but their carbohydrate recognition domains are relatively conserved (2). Previous studies showed that Gal-3 is highly expressed in many types of cancer cells (3–5). Depending on cell type and proliferative status, Gal-3 can exist in the cytoplasm, within the nucleus, on the cell surface, and in the extracellular compartment (6–8). Published data regarding different tumor cells, such as human breast cancer (9, 10) and melanoma (11) cells, have shown that intracellular Gal-3 promotes tumor growth, survival, and metastasis and that extracellular Gal-3 may facilitate metastasis by promoting tumor cell adhesion (12), invasiveness (13), and even immune escape (1, 14), the latter of which is investigated relatively rarely.
More than ten years ago, it was noted that Gal-3 negatively regulates the T cell response and plays an important role in host homeostasis. The most recognizable mechanism by which Gal-3 regulates the immune response is through the induction of apoptosis in T cells. Fukumori et al. (15) reported that the secretion of extracellular Gal-3 from tumor cells can activate apoptosis in both human and murine T cells after its binds to the cell surface glycoconjugate receptors CD7 and CD29, providing new insight into the mechanism by which cancer cells escape the immune system. Wang and co-workers (11) further confirmed this conclusion in both humans and mice by showing that colorectal tumor-reactive T cells became apoptotic in response to Gal-3 stimulation, leading to enhanced tumor growth in vitro and in vivo (11). A human study also demonstrated that Gal-3 was down-regulated significantly in biopsies of inflamed tissue from inflammatory bowel disease patients. However, Gal-3 was expressed at comparably high levels in recovered inflammatory bowel disease patients. A genetic deficiency in Gal-3 rescued the apoptosis phenotype of the T cells and induced autoimmunity. In contrast, exogenous Gal-3 led to reduced proliferation of blood T cells. This finding illustrates that constitutive expression of epithelial Gal-3 may help to prevent inappropriate immune responses, providing solid evidence to support the hypothesis that Gal-3 is an immune regulator (16). On the basis of these findings, blockade approaches against Gal-3 have been explored. It has been reported that treatment with N-acetyllactosamine, a galectin ligand, or an anti-Gal-3 antibody restored the impaired secretion of IFN-γ by tumor-infiltrating cytotoxic T lymphocytes. Moreover, GCS-100, a polysaccharide that may detach Gal-3 from tumor-infiltrating lymphocytes, boosted cytotoxicity and the secretion of IFN-γ, leading to tumor rejection in mice (17). Recently, Guha et al. (18) found that TFD100, a glycopeptide from cod that binds Gal-3 with picomolar affinity, inhibited the apoptosis of activated T cells following induction with either recombinant Gal-3 or prostate cancer patient serum-associated Gal-3 at nanomolar concentrations. Collectively, Gal-3 may work as an immune regulator to induce apoptosis in activated T cells.
Natural killer (NK) cells, which are effector lymphocytes of the innate immune system, provide the first line of defense against tumors. NK cells distinguish between normal healthy cells and abnormal cells using a sophisticated repertoire of cell surface receptors that control their activation, proliferation, and effect functions (19). For example, the natural cytotoxicity receptors (20), including NKp44 (21, 22), NKp46 (23), and NKp30 (24, 25), as well as NKG2D, are involved in the antitumor response (26, 27). Previous studies showed that Gal-3 is involved in the regulation of NK cell activation and function. Data from Dr. Gordana (41) demonstrated that Galectin-3-deficient mice are more resistant to lung metastases of malignant melanoma and that tumor-bearing Gal-3-deficient mice exhibit higher serum levels of IFN-γ and IL-17 than control tumor-bearing mice. Interestingly, in this model, the cytotoxic activity of splenic NK cells, but not cytotoxic T lymphocytes was greatly enhanced in Gal-3-deficient mice, suggesting that the NK cells of tumor-bearing mice are preferentially affected by Gal-3. In contrast with the Gal-3-induced apoptosis of T cells in antitumor immunity, the mechanism of Gal-3 inhibition in NK cell tumor immunity involves shielding the ligands on the tumor cells from NK cell-activating receptors. For example, the NK-activating receptor NKG2D is critical for tumor rejection after recognition of its tumor-associated ligand, major histocompatibility complex class I-related chain A (MICA). Gal-3 can bind the NKG2D binding site of MICA, which is expressed on the tumor cell surface, through the core two O-glycans of MICA, thus shielding the interactive recognition of the bladder tumor cells by the NK cells and severely impairing NK cell activation and silencing the NK cells (28). Because MUC-1, another ligand of the NK cell receptors, also contains the core two O-glycan structure, the binding of Gal-3 to MUC-1 attenuated the interaction between the tumor cells and the NK cells, leading to the tumor cells evading NK cell immunity (29).
In this study, we explored a novel mechanism by which Gal-3 regulates the antitumor immunity of human NK cells. Using the human cervical cancer and breast cancer model, we found that soluble Gal-3 released from tumor cells could specifically bind NKp30, thereby inhibiting the NKp30-mediated cytotoxicity of NK cells. Genetic down- or up-regulation of Gal-3 in tumor cells led to tumor growth inhibition or enhancement in an NK cell-dependent manner, respectively, both in vitro and in vivo. These findings demonstrate that Gal-3 is a soluble inhibitory ligand of NKp30 and may function as an immune regulator to inhibit the NK cell immune responses against tumors, providing a new therapeutic target for tumor treatment.
MATERIALS AND METHODS
Antibodies
Anti-human NKp30 (clone P30-15) was purchased from Biolegend. Anti-human Gal-3 (clone B2C10), PE-conjugated anti-human -NKp30, PE-conjugated anti-human CD107a, FITC/PE-Cy5-conjugated anti-human CD56, FITC-conjugated goat anti-human IgG, and PE-conjugated goat anti-mouse IgG were purchased from BD Biosciences. PE-CY7 -conjugated anti-human NKG2D was obtained from R&D Technologies.
Mice and Cells
All experiments involving mice were approved by the Animal Care and Use Committee at the University of Science and Technology of China. The 6- to 8-week-old NOD-SCID mice were purchased from the Shanghai Laboratory Animal Center (Shanghai, China) and were bred under specific pathogen-free conditions according to the experimental animal guidelines of the University of Science and Technology of China.
Human cervical cancer cell line HeLa cells, human breast cancer cell line MDA-MB-435 cells, and HEK 293T cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS and 2 mm l-glutamate. NK-92 cells were cultured in α-minimum Eagle's medium (Invitrogen) containing 12.5% FBS (Invitrogen), 12.5% equine serum (HyClone), 2 mm l-glutamate, 0.1 mm 2-mercaptoethanol, and 100 units/ml rhIL-2 (Changchun Institute of Biological Products, China). Human NK cells were obtained from the peripheral blood mononuclear cells of healthy donor buffy coats using Ficoll-Paque density gradient centrifugation (Solarbio, China). Non-NK cells were depleted using an NK cell isolation kit according to the instructions of the manufacturer (Miltenyi Biotech). The separated NK cells were cultured in complete α-minimum Eagle's medium (Invitrogen). Cell culture was performed at 37 °C in a 5% CO2 humidified atmosphere.
Soluble NKp30-Fc Fusion Proteins
The sequence encoding the extracellular portions of NKp30 (GenBank accession no. AB055881) was amplified by PCR from cDNA isolated from human NK clones. The NKp30-specific primers were 5′ primer (including an EcoRI restriction site) 5′-CCGGAATTCCTCTGGGTGTCCCAGCCCCCTGAGATTCGTA-3′. The 3′ primer (including the XhoI restriction site) was 5′-CCGCTCGAGGGACTGAAAATACAGGTTTTCCCCTAGCTGAGGATGTTC-3′. The cDNA fragment encoding the Fc regions of human immunoglobulin G1 (GenBank accession no. BC006402) was amplified by RT-PCR from RNA isolated from human peripheral blood lymphocytes. The Fc region-specific primers were 5′-primer (including an XhoI restriction site) 5′-CCGCTCGAGCCCAAATCTTGTGACAAAACT-3′ and 3′-primer (including the XbaI restriction site) 5′-CTAGTCTAGATCATTTACCCGGAGACAGGGAGAGG-3′. The resulting amplified product was cloned into the pcDNA3.0 vector (Invitrogen). Sequencing of the constructs revealed that NKp30 receptor ectodomain cDNA was in-frame with the human Fc genomic DNA and was identical to the reported sequences. CHO-K1 cells were transfected with these expression vectors and G418-selected clones were screened for highest protein production and subcloned further by limited dilution method for high producer clones. Supernatants were collected daily and purified on protein G columns. SDS-PAGE analysis revealed that both Ig fusion proteins were ∼95% pure and of the proper molecular mass (∼55 kDa for NKp30-Fc under reducing conditions).
Depletion and Overexpression of Gal-3
For the depletion of Gal-3, lentiviruses expressing Gal-3 or control shRNAs were prepared. HEK 293T cells grown in a 6-cm dish were transfected with 2 μg of Gal-3 shRNA (cloned into the PLKO.1 vector) or control vector, 2 μg of pREV, 2 μg of pGag/Pol/PRE, and 1 μg of pVSVG. Twenty-four hours after transfection, the cells were cultured in DMEM containing 20% FBS for an additional 24 h. The culture medium containing the lentivirus particles was centrifuged at 1000 × g for 5 min. The viruses in the supernatant were used to infect tumor cells. The knockdown efficiency was evaluated using Western blot and real-time RT-PCR analyses. The shRNA sequence targeting Gal-3 was 5′-CCGGGCTCACTTGTTGCAGTACAATCTCGAGATTGTACTGCAACAAGTGAGCTTTTT-3′.
For the overexpression of Gal-3, HeLa cells were transfected with pCMV6-Gal-3 or the control vector. Twenty-four hours after transfection, the cells were cultured in DMEM containing 20% FBS for an additional 24 h. The cells were then screened with G418 for 4 days. The overexpression efficiency was evaluated using Western blot and real-time RT-PCR analyses.
Immunoprecipitation and Western Blot Analysis
Cells were collected and resolved in lysis buffer containing 1% Triton X-100, 25 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 100 mm NaCl, 10 mm NaF, 1 mm PMSF (Sigma), 2 mm EDTA, and a protease inhibitor mixture (Roche). The cell lysates were preincubated with soluble proteins (final concentration, 10 μg/ml) at 4 °C for 2 h. Protein A/G-agarose was preincubated with or without mAbs (final concentration, 10 μg/ml) at 4 °C for 2 h. Then the protein A/G-agarose was washed twice with cell lysis buffer and added to the cell lysates for 2 h or overnight at 4 °C. For the biotinylation of the proteins, cell monolayers were incubated with 1 mg/ml EZ-Link Sulfo-NHS-Biotin (Pierce) in PBS (pH 8.0) for 30 min at 25 °C, followed by three washes with cold PBS. After extensive washing, the cells were lysed and incubated with streptavidin-coated agarose beads (Pierce) at 4 °C for 2 h to precipitate the cell membrane proteins. The protein-bound beads were washed four times with cell lysis buffer and resuspended in PBS. Finally, the suspension was mixed with 2× SDS loading buffer, boiled, and centrifuged to remove the pellet. The supernatant was separated using SDS-PAGE electrophoresis under reducing or non-reducing conditions on a 12% polyacrylamide gel and then electrotransferred onto PVDF membranes (Millipore) in vertical buffer tanks. The membranes were blocked with 5% nonfat milk in TBST buffer (50 mm Tris-HCl (pH 7.4), 0.9% NaCl, and 0.1% Tween 20) and then incubated with primary antibodies for 2–3 h at room temperature or overnight at 4 °C. After addition of the HRP-conjugated secondary antibodies for 1 h, the bands on the membranes were detected using an enhanced chemiluminescence system (Pierce).
Surface Plasmon Resonance
Surface plasmon resonance analysis was performed on a BIAcore 3000 apparatus at 25 °C. Recombinant human (rh) Gal-3 (R&D Technologies) was covalently immobilized to the carboxyl groups in the dextran layer of a Biacore Sensor Chip CM5. Gal-3 was injected in 0.01 m sodium acetate buffer (pH 5). Solutions containing the different recombinant proteins were injected sequentially over the surface of the immobilized Galectin-3, or buffer was used as a blank. For the regeneration of the surface, we used PBS (pH 7.2), which also served as the running buffer. The flow rate was set at 10 μl/min. The data were analyzed using Biaevaluation 4.1 software.
Flow Cytometric Analysis
For cell surface staining, the cells were harvested, blocked with an anti-FcγR mAb, and incubated with the appropriate concentrations of the recombinant proteins or Abs for 30 min at 4 °C. To stain intracellular proteins, the cells were fixed in 1% paraformaldehyde in PBS/0.05% BSA for 30 min on ice and washed in permeabilization buffer (eBioscience). The cells were permeabilized in permeabilization buffer containing 3% BSA for 30 min at 4 °C, followed by staining with the primary antibodies. All proteins and mAbs were used at a final concentration of 10 μg/ml. All stained cells were gated for analysis using a FACSCalibur cytometer (BD Biosciences), and the data were processed using FlowJo software (TreeStar, Ashland, OR).
ELISA
For assays determining the amount of Gal-3 protein secreted by the tumor cells, a direct ELISA was used. Briefly, 150 μl of supernatant, which was collected at various time points, was added to microtiter plates, and the plates were incubated overnight at 4 °C. After washing, the plates were blocked as described above. An anti-human Gal-3 antibody (diluted 1:500 in 0.3% BSA/0.1% PBST) was then added to the plates for 2 h at 37 °C. The plates were incubated with an HRP-conjugated anti-mouse antibody for 1 h, followed by incubation with tetramethylbenzidine as a substrate for 5–10 min. The colorimetric signal was measured at an optical density of 450 nm.
Cytotoxicity Assay
The cytotoxicity of the NK-92 cells and human NK cells against the target cells was measured using a standard 4-hour 51Cr release assay as described previously (30). Briefly, the target cells were labeled with 51Cr by incubation with Na251CrO3 (200 μCi/106 cells) for 1 h at 37 °C and then washed three to four times with PBS. The labeled target cells were combined with NK cells at different effector:target (E:T) cell ratios. For the blocking assay, the NK-92 cells or primary NK cells were preincubated with Gal-3 protein or an anti-NKp30 antibody for 30 min or 1 h at 4 °C prior to the addition of the target cells to prevent the NKp30 ligands on the target cells from binding the NKp30-activated receptor on the NK cells. In turn, the target cells were preincubated with the NKp30-Fc protein or an anti-Gal-3 antibody for 30 min at 4 °C prior to the addition of the NK cells to prevent the activation of NK cell killing by the NKp30 ligands derived from the target cells. To restore NK cell killing, NKp30-Fc was preincubated with Gal-3 protein for 30 min at 4 °C before the NK cells killed the target cells. Maximum 51Cr release was determined by incubating the target cells with 2% Triton X-100. To measure the spontaneous 51Cr release, the target cells were incubated without effector cells in assay medium alone. All experiments were performed in triplicate. The plates were spun slightly and incubated at 37 °C for 4 h. After 4 h, the cells were centrifuged, and the amount of 51Cr release in 100 μl of the supernatant was counted using a γ counter. The percentage of specific 51Cr release was calculated using the following formula: ([experimental release − spontaneous release] / [maximum release − spontaneous release]) × 100. Spontaneous release did not exceed 10% of the maximum release in all experiments.
Degranulation Assays
To test the NK cell-derived degranulation, NK-92 cells (1 × 105) were incubated with different proteins or protein complexes (10 μg/ml) for 1 h at 37 °C, and tumor cells were incubated with antibody or control mouse IgG (5 μg/ml) for 1 h at 37 °C. Then the NK-92 and untransfected or transfected tumor cells were mixed at an effector:target ratio of 1:1, and the cells were incubated for 2 h at 37 °C in the presence of anti-human CD107a PE-conjugated antibodies, anti-human CD56 PE-Cy5/FITC-conjugated antibodies, and isotype-matched control antibodies. As a positive control, NK-92 cells were incubated with 5 μg/ml phorbol 12-myristate 13-acetate (final concentration of 50 ng/ml, Sigma) and 1 μg/ml ionomycin (final concentration of 10 μg/ml, Sigma). After incubation, the cells were washed and resuspended. CD56+ cells were gated for analysis using a FACSCalibur flow cytometer (BD Biosciences).
Cervical Adenocarcinoma Xenograft Model
HeLa cells (4 × 106) were left untreated or were transfected with the different Gal-3 constructs used to deplete or overexpress the protein. The HeLa tumor cells were resuspended in 200 μl of PBS and injected subcutaneously into NOD-SCID mice to establish tumors. NK-92 cells were injected subcutaneously into the tumor issue area (2 × 106/200 μl/animal) 5 days after tumor inoculation. The tumor volumes were measured on days 10 and 20, and the formation of the tumors was observed on day 20.
Statistical Analysis
Data were expressed as mean ± S.D., and significance was denoted as *, p < 0.05; **, p < 0.01; and ***, p < 0.001. Calculations were performed using GraphPad Prism software with Student's t test and PS-Power and sample size software with power analysis for Student's t test (31).
RESULTS
Identification of Galectin-3 as a Ligand of Human NKp30
NK cells are important components of the innate immune system (32) and participate in the elimination of tumors (33). NKp30 is a human receptor that triggers antitumor NK cell cytotoxicity and cytokine secretion (25). To detect a novel ligand of NKp30, we generated soluble NKp30-Fc fusion proteins. When NKp30-Fc was immunoprecipitated from HeLa cell lysates, an extra band was observed in the NKp30-Fc precipitate (Fig. 1A). This protein band was subjected to in-gel tryptic digestion and analyzed by mass spectrometry, and the results demonstrated that this band corresponded to Gal-3, a member of the β-galactoside-binding lectin family. Immunoblot analysis also showed that NKp30-Fc could interact with Gal-3 in HeLa cells (Fig. 1B). To confirm the direct interaction between NKp30 and Gal-3 and to rule out the possibility of Gal-3 binding to the Fc tag, surface plasmon resonance experiments were performed. The results showed that Gal-3 specifically bound to the extracellular domain of NKp30 because it did not bind to human IgG (Fig. 1C). These results suggest that tumor-derived Gal-3 is a potential ligand of NKp30.
FIGURE 1.
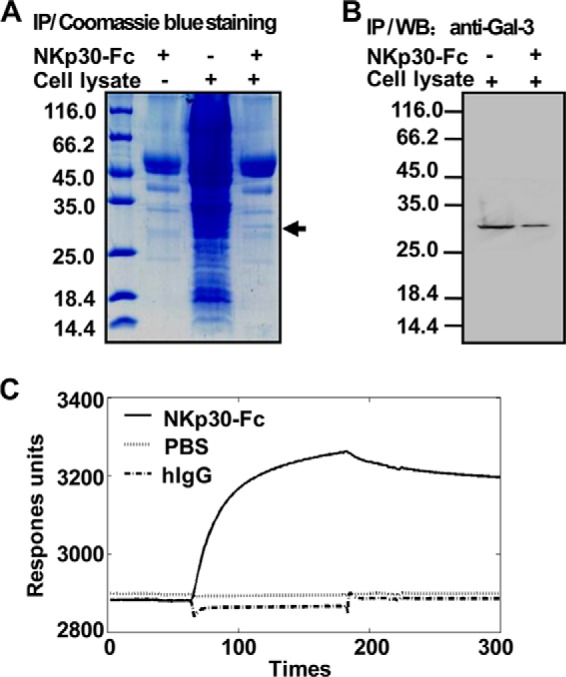
Interaction of Gal-3 with the extracellular domain of NKp30. A, binding of tumor-associated proteins to soluble NKp30-Fc. The NKp30-Fc fusion protein coupled to protein A/G-Sepharose beads were incubated with HeLa cell lysate, and the proteins that bound to NKp30-Fc were resolved by SDS-PAGE and stained with Coomassie Blue. An ∼30-kDa cellular protein that interacts with NKp30-Fc was present in the cell lysate. NKp30-Fc alone served as a negative control. IP, immunoprecipitation. B, binding of tumor-derived Gal-3 to soluble NKp30-Fc. Gal-3 protein or the cell lysate was examined by immunoblot analysis with an anti-Gal-3 mAb in immune complex. WB, Western blot. C, kinetics of the interactions between rhGal-3 and NKp30-Fc. rhGal-3 was covalently immobilized to the carboxyl groups in the dextran layer of a BIAcore sensor chip. Solutions containing NKp30-Fc or human IgG (hIgG) were injected over the surface of immobilized rhGal-3, and PBS buffer was used as a control. Surface plasmon resonance analysis was performed on a BIAcore 3000 apparatus at 25 °C. Under these conditions, binding of immobilized rhGal-3 to soluble NKp30-Fc was observed but binding to human IgG was not. The data are representative of two independent experiments.
Cellular Distribution of Galectin-3 in Human Tumor Cells
Gal-3 is expressed in a variety of forms in tumor cells, depending on the cell type and proliferative status. A previous study showed that Gal-3 can be detected in the nucleus and cytoplasm of the HeLa cell line (34). Here biotinylated membrane-bound proteins on HeLa cells were immunoprecipitated with streptavidin-coated agarose beads, followed by immunoblot analysis with an anti-Gal-3 antibody. Gal-3 was present in the total cell lysate but was not found in the membrane fraction of HeLa cells (Fig. 2A). Flow cytometry also showed that Gal-3 was expressed intracellularly but that it was not found on the membranes of HeLa cells (Fig. 2B). Additionally, it has been reported previously that the concentration of circulating Gal-3 was increased markedly in the sera of patients with different tumor types (35, 36). Therefore, an ELISA assay was performed to identify the soluble Gal-3 released from tumor cells. As shown in Fig. 2, C and D, there was a high level of Gal-3 in the HeLa and MDA-MB-435 cells culture supernatant, and this level peaked at 12 and 24 h, respectively. The results showed that Gal-3 was present in a soluble form and could be released by tumor cells.
FIGURE 2.
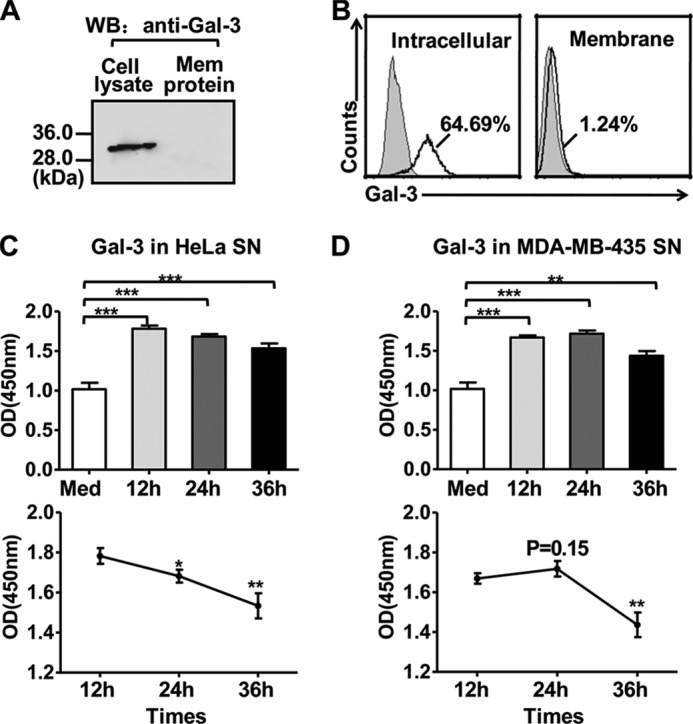
Gal-3 distribution in tumor cells. A and B, Gal-3 distribution (intracellular, surface) in HeLa cells was examined by immunoprecipitation (A) or flow cytometry (B). A, HeLa cells were biotinylated to label the cell surface proteins, and the biotinylated membrane proteins were then precipitated using streptavidin-coated agarose beads. Gal-3 from the cell lysate (left lane) or the membrane protein fraction (right lane) was detected using immunoblot analysis with an antibody against Gal-3. WB, Western blot. B, HeLa cells were treated with permeabilization buffer (left panel) or without (right panel), followed by incubation with an anti-Gal-3 mAb, and stained with a fluorescence-conjugated polyclonal antibody. C and D, Gal-3 is released from tumor cells. ELISA plates were coated with 100 μl of culture supernatants (SN) collected from HeLa (C) or MDA-MB-435 (D) cells 12, 24, or 36 h after cells were recovered in fresh medium. Control medium served as a negative control. Gal-3 was detected using an anti-Gal-3 mAb and stained with anti-mouse HRP-linked secondary antibody. The data represent the absorbance at 450 nm after normalization to the background (nonspecific binding of the antibody to the plate). The data are representative of two independent experiments and were analyzed using Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.0001.
Galectin-3 Specifically Binds to NKp30 on NK Cells
Flow cytometry analysis showed that Gal-3 protein, compared with BSA, was significantly bound to the NK cells (Fig. 3A), resulting in a much higher mean fluorescence intensity (MFI, Fig. 3B). We next examined whether Gal-3 directly interacted with the NKp30 receptor. Total NK cell lysate was incubated with Gal-3 and coprecipitated with an anti-Gal-3 antibody or an immobilized NKp30 antibody, followed by immunoblot analysis for NKp30 or Gal-3. The results showed that Gal-3 could specifically bind NKp30 from NK cells (Fig. 3C).
FIGURE 3.
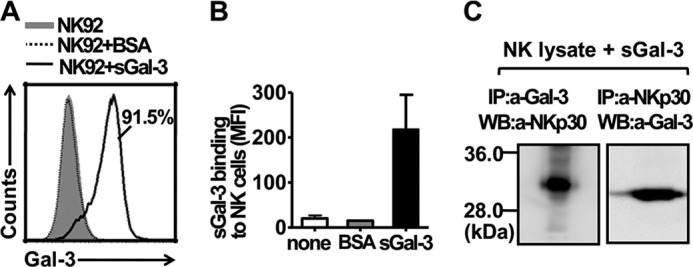
Binding of Gal-3 to NKp30 on NK cells. A, binding of Gal-3 to NK-92 cells. An anti-Gal-3 mAb was preincubated with the irrelevant protein BSA or soluble Gal-3, with or without an isotype-matched control antibody (gray histogram), and the solution was then incubated with NK-92 cells and stained with a fluorescence-conjugated polyclonal antibody. B, the binding ratio of BSA or Gal-3 to the NK-92 cells was determined by statistical analysis of the mean fluorescence intensity (MFI) from two independent experiments, as described in A. C, soluble Gal-3 (sGal-3) was precipitated with NKp30 from NK cells. NK-92 cell extracts were incubated with soluble Gal-3, and the protein complex was immunoprecipitated (IP) with an anti-Gal-3 mAb or an anti-NKp30 mAb immobilized on protein A/G-Sepharose beads. Immunoblot analysis was performed for NKp30 using an anti-NKp30 mAb (left panel) or for Gal-3 using an anti-Gal-3 mAb (right panel). WB, Western blot.
Galectin-3 Inhibits the NKp30-mediated Cytolysis of NK Cells
Because it has been reported that Gal-3 might block the binding of NKG2D to MICA (28), we selected a NK-92 cell clone that does not express NKG2D to exclude the influence of Gal-3 interacting with NKG2D (Fig. 4A). As shown in Fig. 4, the addition of Gal-3 into the NK-92-HeLa coculture system significantly reduced the expression of CD107a, a marker of NK cell degranulation, during the initiation of cytolysis, whereas CD107a expression was restored by preincubating the Gal-3 with the NKp30-Fc fusion protein (Fig. 4, B and C). This result was further confirmed using a 4-h 51Cr release assay to examine the direct cytolysis of the tumor cells by the NK cells. The results showed that the susceptibility of the tumor cells to NK-92 (NKG2D-NK92 clone) cell cytolysis was impaired by the addition of Gal-3, whereas this effect of Gal-3 was attenuated by preincubating the Gal-3 with the NKp30-Fc fusion protein (Fig. 4D). Moreover, the cytotoxicity of primary human NK cells was also impaired by the addition of Gal-3, and these results were the opposite of what was observed following incubation with an anti-Gal-3 antibody (Fig. 4E). Not only HeLa cells but also MDA-MB-435 cells could induce NK-92 cells to express CD107a, which was increased significantly in the presence of anti-Gal-3 antibody (Fig. 4F). Therefore, the results suggested that the Gal-3 that is released from tumor cells could impair NK cell-mediated cytotoxicity by directly antagonizing NKp30 function.
FIGURE 4.
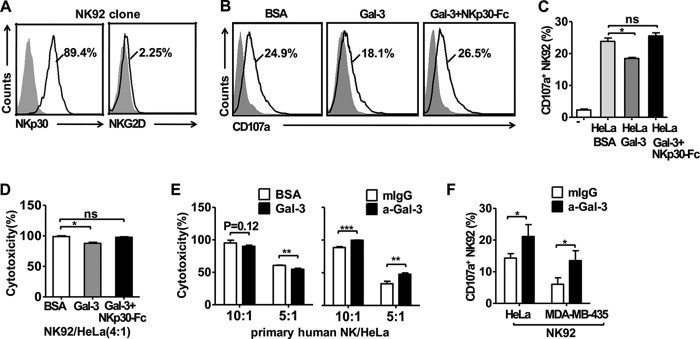
Gal-3 inhibits NK cell cytotoxicity by binding to NKp30. A, selection of the NKp30-expressing, but not NKG2D-expressing, NK-92 cell clone. NK-92 cells were stained with PE-conjugated anti-NKp30 antibody, PE-CY7-conjugated anti-NKG2D antibody, or an isotype control (gray histograms). The data are representative of two independent experiments. B and C, CD107a surface expression was measured by flow cytometry after coincubation of NK-92 cells with HeLa cells in the presence of BSA, Gal-3, or Gal-3 plus NKp30-Fc. The gray histograms represent the isotype control. D, blocking soluble Gal-3 restored the cytotoxicity of the NK-92 cells. At 4 h, the cytotoxicity assay was performed in the presence of BSA, Gal-3, or Gal-3 plus NKp30-Fc at a 4:1 (E:T) ratio. The cytotoxicity of NK-92 cells against HeLa cells was normalized to 100% in the presence of BSA. E, the cytotoxicity of primary human NK cells against HeLa cells was assayed. The effector NK cells were incubated with Gal-3 or BSA or with the anti-Gal-3 mAb or control mIgG1κ mAb for 1 h. Then, HeLa cells (target cells) labeled with 51Cr were added at the indicated E:T ratios. A 4-h cytotoxicity assay was performed, and the inhibition of NK cell cytotoxicity by soluble Gal-3 is indicated. The cytotoxicity of human NK cells against HeLa cells was normalized to 100% at the most E:T (10:1) ratios. The data of the cytotoxicity assay are representative of two independent experiments. F, anti-Gal-3 antibody restored the CD107a surface expression of NK-92 cells. Flow cytometry was performed to measure CD107a surface expression after coincubation of NK-92 cells with HeLa cells or MDA-MB-435 cells in the presence of anti-Gal-3 (a-Gal-3) mAb or control mIgG1κ. The data are representative of two independent experiments and were analyzed using Student's t test. *, p < 0.05; **, p < 0.01;***, p < 0.0001; n.s., not significant.
Galectin-3 Is an Inhibitory Ligand That Blocks Human NK Cell Activity against Tumors in Vitro and in Vivo
We constructed a Gal-3-specific lentiviral-based shRNA to knock down the endogenous expression of Gal-3 in HeLa tumor cells, and the results showed that genetic down-regulation of Gal-3 (shGal-3) resulted in the HeLa cells becoming more sensitive to NK cell lysis (Fig. 5A). In contrast, HeLa cells that overexpressed Gal-3 (exGal-3) became less sensitive to NK cell killing (Fig. 5B). This result was also found using Gal-3-silenced MDA-MB-435 cells to examine the CD107a expression of NK-92 cells (Fig. 5C). Therefore, the evidence suggests that Gal-3 expression of tumor cells directly affects NK cell function.
FIGURE 5.
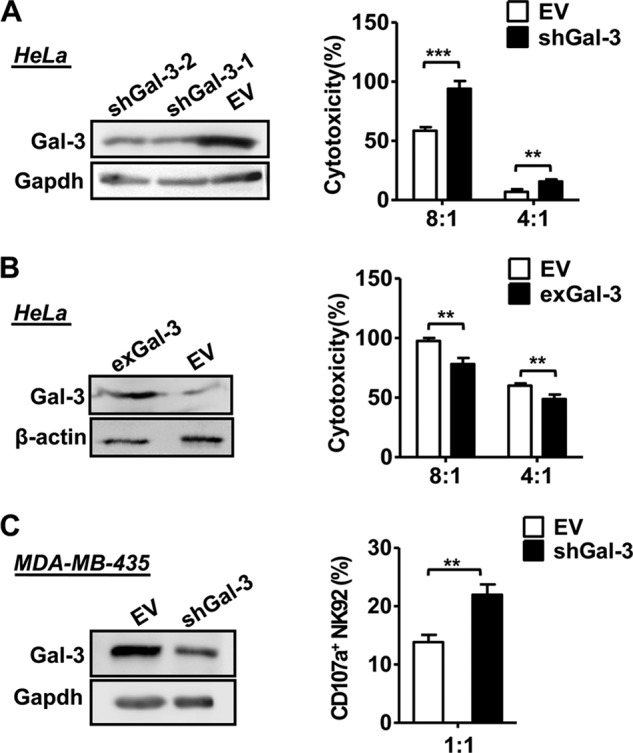
Effect of Galectin-3 down-regulation and overexpression on NK cytotoxicity. HeLa cells were transfected with an empty vector (EV), Gal-3-specific lentivirus-based shRNA (shGal-3, A), or Gal-3 expression vector (exGal-3, B), and then the cell lysate was prepared and analyzed for Gal-3 expression using immunoblot analysis. GAPDH (A) and β-actin (B) were used as controls. Then transfected HeLa cells were incubated with NK-92 cell clones at effector:target ratios of between 8:1 and 4:1, and the lysis of the target cells was determined using a 4-h europium release assay. NK cell cytotoxicity was normalized to 100% at the most E:T (8:1) ratios. C, MDA-MB-435 cells were transfected with an empty vector or Gal-3-specific lentivirus-based shRNA, and immunoblot analysis was performed as described previously. Flow cytometry was performed to measure CD107a surface expression after coincubation of NK-92 cells with empty vector or shGal-3 MDA-MB-435 cells. The data are representative of two independent experiments and were analyzed using Student's t test. **, p < 0.01; ***, p < 0.0001.
It has been reported that Gal-3 expression by tumor cells might intrinsically contribute to tumor progression (9–11). Here we wanted to elucidate whether Gal-3 negatively regulated NK cell-mediated antitumor immunity in vivo. Therefore, we used NOD-SCID mice bearing xenografts of Gal-3-silenced HeLa (shGal-3) or Gal-3-overexpressing HeLa (exGal-3) tumor cells, and we treated these mice using adoptive immunotherapy with human NK-92 cells. As shown in Fig. 6A, the NOD-SCID mice were left untreated or were injected subcutaneously with HeLa cells (exGal-3 or shGal-3), followed by intratumoral injection of NKG2D-NK-92 cells (Fig. 4A) during the early stage (day 5) of tumor growth. Tumor growth was then observed on days 10 and 20. We found that the tumor volume was reduced after NK treatment in the WT HeLa-bearing mice on days 10 and 20 (Fig. 6, B and C), indicating that the HeLa tumor was sensitive to NK cell therapy. Importantly, the shGal-3 HeLa tumors became much more sensitive to NK attack, whereas the exGal-3 HeLa tumors became more resistant to NK attack on days 10 and 20 (Fig. 6, B and C). These results further suggested that Gal-3 secreted by the tumor antagonized human NK cell attack against the tumor in vivo. Previous reports also demonstrated that Gal-3 expression directly improved tumor growth in xenograft mice without NK cell therapy (9–11). However, the efficiency of NK cell therapy was still attenuated by Gal-3 expression from the xenograft tumor (data not shown). Taken together, these results imply that Gal-3 may function as an immune regulator to inhibit NK cell function against the tumor and that inhibition of Gal-3 expression may serve as a therapeutic target for cancer therapy.
FIGURE 6.

Gal-3 blocks human NK cell activity against tumors in vitro and in vivo. A, xenograft tumor model. Untreated or transfected (exGal-3 or shGal-3, respectively) HeLa tumor cells were injected intraperitoneally into NOD-SCID mice, followed by injection with PBS or NK cells into the tumor tissue area 5 days after tumor inoculation. B and C, tumor volumes were measured on days 10 and 20 (B), and the formation of tumors was observed on day 20 (C) after tumor inoculation. The data are representative of two independent experiments with five animals in each group, and the maximum and minimum tumor volumes were excluded from the statistics. The data were analyzed using Student's t test and power analysis for t test (Pwr). *, p < 0.05; **, p < 0.01; ***, p < 0.0001.
DISCUSSION
Using human cervical cancer and breast cancer cell lines and the xenograft tumor NOD-SCID model, we showed that tumors can escape from NK cell attack by releasing soluble Gal-3, which specifically binds to NKp30 and inhibits the NKp30-mediated cytotoxicity of NK cells. Genetic up- or down-regulation of Gal-3 in tumor cells led to tumor promotion or inhibition, respectively, in an NK cell-dependent manner both in vitro and in vivo. Our results revealed that Gal-3 is a soluble ligand of NKp30 and can function as an immune regulator to mediate tumor escape from NK cell immunity.
Previous studies have shown that Gal-3 may facilitate tumor metastasis through immune escape. For example, data from Zubieta et al. (37) demonstrate that Gal-3 expression correlates with the apoptosis of tumor-associated lymphocytes in human melanoma biopsies. A recent study also demonstrated that Gal-3 secreted from tumors directly interferes with the formation of the immune synapses of antitumor T cells and, thereby, inhibits tumor-reactive CD8+ T cells, leading to fast tumor growth in a mouse model of colorectal cancer (11). Until now, the mechanisms by which Gal-3 induces immune suppression were grouped into two categories. One category includes the stimulation of apoptosis by the binding of Gal-3 to antitumor T cells (11, 37), and the other includes the shielding of ligands on the surface of tumor cells from the NK cell-activating receptors (28, 29). NK cells are critically important in destroying tumor cells, and their antitumor function is performed mainly through NK cell receptor-tumor ligand interactions, resulting in the release of cytotoxic granules containing perforin and granzymes (26, 27). For instance, tumor cells expressing ligand MICA of the NK-activating receptor NKG2D stimulated tumor immunity mediated by NK cells (38). Interestingly, a study from Tsuboi et al. (28) demonstrated that Gal-3 could bind the NKG2D-binding site of MICA and reduce the affinity of MICA for NKG2D in bladder tumor cells, thereby evading NK cell immunity. This finding suggests that the Gal-3 released by tumor cells may possibly alter the NK cell responses by modulating their recognition of the tumor.
In our study, a new mechanism was observed in which tumor-associated Gal-3, as a soluble ligand of NKp30, directly bound to NKp30 and blocked NKp30-mediated cytotoxicity of NK cells. Similar to NKG2D, NKp30, which is an important NK-activating receptor, is also involved in NK lytic activity against tumor cells by interacting with their physiological ligands. Several tumor ligands of NKp30 have been found. For example, the nuclear factor HLA-B-associated transcript 3 (BAT3) is released from tumor cells in membrane vesicles, such as exosomes (39), and B7-H6 is expressed selectively on some tumor cells (40). Both of these ligands can engage NKp30 on NK cells and induce NKp30-dependent cell activation and cytotoxicity. In contrast to these ligands, soluble Gal-3, which was released from tumors in our study, could directly inhibit NK cell activation by binding to NKp30. To exclude the influence of Gal-3 on MICA/NKG2D, we selected a NKG2D−NK-92 cell clone, and we found that Gal-3 absolutely inhibited NK cells in the absence of NKG2D (Fig. 4A), demonstrating that the NKp30-mediated cytolysis of NK cells was impaired in the presence of Gal-3.
Our in vivo results clearly demonstrated the regulatory effect of Gal-3 on the antitumor NK cells. In our study, the cytotoxicity of NK cells was almost completely lost when a blocking anti-NKp30 antibody was added to the NK-HeLa coculture system in vitro because of the complete blockade of all NKp30-ligand interactions (data not shown). These results demonstrate that NKp30 is critical for the antitumor capability of NK cells. Moreover, when Gal-3 was depleted or overexpressed in human tumor cells, the antitumor function of the adoptively transferred human NKG2D-NK cells was enhanced or reduced, respectively, further indicating that Gal-3 inhibits NK cell function by binding to NKp30 (Fig. 6). We demonstrated that, after incubation of the NKp30-Fc protein with Gal-3 in vitro, the NKp30-Fc protein could compete with Gal-3 and attenuate the Gal-3 effect on NK cells. However, we inferred that the in vivo data did not support this conclusion because NKp30-Fc could bind to all of its activating and inhibiting ligands, leading the NK cells to become unresponsive to both positive and negative stimulation. In our study, the in vivo results showed that Gal-3 secreted from the tumor cells contributes to tumor escape from the NK-mediated innate immune defense. Taken together with the results shown in Fig. 4, it is likely that the effect of the Gal-3-impaired, NK cell-mediated tumor rejection was through its interaction with the NKp30 receptor on the NK cells.
Because it was established that Gal-3 interacts with cross-linked glycan molecules on the cell surface, such as MICA, to attenuate the interaction between the tumor cells and the immune cells, we observed that NKp30, as an essential glycoprotein, also contains two predicted N-glycosylation sites (Asn-42 and Asn-121) in its ectodomain. Taken together with a study from Tsuboi et al. (28), it seems that the tumor-released Gal-3 may reduce the tumor cell sensitivity of NK cells by binding glycosylated NKp30, thus interfering with the binding of NKp30 to its ligands on tumor cells and resulting in the evasion of the tumor cells from NK cell attack in the human cervical cancer model. This speculation should be examined further. Taken together, although the functional details of how Gal-3 inhibits NK cell function by binding to NKp30 remain unknown, our data, for the first time, reveal that the Gal-3 secreted from the tumor works as a soluble inhibitory ligand to compromise NK cells, which helps the tumor escape from immune attack.
Footnotes
- Gal-3
- Galectin-3
- NK cell
- natural killer
- MICA
- major histocompatibility complex class I-related chain A
- NOD
- non-obese diabetic
- SCID
- severe combined immunodeficiency
- hr
- human recombinant
- E:T
- effector:target
- PE
- phycoerythrin.
REFERENCES
- 1. Liu F. T., Rabinovich G. A. (2005) Galectins as modulators of tumour progression. Nat. Rev. Cancer 5, 29–41 [DOI] [PubMed] [Google Scholar]
- 2. Sturm A., Lensch M., André S., Kaltner H., Wiedenmann B., Rosewicz S., Dignass A. U., Gabius H. J. (2004) Human galectin-2: novel inducer of T cell apoptosis with distinct profile of caspase activation. J. Immunol. 173, 3825–3837 [DOI] [PubMed] [Google Scholar]
- 3. Lahm H., André S., Hoeflich A., Fischer J. R., Sordat B., Kaltner H., Wolf E., Gabius H. J. (2001) Comprehensive galectin fingerprinting in a panel of 61 human tumor cell lines by RT-PCR and its implications for diagnostic and therapeutic procedures. J. Cancer Res. Clin. Oncol. 127, 375–386 [DOI] [PubMed] [Google Scholar]
- 4. Lotan R., Matsushita Y., Ohannesian D., Carralero D., Ota D. M., Cleary K. R., Nicolson G. L., Irimura T. (1991) Lactose-binding lectin expression in human colorectal carcinomas: relation to tumor progression. Carbohydr. Res. 213, 47–57 [DOI] [PubMed] [Google Scholar]
- 5. Miyazaki J., Hokari R., Kato S., Tsuzuki Y., Kawaguchi A., Nagao S., Itoh K., Miura S. (2002) Increased expression of galectin-3 in primary gastric cancer and the metastatic lymph nodes. Oncol. Rep. 9, 1307–1312 [PubMed] [Google Scholar]
- 6. Moutsatsos I. K., Wade M., Schindler M., Wang J. L. (1987) Endogenous lectins from cultured cells: nuclear localization of carbohydrate-binding protein 35 in proliferating 3T3 fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 84, 6452–6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perillo N. L., Marcus M. E., Baum L. G. (1998) Galectins: versatile modulators of cell adhesion, cell proliferation, and cell death. J. Mol. Med. 76, 402–412 [DOI] [PubMed] [Google Scholar]
- 8. Sato S., Hughes R. C. (1994) Regulation of secretion and surface expression of Mac-2, a galactoside-binding protein of macrophages. J. Biol. Chem. 269, 4424–4430 [PubMed] [Google Scholar]
- 9. Honjo Y., Nangia-Makker P., Inohara H., Raz A. (2001) Down-regulation of galectin-3 suppresses tumorigenicity of human breast carcinoma cells. Clin. Cancer Res. 7, 661–668 [PubMed] [Google Scholar]
- 10. Moon B. K., Lee Y. J., Battle P., Jessup J. M., Raz A., Kim H. R. (2001) Galectin-3 protects human breast carcinoma cells against nitric oxide-induced apoptosis: implication of galectin-3 function during metastasis. Am. J. Pathol. 159, 1055–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng W., Wang H. Y., Miyahara Y., Peng G., Wang R. F. (2008) Tumor-associated galectin-3 modulates the function of tumor-reactive T cells. Cancer Res. 68, 7228–7236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ochieng J., Warfield P., Green-Jarvis B., Fentie I. (1999) Galectin-3 regulates the adhesive interaction between breast carcinoma cells and elastin. J. Cell. Biochem. 75, 505–514 [DOI] [PubMed] [Google Scholar]
- 13. Le Marer N., Hughes R. C. (1996) Effects of the carbohydrate-binding protein galectin-3 on the invasiveness of human breast carcinoma cells. J. Cell. Physiol. 168, 51–58 [DOI] [PubMed] [Google Scholar]
- 14. Thijssen V. L., Poirier F., Baum L. G., Griffioen A. W. (2007) Galectins in the tumor endothelium: opportunities for combined cancer therapy. Blood 110, 2819–2827 [DOI] [PubMed] [Google Scholar]
- 15. Fukumori T., Takenaka Y., Yoshii T., Kim H. R., Hogan V., Inohara H., Kagawa S., Raz A. (2003) CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 63, 8302–8311 [PubMed] [Google Scholar]
- 16. Müller S., Schaffer T., Flogerzi B., Fleetwood A., Weimann R., Schoepfer A. M., Seibold F. (2006) Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm. Bowel Dis. 12, 588–597 [DOI] [PubMed] [Google Scholar]
- 17. Demotte N., Wieërs G., Van Der Smissen P., Moser M., Schmidt C., Thielemans K., Squifflet J. L., Weynand B., Carrasco J., Lurquin C., Courtoy P. J., van der Bruggen P. (2010) A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 70, 7476–7488 [DOI] [PubMed] [Google Scholar]
- 18. Guha P., Kaptan E., Bandyopadhyaya G., Kaczanowska S., Davila E., Thompson K., Martin S. S., Kalvakolanu D. V., Vasta G. R., Ahmed H. (2013) Cod glycopeptide with picomolar affinity to galectin-3 suppresses T-cell apoptosis and prostate cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 110, 5052–5057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spits H., Blom B., Jaleco A. C., Weijer K., Verschuren M. C., van Dongen J. J., Heemskerk M. H., Res P. C. (1998) Early stages in the development of human T, natural killer and thymic dendritic cells. Immunol. Rev. 165, 75–86 [DOI] [PubMed] [Google Scholar]
- 20. Moretta A., Bottino C., Vitale M., Pende D., Cantoni C., Mingari M. C., Biassoni R., Moretta L. (2001) Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19, 197–223 [DOI] [PubMed] [Google Scholar]
- 21. Cantoni C., Bottino C., Vitale M., Pessino A., Augugliaro R., Malaspina A., Parolini S., Moretta L., Moretta A., Biassoni R. (1999) NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med. 189, 787–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vitale M., Bottino C., Sivori S., Sanseverino L., Castriconi R., Marcenaro E., Augugliaro R., Moretta L., Moretta A. (1998) NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 187, 2065–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pessino A., Sivori S., Bottino C., Malaspina A., Morelli L., Moretta L., Biassoni R., Moretta A. (1998) Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 188, 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nalabolu S. R., Shukla H., Nallur G., Parimoo S., Weissman S. M. (1996) Genes in a 220-kb region spanning the TNF cluster in human MHC. Genomics 31, 215–222 [DOI] [PubMed] [Google Scholar]
- 25. Pende D., Parolini S., Pessino A., Sivori S., Augugliaro R., Morelli L., Marcenaro E., Accame L., Malaspina A., Biassoni R., Bottino C., Moretta L., Moretta A. (1999) Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 190, 1505–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bottino C., Castriconi R., Moretta L., Moretta A. (2005) Cellular ligands of activating NK receptors. Trends Immunol. 26, 221–226 [DOI] [PubMed] [Google Scholar]
- 27. Lanier L. L. (2005) NK cell recognition. Annu. Rev. Immunol. 23, 225–274 [DOI] [PubMed] [Google Scholar]
- 28. Tsuboi S., Sutoh M., Hatakeyama S., Hiraoka N., Habuchi T., Horikawa Y., Hashimoto Y., Yoneyama T., Mori K., Koie T., Nakamura T., Saitoh H., Yamaya K., Funyu T., Fukuda M., Ohyama C. (2011) A novel strategy for evasion of NK cell immunity by tumours expressing core2 O-glycans. EMBO J. 30, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki Y., Sutoh M., Hatakeyama S., Mori K., Yamamoto H., Koie T., Saitoh H., Yamaya K., Funyu T., Habuchi T., Arai Y., Fukuda M., Ohyama C., Tsuboi S. (2012) MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int. J. Oncol. 40, 1831–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng X., Wang Y., Wei H., Sun R., Tian Z. (2009) LFA-1 and CD2 synergize for the Erk1/2 activation in the natural killer (NK) cell immunological synapse. J. Biol. Chem. 284, 21280–21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dupont W. D., Plummer W. D., Jr. (1990) Power and sample size calculations: a review and computer program. Control. Clin. Trials 11, 116–128 [DOI] [PubMed] [Google Scholar]
- 32. Sun H., Sun C., Tian Z., Xiao W. (2013) NK cells in immunotolerant organs. Cell Mol. Immunol. 10, 202–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng M., Chen Y., Xiao W., Sun R., Tian Z. (2013) NK cell-based immunotherapy for malignant diseases. Cell Mol. Immunol. 10, 230–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ruebel K. H., Jin L., Qian X., Scheithauer B. W., Kovacs K., Nakamura N., Zhang H., Raz A., Lloyd R. V. (2005) Effects of DNA methylation on galectin-3 expression in pituitary tumors. Cancer Res. 65, 1136–1140 [DOI] [PubMed] [Google Scholar]
- 35. Iurisci I., Tinari N., Natoli C., Angelucci D., Cianchetti E., Iacobelli S. (2000) Concentrations of galectin-3 in the sera of normal controls and cancer patients. Clin. Cancer Res. 6, 1389–1393 [PubMed] [Google Scholar]
- 36. Vereecken P., Zouaoui Boudjeltia K., Debray C., Awada A., Legssyer I., Sales F., Petein M., Vanhaeverbeek M., Ghanem G., Heenen M. (2006) High serum galectin-3 in advanced melanoma: preliminary results. Clin. Exp. Dermatol. 31, 105–109 [DOI] [PubMed] [Google Scholar]
- 37. Zubieta M. R., Furman D., Barrio M., Bravo A. I., Domenichini E., Mordoh J. (2006) Galectin-3 expression correlates with apoptosis of tumor-associated lymphocytes in human melanoma biopsies. Am. J. Pathol. 168, 1666–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Friese M. A., Platten M., Lutz S. Z., Naumann U., Aulwurm S., Bischof F., Bühring H. J., Dichgans J., Rammensee H. G., Steinle A., Weller M. (2003) MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 63, 8996–9006 [PubMed] [Google Scholar]
- 39. Pogge von Strandmann E., Simhadri V. R., von Tresckow B., Sasse S., Reiners K. S., Hansen H. P., Rothe A., Böll B., Simhadri V. L., Borchmann P., McKinnon P. J., Hallek M., Engert A. (2007) Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 27, 965–974 [DOI] [PubMed] [Google Scholar]
- 40. Brandt C. S., Baratin M., Yi E. C., Kennedy J., Gao Z., Fox B., Haldeman B., Ostrander C. D., Kaifu T., Chabannon C., Moretta A., West R., Xu W., Vivier E., Levin S. D. (2009) The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 206, 1495–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Radosavljevic G., Jovanovic I., Majstorovic I., Mitrovic M., Lisnic V. J., Arsenijevic N., Jonjic S., Lukic M. L. (2011) Deletion of galectin-3 in the host attenuates metastasis of murine melanoma by modulating tumor adhesion and NK cell activity. Clin. Exp. Metastasis 28, 451–462 [DOI] [PubMed] [Google Scholar]