

Background: Artesunate is capable of inducing cell death in cancer cells.
Results: Artesunate accumulates in lysosomes and promotes lysosomal function and ferritin degradation, leading to mitochondrial reactive oxygen species production and eventually cell death.
Conclusion: Intracellular iron and ferritin degradation is essential for artesunate-induced lysosomal activation and cell death.
Significance: This work reveals a novel mechanism underlying artesunate-induced cell death.
Keywords: Cell Death, Ferritin, Iron, Lysosome, Reactive Oxygen Species (ROS), Artesunate
Abstract
Artesunate (ART) is an anti-malaria drug that has been shown to exhibit anti-tumor activity, and functional lysosomes are reported to be required for ART-induced cancer cell death, whereas the underlying molecular mechanisms remain largely elusive. In this study, we aimed to elucidate the molecular mechanisms underlying ART-induced cell death. We first confirmed that ART induces apoptotic cell death in cancer cells. Interestingly, we found that ART preferably accumulates in the lysosomes and is able to activate lysosomal function via promotion of lysosomal V-ATPase assembly. Furthermore, we found that lysosomes function upstream of mitochondria in reactive oxygen species production. Importantly, we provided evidence showing that lysosomal iron is required for the lysosomal activation and mitochondrial reactive oxygen species production induced by ART. Finally, we showed that ART-induced cell death is mediated by the release of iron in the lysosomes, which results from the lysosomal degradation of ferritin, an iron storage protein. Meanwhile, overexpression of ferritin heavy chain significantly protected cells from ART-induced cell death. In addition, knockdown of nuclear receptor coactivator 4, the adaptor protein for ferritin degradation, was able to block ART-mediated ferritin degradation and rescue the ART-induced cell death. In summary, our study demonstrates that ART treatment activates lysosomal function and then promotes ferritin degradation, subsequently leading to the increase of lysosomal iron that is utilized by ART for its cytotoxic effect on cancer cells. Thus, our data reveal a new mechanistic action underlying ART-induced cell death in cancer cells.
Introduction
Artesunate (ART),3 a water-soluble derivative of artemisinin, has been widely used for treatment of malaria (1). ART, together with other derivatives of artemisinin, was first shown to exhibit toxicity to Ehrlich ascites tumor cells (2). At present, there is an increasing amount of evidence suggesting that ART has an anti-cancer function (3). For instance, ART has been shown to induce apoptosis and necrosis in multiple human cancer cells (4, 5). The anti-cancer effects of ART also include the following: cell cycle arrest (6), inhibition of angiogenesis (7), and reduction of cell invasion and metastasis (8). Moreover, the anti-cancer potential of ART has also been tested in a number of animal cancer models (4, 6, 9). However, the exact mechanisms underlying ART-mediated cell death have not been fully elucidated.
Lysosomes are the key intracellular organelles that perform the digestive function via the endocytic or autophagic pathway (10). Lysosomal hydrolases and integral lysosomal membrane proteins are essential for lysosomal function (11). The internal pH of lysosomes is acidic (below pH 5), which is mainly controlled by the vacuolar H+-ATPase (V-ATPase) complex (12). On the other hand, lysosomes are known to play a critical role in autophagy; at the maturation/degradation stage, autophagosomes fuse with lysosomes to form autolysosomes for degradation (13, 14). At present, the effects of ART on autophagy and lysosome remain controversial. It has been reported that ART inhibits autophagy via inhibition of lysosomal turnover without affecting lysosomal functions in breast cancer cells (15). In contrast, one recent report showed that ART promotes autophagy via up-regulating the expression of Beclin 1, which was also studied in breast cancer cells (16). Moreover, ART-induced cancer cell death was shown to be dependent on lysosomes, although the mechanism is unclear (15).
Iron is an essential nutrient, and its intracellular availability is tightly regulated. One of the key regulatory mechanisms involves ferritin (17). Ferritin functions as the major iron storage protein in mammals and consists of 24 protein subunits that can store up to 4,500 atoms of iron per ferritin (18). There are two subunits of ferritin, ferritin heavy chain (FTH) and ferritin light chain (FTL). FTH has a ferroxidase function that can catalyze extracellular iron into nontoxic ferric iron form and store this form of iron in the ferritin complex (19). Ferritin is stable in iron-rich conditions, whereas it is rapidly degraded under conditions of iron starvation (20). Ferritin has been reported to be degraded either in the lysosomes or by the proteasomes depending on the cellular stimulants (21). Recently, the nuclear receptor coactivator 4 (NCOA4) was identified as the cargo for the ferritin degradation via autophagic pathway, and the autophagic turnover of ferritin is termed ferritinophagy (22). To date, the effect of ART on ferritin and the link between free iron ions and lysosomal function in ART-induced cell death have not been well studied.
In this study, we aimed to clarify the effect of ART on autophagy and lysosomal function as well as to elucidate the underlying molecular mechanisms of how lysosome is involved in ART-induced cell death. Here, we utilized a blue fluorescence-tagged ART to investigate its localization, and our results clearly showed that ART accumulates in lysosome. Interestingly, we found that ART promotes lysosomal function via engaging lysosomal V-ATPase. More importantly, we provided clear evidence demonstrating that ferritin degradation via lysosomes is essential for ART-induced cell death. The results from our study shed new light on the molecular mechanisms underlying ART-induced cell death and support the development of this important anti-malaria drug as a cancer therapeutic agent.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa and HepG2 cells were obtained from American Type Culture Collection. The GFP-LC3-expressing stable HeLa cells were provided by Dr. N. Mizushima (Tokyo Medical and Dental University, Japan), TSC2-WT and TSC2-KO MEFs were obtained from Dr. D. J. Kwiatkowski (23). All cell lines were maintained in DMEM (Sigma-Aldrich, D1152) containing 10% fetal bovine serum (HyClone, SV30160.03) in a 5% CO2 atmosphere at 37 °C.
Western Blotting
After the indicated times of designated treatments, cells were collected and rinsed with PBS. The whole cell lysates were prepared in the Laemmli buffer (62.5 mm Tris-HCl, pH 6.8, 20% glycerol, 2% SDS, 2 mm DTT, phosphatase inhibitor, and proteinase inhibitor mixture). Protein concentrations were determined by the DCTM protein assay (Bio-Rad, 162-0177). An equal amount of protein was resolved by SDS-PAGE and transferred onto PVDF membrane (Bio-Rad). After blocking with StartingBlock blocking buffers (Thermo Scientific, 37538), the membrane was probed with designated first and second antibodies, developed with the enhanced chemiluminescence method (Thermo Scientific, 34076), and visualized using Kodak Image Station 4000R (Eastman Kodak Co.). Antibodies were obtained as follows: anti-microtubule-associated protein 1 light chain 3 (LC3) antibody (Sigma-Aldrich, L7543), anti-ATG7 antibody (ProScience, 3617), anti-tubulin (Sigma-Aldrich, T6199), anti-FLAG (Sigma-Aldrich, F3165), anti-β-actin (Sigma-Aldrich, A5441), anti-V-ATPase D1 (Santa Cruz Biotechnology, Inc., SC-69105), anti-V-ATPase B2 (Santa Cruz Biotechnology, SC-166122), anti-iron-responsive protein 2 (IRP2) (Santa Cruz Biotechnology, SC-33682), anti-FTH (Abcam, ab65080), anti-FTL (Abcam, ab69090), anti-TFEB (Bethyl Laboratories, A303-673A), and anti-NCOA4 (Sigma, SAB1404569). All of the other antibodies were purchased from Cell Signaling Technology: anti-lysosome-associated membrane protein 1 (LAMP1) antibody (catalog nos. 3243S and 9091S), anti-phospho-S6 (catalog no. 2211), anti-S6 (catalog no. 2217), caspase 3 (catalog no. 9662), and anti-PARP-1 (catalog no. 9542).
Confocal Imaging
Briefly, cells were first cultured on 8-well Lab-TekTM chambered coverglass (Thermo Scientific, 155411) overnight, followed by designated treatment. All of the confocal images were obtained with 60× oil objective (numerical aperture 1.4) lenses of Olympus Fluoview FV1000. The images were processed with FV10-ASW 3.0 Viewer software.
Detection of Cell Death
Cell death was estimated by morphological changes under phase-contrast microscopy and quantified by a propidium iodide (PI; 5 μg/ml) exclusion assay coupled with flow cytometry (BD Biosciences). Western blotting was also used to indicate the cell death via PARP-1 and caspase-3 cleavages.
Detection of the Intracellular Localization of ART
The cells were treated with blue fluorescent ART (20 μm, LynxTag-ARTTMASBlue, from BioLynx Technologies) with or without bafilomycin A1 (BAF) (50 nm) in full DMEM for 30 min. Subsequently, 50 nm LysoTracker Red DND-99 (LTR; Invitrogen, L7528) or MitoTracker Red CMXRos (MTR; Invitrogen, M7512) was added for 30 min. The cells were washed with PBS twice, and DMEM full medium was added into the well, followed by observation under the confocal microscope.
LTR, LysoTracker Green, and MTR Staining
After the designated treatments, cells were incubated with 50 nm LTR or LysoTracker Green DND-26 (LTG; Invitrogen, L7526) reagents in full DMEM for 30 min for labeling and tracking acidic organelles in live cells. For labeling mitochondria, the cells were incubated with 50 nm MTR in PBS for 15 min and then washed twice with PBS followed by incubation of full DMEM for the imaging or collection for flow cytometry. The cells in the chambered coverglass were observed under a confocal microscope. The cells from the 24-well plate were collected, and the fluorescence intensities of 10,000 cells/sample were measured by flow cytometry using the BD FACS cytometer (BD Biosciences). We recorded the fluorescence of LTR and MTR using the FL-2 channel of FACS (BD Biosciences).
Magic Red Cathepsin B and L Activity Assay
Lysosomal function was also estimated by the cathepsin B and L enzymatic activity. After designated treatment, cells were further loaded with Magic RedTM cathepsin B (Immunochemistry Technologies, 938) or cathepsin L (Immunochemistry Technologies, 942) reagents for 30 min. The cells in the chambered coverglass were observed under a confocal microscope. The cells from the 24-well plate were collected, and the fluorescence intensities of 10,000 cells per sample were measured by flow cytometry. We recorded the fluorescence of Magic Red using the FL-2 channel of FACS.
Determination of Protein Proteolysis Using DQ Red BSA Staining
Lysosomal protein proteolysis was estimated by DQ Red BSA (Invitrogen, D12051) staining. Cells were first incubated with DQ Red BSA for 1 h and then washed with PBS two times, followed by the designated treatment. The cells in the chambered coverglass were observed under a confocal microscopy. The cells from the 24-well plate were collected, and the fluorescence intensities of 10,000 cells/sample were measured by flow cytometry. We recorded the fluorescence of DQ Red BSA using the FL-3 channel of FACS.
Immunofluorescence Staining
Cells were first cultured in an 8-well chambered coverglass overnight. After the designated treatment, cells were first fixed with 4% paraformaldehyde in PBS for 15 min at 37 °C and then permeabilized with 0.01% saponin in PBS for 10 min. After blocking with 1% BSA in PBS for 30 min, cells were incubated with V-ATPase B2 primary antibody (Santa Cruz Biotechnology, SC-166122) in a 1:100 dilution overnight at 4 °C. On the second day, the cells were then incubated with LAMP1 in a 1:100 dilution (Cell Signaling Technology, 9091S) for 3 h at room temperature, followed by Alexa Fluor 488 goat anti-mouse secondary antibody (Invitrogen, A-11029) and Alexa Fluor 555 donkey anti-rabbit secondary antibody (Invitrogen, A-31572). The cells were examined using a confocal microscope, and representative cells were selected and photographed.
Use of in Situ Proximity Ligation Assay to Check the Interaction between V1 and V0 in Situ
The use of two different proximity ligation assay probes with amplifiable DNA reporter enabled us to visualize and quantify the protein-protein interactions in situ (24). The HeLa cells were first seeded in a 16-well chamber. Treated cells were first fixed with 4% paraformaldehyde for 15 min at 37 °C and then permeabilized with 0.01% saponin in PBS for 10 min, followed by blocking with 1% BSA in PBS for 30 min. Cells were then incubated with anti V-ATPase V1 domain subunit B2 (V1B2) and anti-V-ATPase V0 domain subunit D1 (V0D1) in a 1:100 dilution, incubated overnight at 4 °C. The chamber was then performed with the procedure based on the manufacturer's instructions (Olink Bioscience).
Small Interfering RNA (siRNA) and Transient Transfection
The scrambled RNAi oligonucleotides (Dharmacon, ON-TARGETplus Non-targeting Pool, D-001810–10-05) and siRNAs targeting ATG7 (Dharmacon, SMARTpool, ON-TARGETplus human ATG7, L-020112-00-0005; target sequences: CCAACACACUCGAGUCUUU, GAUCUAAAUCUCAAACUGA, GCCCACAGAUGGAGUAGCA, and GCCAGAGGAUUCAACAUGA), TFEB (Dharmacon, SMARTpool, ON-TARGETplus human TFEB, L-009798-00-0005; target sequences: CAACAGUGCUCCCAAUAGC, GCAGCCACCUGAAUGUGUA, UGAAAGGAGACGAAGGUUC, and GCAGAUGCCCAACACGCUA), and NCOA4 (Dharmacon, SMARTpool, ON-TARGETplus human NCOA4, L-010321-00-0005; target sequences: CAGAUUCACAGUUGCAUAA, ACAAAGAUCUAGCCAAUCA, ACAAGUGGCUGCUUCGAAA, and GAGAAGUGGUUAUAUCGAA) were transfected into HeLa cells using the DharmaFECT 4 Transfection Reagent (Dharmacon, T-2001-02) according to the manufacturer's protocol. After 48 h, the cells were subjected to the designated treatment. For plasmid transfection, HeLa cells were transiently transfected with pcDNA or FTH-FLAG plasmid using LipofectamineTM 2000 according to the manufacturer's protocol. After 24 h, the cells were treated as indicated.
Measurement of Reactive Oxygen Species (ROS) Production
CM-H2DCFDA (Invitrogen, C6827) and MitoSOXTM Red (MSR; Invitrogen, M36008) were chosen for the detection of intracellular ROS and mitochondrial superoxide production, respectively. When CM-H2DCFDA passively diffuses into cells, its acetate groups are cleaved by intracellular esterases and subsequently oxidized by ROS and yield a fluorescent adduct, CM-DCF (25). MSR is a fluoroprobe for detection of superoxide in the mitochondria of live cells (26). Briefly, cells were first cultured in a Lab-TekTM chambered coverglass or 24-well plate overnight. After the designated treatments, cells were incubated with 5 μm MSR or 1 μm CM-H2DCFDA in PBS for 10 min. Then the MSR or CM-H2DCFDA was removed, and the cells were washed with PBS twice. The cells in the coverglass were incubated in full medium and observed under a confocal microscope. The cells in the 24-well plate were collected, and fluorescence intensity was measured. We recorded the fluorescence of CM-DCF using the FL-1 channel and MSR with the FL-2 channel of FACS (BD Biosciences).
Luciferase Assays
TFEB luciferase vector was provided by Dr. A. Ballabio (27). The transient transfection of the TFEB luciferase vector was done in HeLa cells using LipofectamineTM 2000 transfection reagent according to the manufacturer's protocols. Renilla luciferase vector was used as a transfection control. The luciferase activity was measured at 48 h after transfection using the Dual-Luciferase reporter assay system (Promega, E1960) based on the protocol provided by the manufacturer. Briefly, following the treatments, the cell lysate was collected from each well after the addition of cell lysis reagent. After the addition of luciferase assay substrate, the firefly luciferase activity was determined using a luminometer (Promega), and the Renilla luciferase activity was then measured by adding the Stop & Glo substrate.
Reverse Transcription and Quantitative Real-time PCR
RNA was extracted with the RNeasy kit (Qiagen, 217004). A reverse transcription reaction was performed using 1 μg of total RNA with iScriptTM Reverse Transcription Supermix for RT-qPCR (Bio-Rad, 170-8841). The mRNA expression levels were determined by real-time PCR using SsoFast EvaGreen Supermix (Bio-Rad, 172-5201AP) and the CFX96 Touch Real-time PCR Detection System (Bio-Rad). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control of RNA integrity. Real-time PCR was performed in triplicate. The primers used for FTH and FTL were purchased from Qiagen: Hs_FTH1_1_SG QuantiTect primer assay (QT00072681) and Hs_FTL_1_SG QuantiTect Primer Assay (QT00055860).
Statistical Analysis
All Western blot data and image data presented are representative of three independent experiments. The numeric data except for quantitative RT-PCR data are presented as mean ± S.D. from three independent experiments and analyzed using Student's t test. Quantitative RT-PCR data are presented as mean ± S.D. from two independent experiments (each in triplicate).
RESULTS
ART Induces Apoptotic Cell Death in Human Cancer Cells
ART has been shown to inhibit cell growth, induce radiosensitivity, and enhance TRAIL-induced apoptosis in human cervical cancer HeLa cells (28–30). In this study, we first examined the cytotoxicity of ART on HeLa cells. We found that ART (50 μm for 48 h) caused evident cell death in HeLa cells, as assessed by morphological changes (Fig. 1A, top), Hoechst staining for chromatin condensation (Fig. 1A, bottom), and quantitatively through PI-live cell exclusion test (Fig. 1, B and C). ART-induced cell death was also found to be dose-dependent in HeLa cells (Fig. 1D). In addition to its toxicity, treatment with ART alone or combined with N-benzyloxycarbonyl-Phe-Ala-fluoromethylketone (Z-VAD-fmk) was also shown to reduce the number of cells (Fig. 1A), which is consistent with the earlier findings that ART inhibits cancer cell proliferation (6, 16). To further examine the type of cell death induced by ART, we tested the protective effect of the general caspase inhibitor Z-VAD-fmk on ART-mediated cell death. As shown in Fig. 1, A–C, the cell death induced by ART was dramatically protected by Z-VAD-fmk. Consistently, we also observed cleavage of both caspase-3 (17 kDa) and PARP-1 (89 kDa), both classical markers of apoptotic cell death in HeLa cells treated with ART (Fig. 1E). The caspase-3 and PARP cleavage can also be blocked by Z-VAD-fmk (Fig. 1F). Similar results were also observed in HepG2 cells (data not shown).
FIGURE 1.

ART induces apoptosis in cancer cells. A, HeLa cells were treated with ART (50 μm) with or without Z-VAD-fmk (20 μm) for 48 h. Morphological changes of ART-induced cell death were observed under a light microscope (top) or by using a fluorescent microscope after incubation with Hoechst dye for 30 min (bottom). Scale bar, 200 μm. B, HeLa cells were treated as indicated in A, and the cell death was quantified by flow cytometry using PI (5 μg/ml) staining (R1 area, representative of viable cells). C, statistical analysis of the percentage of viable cells of three independent experiments performed as in B (mean ± S.D. (error bars)) (**, p < 0.01, Student's t test). D, HeLa cells were treated with the indicated dose of ART, and cell death was quantified by flow cytometry after PI staining as described earlier (**, p < 0.01, Student's t test). E, HeLa cells were collected after treatment with ART (50 μm) for the indicated period of time, and Western blotting was then performed for the detection of various indicated markers of apoptosis. β-Actin and tubulin served as loading controls. F, HeLa cells were treated with ART (50 μm) alone or combined with Z-VAD-fmk (20 μm) together for 24 h followed by Western blotting to detect the apoptosis markers.
ART Accumulates in the Lysosomal Fraction and Activates Lysosomal Function
In order to understand the molecular mechanisms underlying ART-mediated cell death, we utilized LynxTag-ARTTM ASBlue, a blue fluorescence-tagged ART, to investigate its cellular localization. As shown in Fig. 2, ART was found to be predominantly localized in lysosomes, as evidenced by its co-staining with LTR (Fig. 2A) and LAMP1 (Fig. 2B). There was little colocalization of ART with MTR (Fig. 2C). Altogether, the data showed that ART accumulates in the lysosomes. Notably, treatment with BAF, an inhibitor of lysosomal acidification via inhibition of the lysosomal V-ATPase activity (31), abolished LTR staining of the lysosomes (Fig. 2A), whereas accumulation of ART was not affected by BAF treatment (Fig. 2B), suggesting that ART accumulation in lysosomes is independent of lysosomal pH. An earlier study has indicated that functional lysosomes are required for ART-induced cell death (15). Here, we investigated the effects of ART on lysosomal function determined by the following assays: (i) lysosomal pH indicated by LTR and LTG, (ii) cathepsin enzyme activities measured by Magic Red fluorescence intensity with both confocal microscopy and flow cytometry, and (iii) lysosomal proteolysis capability using the DQ Red BSA reagent. As shown in Fig. 3, A–C and F, ART enhanced LTR and LTG stainings significantly, suggesting that ART treatment decreases lysosomal pH. Moreover, the decrease of lysosomal pH is in a time-dependent manner, as evidenced by the time course increase of the LTR staining (Fig. 3B). ART also increased the Magic Red cathepsin L fluorescence intensity when observed under confocal microscopy (Fig. 3C), indicating that ART is able to enhance the cathepsin L enzymatic activity in HeLa cells. Similar results were also observed in HepG2 cells (Fig. 3, D and E). Finally, we found that ART treatment increased the fluorescence intensity of the DQ Red BSA (Fig. 3F), which indicates that ART promotes the lysosomal proteolysis. To check whether the increase of the above staining is due to the increase of lysosome number, we examined the changes of LAMP1 protein level treated with ART. We first performed immunofluorescence staining of LAMP1, and the result showed that there was a cluster of lysosomes in ART-treated cells (Fig. 3G), which is consistent with the earlier study (15). We then performed statistical analysis of the LAMP1 signal, and there was no significant difference between the control group and ART-treated cells (Fig. 3H). To further confirm our findings, we then checked the temporal pattern of LAMP1, and there was no increase of LAMP1 protein level in ART-treated cells for up to 18 h (Fig. 3I), indicating that ART treatment does not increase the number of lysosomes. Taken together, our data suggest that ART activates lysosomal function.
FIGURE 2.
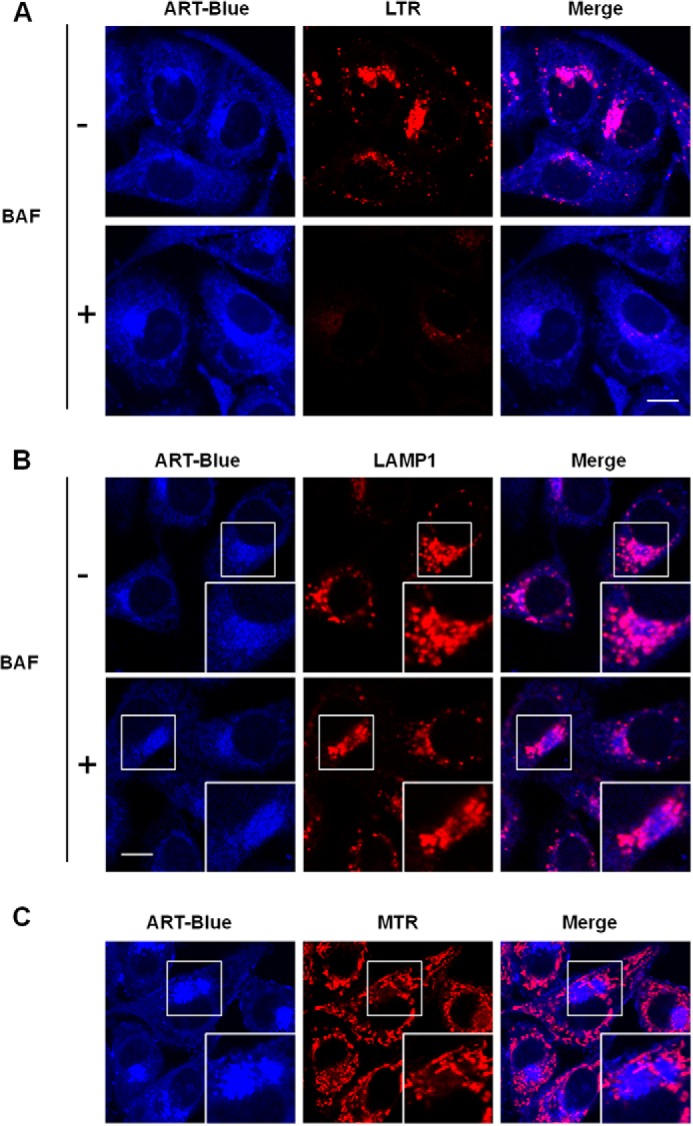
ART accumulates in the lysosomes. A, HeLa cells were treated with blue fluorescent ART (20 μm) or combined with BAF (50 nm) for 30 min, followed by LTR incubation for 30 min. B, HeLa cells were treated as indicated in A and then immunostained with LAMP1 (red). C, HeLa cells were treated with blue fluorescent ART for 30 min and then incubated with MTR. All images were analyzed using confocal microscopy. Scale bar, 10 μm.
FIGURE 3.

ART activates lysosomal function. A, HeLa cells were first treated with ART (50 μm) for 12 h, followed by LTR labeling for 30 min, and further analyzed using confocal microscopy. B, HeLa cells were loaded with LTR following time course drug treatment, and fluorescence intensity of 10,000 cells/sample was measured by flow cytometry. C, HeLa cells were treated as indicated in A and then incubated with Magic Red cathepsin L and LTG for 30 min and observed under a confocal microscope. D, HepG2 cells were treated as in A and then labeled with LTR described earlier in A. E, HepG2 cells were treated as in A, followed by a Magic Red cathepsin B and L assay for 45 min. Fluorescence intensity of 10,000 cells/sample was measured using flow cytometry. F, HeLa cells were first loaded with DQ Red BSA for 1 h and then treated with ART (50 μm) for 18 h and incubated with LTG for 30 min. G, HeLa cells were treated with ART for 18 h and then immunostained for LAMP1. H, LAMP1 signals of 80 cells from three independent experiments were quantified by ImageJ (Student's t test, #, no significance). I, HeLa cells were treated with ART for the indicated time, and then Western blot was performed to detect the LAMP1 protein level. Scale bar, 10 μm. Error bars, S.D.
Activation of Lysosomal Function by ART Involves V-ATPase
Earlier studies from our laboratory have shown that mammalian target of rapamycin complex 1 or mechanistic target of rapamycin complex 1 (mTORC1), a negative upstream regulator of autophagy, inhibits lysosomal function in the course of autophagy (32). ART is known to inhibit the phosphoinositide 3-kinase/Akt (PI3K/Akt) pathway (33), and PI3K/Akt is a positive regulator of mTORC1 (34). Therefore, there is a possibility that ART activates lysosomal function via suppression of the PI3K/Akt-mTORC1 pathway. As expected, mTORC1 activity was decreased, as evidenced by the loss of S6 protein phosphorylation with prolonged treatment of ART (for 12 h) (Fig. 4A). Because evident lysosomal activation was found from 3 h onward in ART-treated cells (Fig. 3B), it appears that mTORC1 suppression observed in ART-treated cells occurs later than lysosomal activation, thus excluding the possibility that lysosomal activation is due to the mTOR inhibition. To further confirm this finding, we utilized Tsc2+/+ and Tsc2−/− mouse embryonic fibroblasts (MEFs). In Tsc2−/− MEFs, ART failed to inhibit the phosphorylation of S6 (Fig. 4B), whereas it was still capable of increasing cathepsin B enzymatic activity (Fig. 4C), suggesting that mTORC1 inhibition may not play a role in ART-mediated lysosomal activation. It has been reported that transcription factor EB (TFEB) plays a critical role in the biogenesis and function of lysosomes (27). We then tried to look into the role of TFEB in the course of lysosomal activation. ART increased the TFEB transcriptional activity in HeLa cells, as indicated by the increase of luciferase activity at 24 h (Fig. 4D). However, knockdown of TFEB did not affect lysosomal activation induced by ART (Fig. 4, E and F), suggesting that TFEB was not directly involved in the activation of lysosomal function by ART.
FIGURE 4.

ART increases lysosomal function via increasing V-ATPase assembly. A, HeLa cells were treated with 50 μm ART at the indicated time point. Western blot was then performed for the detection of S6 protein phosphorylation. B, Tsc2+/+ MEFs and Tsc2−/− MEFs were treated with ART (50 μm) for the indicated period of time and immunoblotted for the indicated markers. C, Tsc2+/+ MEFs and Tsc2−/− MEFs were treated with ART (50 μm) for 24 h and then incubated with Magic Red cathepsin B for 30 min. The fluorescence intensity was analyzed using flow cytometry. D, HeLa cells were transiently transfected with a TFEB luciferase report vector together with Renilla luciferase vector as a transfection control. Cells were then treated with ART (50 μm) as indicated for a time course assay (**, p < 0.01, Student's t test). E, HeLa cells were transiently transfected with TFEB siRNA or scrambled siRNA as control for 48 h. F, after they were transiently transfected with TFEB siRNA or scrambled siRNA for 48 h, HeLa cells were treated with ART for 24 h and then incubated with Magic Red cathepsin B. Fluorescence intensity was quantified by flow cytometry. G, HeLa cells were treated with ART for 12 h and then coimmunostained for V-ATPase B2 (green) and LAMP1 (red). Scale bar, 10 μm. H, HeLa cells were treated as in G, and then a proximity ligation assay was performed for the detection of interaction between two subunits of V-ATPase: V1B2 and V0D1. Scale bar, 10 μm. Error bars, S.D.
We next investigated the role of lysosomal V-ATPase in the lysosomal activation induced by ART. V-ATPase is required for acidification of lysosomes, and it is activated when the peripheral V1 and integral V0 domains assemble into a single complex on the lysosome membrane (12). We therefore examined the assembly status of the V1 and V0 domain in ART-treated cells. As shown in Fig. 4G, there was an increase of subunit B2 of V1 domain accumulation on the lysosomes in the ART-treated group detected by immunofluorescence staining. In order to confirm such findings, we performed the proximity ligation assay, and our data clearly showed an increased proximity between V1B2 and V0D1 upon ART treatment (Fig. 4H). Altogether, the data from this part suggest that ART is able to enhance lysosomal function via increasing V-ATPase assembly and activity independent of mTORC1.
Lysosomal Activation Is Independent of ART-induced Autophagy
Because ART is able to promote lysosomal function and inhibit mTORC1 activity, we then tested the effect of ART on autophagy in HeLa cells. We first used HeLa cells with stable expression of GFP-LC3 protein. LC3, microtubule-associated protein 1 light chain 3, the homologue of Atg8 in yeast, is thought to be engaged in the expansion of autophagosomal membranes (35). The lipidated and membrane-bound LC3-II is considered an indicator for autophagosome (36). As shown in Fig. 5, A and B, ART markedly increased the average area of GFP-LC3 puncta and changed their distribution pattern (clustered to the perinuclear region), which is consistent with the earlier finding (15). To confirm whether ART increases the autophagic flux, we performed a combined treatment of ART with vinblastine, a microtubule-depolymerizing agent that inhibits the maturation of autophagic vacuoles (37). As shown in Fig. 5, A–D, the presence of vinblastine significantly increased the autophagic markers (both GFP-LC3 puncta and LC3-II protein level), indicating that ART increases autophagic flux in HeLa cells. To understand the role of autophagy in ART-mediated lysosomal activation and cell death, we performed siRNA knockdown of ATG7, one of the key autophagy-related genes (ATGs) in mediating autophagosome formation (38). As shown in Fig. 5E, knockdown of ATG7 abolished LC3-II formation and ART-induced autophagy. However, knockdown of ATG7 failed to have any evident effect on lysosomal function in ART-treated HeLa cells (Fig. 5F), indicating that lysosomal activation by ART is independent of autophagy.
FIGURE 5.

ART induces autophagy. A, HeLa cells with stable expression of GFP-LC3 were treated with 50 μm ART or combined with vinblastine (VIN; 1 μm) for 16 h. Autophagosomes were inferred by the presence of GFP-LC3 puncta under confocal microscopy. Scale bar, 10 μm. B, stable GFP-LC3 HeLa cells were treated as in A, fluorescence images were taken using confocal microscope, and the area of GFP-LC3 puncta was quantified using ImageJ. Data (mean ± S.D. (error bars)) are representative of three independent experiments (*, p < 0.05, Student's t test). C, HeLa cells were treated as in A, and Western blot was then performed for the detection of LC3-II abundance. D, the band density of LC3-II protein was quantified using ImageQuant TL (GE Healthcare). Data (mean ± S.D.) are representative of three independent experiments (*, p < 0.05, Student's t test). E, HeLa cells were transfected with ATG7 siRNA or scrambled siRNA for 48 h. F, after 48 h of siRNA transfection, HeLa cells were treated with ART for 24 h, followed by cathepsin B and L enzymatic activities determined using Magic Red staining coupled with flow cytometry.
ART-induced Oxidative Stress and Cell Death Are Lysosome-dependent
It has been reported that ART is able to increase intracellular ROS level as an important cell death mechanism (39). We first used CM-H2DCFDA to measure the intracellular ROS. ART treatment increased the ROS production, as evidenced by the increased CM-DCF fluorescence intensity (Fig. 6A), indicating that ART treatment increases ROS production. The addition of antioxidant N-acetylcysteine (NAC), which has been shown to directly react with hydrogen peroxide and superoxide (40), was able to inhibit the production of ROS induced by ART (Fig. 6A). Because mitochondria are the main site of ROS formation and superoxide is the precursor of most other ROS, such as hydrogen peroxide (41), we next utilized MSR to detect the superoxide production in mitochondria. As shown in Fig. 6B, ART treatment significantly increased the mitochondrial ROS level. The addition of NAC partially prevented ART-mediated mitochondrial ROS production (Fig. 6B). Interestingly, production of mitochondrial ROS was almost completely abolished by BAF (Fig. 6, C and D), indicating the possibility that lysosome functions upstream of mitochondria in ROS production in ART-treated cells. As expected, both NAC and BAF significantly protected ART-induced cell death (Fig. 6E). To further confirm the involvement of lysosomes in cellular ROS production upon ART treatment, we examined the changes of lysosomal function using NAC or BAF. As shown in Fig. 6F, BAF was able to completely inhibit ART-induced lysosomal activation, whereas NAC failed to have any effect, further confirming the earlier notion that lysosome acts upstream of mitochondrial ROS production. Taken together, these data indicate that lysosomes function upstream of mitochondria in ROS production in ART-treated cells.
FIGURE 6.

Mitochondrial ROS production induced by ART can be inhibited by lysosomal inhibitor. A, HeLa cells were treated with ART with or without NAC for 24 h, followed by incubation with CM-H2DCFDA. The fluorescence intensity was analyzed by flow cytometry. B, HeLa cells were treated as in A, followed by incubation with MSR. The fluorescence intensity was analyzed by flow cytometry. C, HeLa cells were treated with ART with or without BAF (50 nm) for 24 h, followed by incubation with LTG for 30 min and then MSR in PBS for 15 min. The cells were then observed under a confocal microscope. Scale bar, 10 μm. D, HeLa cells were treated as in C, and fluorescence intensity of MSR was recorded by flow cytometry. E, HeLa cells were treated with ART with or without BAF (50 nm) or NAC for 48 h. The cell viability was quantified by flow cytometry using a PI exclusion assay. Data (mean ± S.D. (error bars)) are representative of three independent experiments (*, p < 0.05; **, p < 0.01, Student's t test). F, HeLa cells were treated with ART with or without BAF as indicated for 12 h, and Magic Red cathepsin B and L activities were then determined by flow cytometry.
Lysosomal Activation, ROS Production, and Cell Death Induced by ART Is Dependent on Lysosomal Iron
Because ferrous iron is essential for the activity of ART (39, 42) and lysosomes are known to contain high concentrations of ferrous iron (43), we utilized deferoxamine mesylate (DFO), a lysosomal iron chelator, to investigate the role of lysosomal iron in ART-induced cell death. DFO is known as a lysosomal iron chelator because of its poor membrane permeability, and thus it enters into cells through endocytosis and mostly localizes to lysosomes (44–46). Here, we first observed that DFO inhibited ART-induced lysosomal activation, as evidenced by the following: (i) inhibition of lysosomal acidification induced by ART (Fig. 7A), (ii) blockage of ART-mediated lysosomal enzymatic activation (Fig. 7B), and (iii) suppression of ART-enhanced lysosomal proteolysis (Fig. 7C). Moreover, mitochondrial ROS production induced by ART was also inhibited by DFO (Fig. 7D). Consequently, DFO was shown to completely block ART-induced cell death (Fig. 7E). In summary, these data indicate that lysosomal iron plays a critical role in ART-induced lysosomal activation, mitochondrial ROS production, and cell death.
FIGURE 7.

Chelating of lysosome iron is able to inhibit the cell death and lysosome activation induced by ART. A, HeLa cells were treated with 50 μm ART alone or combined with DFO (100 μm) for 16 h, and lysosomal pH was indicated by LTR fluorescence intensity. B, HeLa cells were treated as indicated in A. Magic Red cathepsin B and L activities were determined as in Fig. 4C. C, HeLa cells were first loaded with DQ Red BSA for 1 h and then treated as indicated in A. The DQ Red BSA fluorescence intensity was analyzed by flow cytometry. D, HeLa cells were treated as indicated in A, and mitochondrial ROS were observed as indicated in Fig. 6C. E, HeLa cells were treated with 50 μm ART alone or combined with DFO (100 μm) for 48 h. The cell viability was quantified as indicated in Fig. 1B. Data (mean ± S.D. (error bars)) are representative of three independent experiments (**, p < 0.01, Student's t test).
ART Promotes Ferritin Degradation in the Lysosomes
The iron level within lysosomes is high because lysosomes release the iron from transferrin or degrade iron-containing proteins, such as ferritin, in the acidic environment (43). Ferritin is the major iron storage protein in cells (18), and it has been shown to be degraded in the lysosomes under iron-depleted conditions in cancer cells (20). On the other hand, it has also been shown that ferritin can be degraded by the proteasome (21). Because our results showed that ART accumulates in lysosomes and activates lysosomal function (Figs. 2 and 3), we hypothesized that ART may promote ferritin degradation in lysosomes. To test this hypothesis, we first examined the changes of both FTH and FTL in cells treated with ART. We observed a time- and dose-dependent reduction of their protein level (Fig. 8, A and B). To check whether the decreased ferritin protein level is due to enhanced protein degradation, we first measured the mRNA level of ferritin. As shown in Fig. 8C, there was a time-dependent increase of mRNA level in ART-treated cells, suggesting that the reduced ferritin protein level by ART is unlikely via suppression of transcription. Although we observed a decrease of ferritin mRNA level in DFO-treated cells, reduction of the mRNA level (after 6 h) occurred later than that of protein (from 3 h onward) (data not shown), indicating that suppression of transcription is unlikely to be an important mechanism for the reduced ferritin protein level in DFO-treated cells. We next inhibited the lysosomal function by lysosomal inhibitor BAF and found that both ART and DFO-reduced ferritin protein level were completely blocked (Fig. 8, D and E). In contrast, proteasome inhibitor MG132 cannot fully inhibit ferritin degradation induced by ART (Fig. 8D). These results thus indicate that ART-induced ferritin degradation is mainly performed by the lysosomes. To check the intracellular iron level cells treated with ART or BAF, we then examined the levels of IRP2. IRP2 is stabilized in response to low iron level, whereas it is degraded under high iron levels (47). As shown in Fig. 8F, BAF significantly increased the IRP2 protein level, which is similar to that of the DFO-treated group, suggesting that BAF decreases the intracellular iron level. We also observed that ART increased the IRP2 level, confirming the notion that the bioactivity of ART requires iron.
FIGURE 8.

Degradation of ferritin induced by ART is inhibited by lysosomal inhibitor. A, HeLa cells were treated with 50 μm ART at the indicated time point. Western blotting was then performed for the detection of FTH and FTL abundance. B, HeLa cells were treated with ART at the indicated dose for 24 h. C, HeLa cells were treated with 50 μm ART at the indicated time point, and the FTH (left) and FTL (right) mRNA levels were measured by quantitative RT-PCR. Values are expressed as -fold increase compared with the control group. Data are presented as mean ± S.D. (error bars) from two independent experiments (each in triplicate) (*, p < 0.05; **, p < 0.01 compared with their respective group in the control cells, Student's t test). D, HeLa cells were treated with BAF (50 nm), DFO (100 μm), and MG132 (2 μm) with or without ART (50 μm) for 24 h. E, HeLa cells were pretreated with DFO for 2 h and then incubated with 50 nm BAF for 6 h. Western blot was then performed for detection of FTH and FTL abundance. F, HeLa cells were treated with ART, BAF, and DFO for the indicated times. IRP2 level was measured by Western blot.
Lysosomal Delivery and Degradation of Ferritin Is Required for ART-induced Cell Death
To investigate the role of ferritin degradation in ART-induced cell death, we overexpressed FLAG-tagged FTH, which has been shown to decrease the cytosolic iron level and to increase the resistance to oxidative stress (48). As shown in Fig. 9A, there was no reduction of FLAG-FTH protein level in ART-treated cells. Also, overexpression of FTH significantly protected ART-induced cell death, especially in higher concentrations of ART-treated cells (Fig. 9B), suggesting that ferritin degradation plays an important role in ART-induced cell death. A recent report has identified NCOA4 as a cargo receptor mediating the delivery and degradation of ferritin in lysosomes (22). Here we used RNAi to knock down NCOA4 and then checked ART-induced cell death. As shown in Fig. 9C, knockdown of NCOA4 blocked ferritin degradation (both heavy chain and light chain). Consistently, knockdown of NCOA4 offered significant protection against ART-induced cell death in HeLa cells (Fig. 9D). Altogether, these data suggest that it is not the lysosomes per se required for ART-induced cell death, but delivery and degradation of ferritin in lysosomes is required for ART-induced cell death.
FIGURE 9.

Ferritin degradation is required for ART-induced cell death. A, HeLa cells were transiently transfected with pcDNA or FLAG-tagged FTH plasmid (FLAG-FTH) for 48 h, followed by ART (50 μm) treatment for 24 h. B, HeLa cells were transiently transfected with FLAG-FTH for 48 h and then treated with ART for 48 h. The cell viability was quantified by flow cytometry after PI staining. C, HeLa cells were transiently transfected with scrambled siRNA or NCOA4 siRNA for 48 h. D, HeLa cells were transiently transfected with scrambled siRNA and NCOA4 siRNA for 48 h and then treated with ART for 48 h. The cell viability was quantified by flow cytometry using a PI live cell exclusion assay (*, p < 0.05; **, p < 0.01, Student's t test). E, illustration showing the mode of action by ART on cell death. Error bars, S.D.
DISCUSSION
One important finding from our study is the activation of lysosomal functions by ART, based on two main observations. First, ART accumulates in the lysosomes (Fig. 2); second, ART increases lysosomal acidification, cathepsin enzyme activity, and protein degradation via promoting lysosomal V-ATPase assembly (Figs. 3 and 4). Accumulation of ART in lysosomes is indeed consistent with the findings in the malaria parasite, in which artemisinin was found to be accumulated in the parasite's digestive food vacuole (49). Although one earlier study showed that acridine-based fluorescence-labeled artemisinin did not localize into the parasite food vacuole (50), it was later found that this inconsistency was due to the quenching of fluorescence of this acridine-based fluorophore in the acidic food vacuole (51). At present, the exact mechanism for the lysosomal accumulation by ART is not known. Nevertheless, our data indicate that it is lysosomal pH-independent.
In an attempt to understand the underlying mechanisms of ART-mediated lysosomal activation, we investigated the possible role of lysosomal V-ATPase and found that the assembly status of the V1 domain and V0 domain of V-ATPase was increased by ART treatment (Fig. 4, G and H). The V-ATPase complex contains two domains: peripheral V1 domain and integral V0 domain. The V1 domain consists of eight different subunits (A–H) and is responsible for ATP hydrolysis, whereas the V0 domain, composed of six subunits, drives proton from cytoplasm to the lumen (52, 53). Based on the reversible association model, V-ATPase is active when the two domains assemble into a single complex on the lysosome membrane (12, 54). Therefore, ART may increase lysosomal V-ATPase activity via promoting the assembly status of the two domains. Apparently, our data differ from an earlier report in which ART did not affect lysosomal pH and cathepsin B activity (15). One possibility underlying the discrepancy between these two studies is the different cell culture conditions used; in our study, cells were treated with ART in full medium (high glucose DMEM with 10% FBS), whereas Hamacher-Brady et al. (15) treated cells in Krebs-Henseleit solution, a defined glucose-containing basic salt solution. Because lysosomal function is known to be up-regulated in the course of autophagy (32, 55), it is likely that lysosomal activity was increased when the cells were cultured in Krebs-Henseleit solution, thus making it hard to detect the further activation of lysosomal function by ART in this context.
The effect of ART on autophagy was first reported by Hamacher-Brady et al. (15). They found that ART inhibits autophagy via disrupting endolysosomal trafficking. In their study, they showed that (i) ART induces perinuclear clustering of autophagosomes, early and late endosomes, and lysosomes; (ii) lysosomal iron is upstream of mitochondrial outer membrane permeabilization; and (iii) lysosomal inhibitors are able to inhibit ART-induced cell death. We also found similar results (Figs. 3G, 5A, 6E, and 7E). However, we found that ART is able to induce autophagy, based on the observations that ART increased autophagosomes formation and enhanced autophagic flux (Fig. 5, A–D). We also demonstrate that ART induces autophagy via a dual mechanism: suppression of mTORC1 activity (Fig. 4, A and B) and activation of lysosomal function (Fig. 3). With regard to the discrepancy, it is believed that, in addition to the different contexts of the experiments (cell type, culture medium, etc.), another possible cause is a difference in data interpretation. For example, the clustered autophagosomes, early and late endosomes, and lysosomes in response to ART were interpreted as evidence for the inhibitory effect of ART on autophagy (15). However, it has been shown that during starvation-mediated autophagy, there is a perinuclear clustering of lysosomes driven by changes of intracellular pH (56). Therefore, we believe that this perinuclear clustering of lysosomes is, in fact, an indication of autophagy induction. Indeed, one recent report showed that ART induces autophagy via up-regulation of Beclin 1 expression (16). Moreover, our findings are consistent with three other reports suggesting that DHA was able to induce autophagy in cancer cells (57–59). DHA is a derivative of artemisinin, with a mechanistic action similar to that of ART (50, 60). Therefore, it is reasonable to postulate that ART might have a similar effect on autophagy as DHA.
It has been well established that ROS is the major cause of ART-induced cell death (39, 61, 62). Several mechanisms have been proposed for ART-induced oxidative stress. On one hand, it has been shown that the mitochondrial electron transport chain is involved in ROS generation induced by ART (39). On the other hand, ART was able to inhibit the expression and activities of the antioxidant superoxide dismutase in ovalbumin-challenged mice (63). In our study, we observed that increase of mitochondrial ROS (measured by MitoSOX Red) occurred at an earlier time point (6 h), prior to ART-induced cell death (after 18 h) (data not shown). In addition, the antioxidant NAC reduced ROS level and showed a protective effect on ART-induced cell death (Fig. 6E). All of these observations support the notion that oxidative stress contributes to cell death. As discussed earlier, the endoperoxide bridge of ART is broken by ferrous iron, and then ART becomes free radicals to target cellular organelles, such as mitochondria, and eventually leads to cell death (64). Consistent with earlier findings (15), we also found that DFO, the lysosomal iron chelator, was able to reduce ROS level and cell death (Fig. 7, D and E), implicating lysosomal iron as a critical mediator in ART-induced mitochondrial ROS production and cell death. Because iron released from ferritin degradation inside the lysosome can be utilized by the cells (20), it is possible that enhanced lysosomal degradation of ferritin induced by ART leads to the transient increase of cytosolic ferrous iron, which then affects the mitochondria, leading to enhanced mitochondrial ROS production.
To understand the involvement of lysosomes in iron-related bioactivity of ART, we examined the changes of ferritin, which consists of FTH and FTL. On one hand, ferritin protein can be regulated at the post-transcriptional level, in which IRP binds with ferritin 5′-iron-responsive element to repress its translation under iron-depleted conditions (17). On the other hand, there is evidence showing that DFO induces ferritin degradation through the lysosomal pathway (20, 21, 65). In this study, we also observed that BAF completely blocked ART- and DFO-mediated FTH and FTL degradation (Fig. 8, D and E), suggesting that ART- or DFO-mediated reduction of ferritin protein level is probably via lysosome-dependent degradation. Notably, although DFO induces ferritin degradation, the released iron from ferritin degradation is effectively chelated by DFO, leading to the protective effect of DFO on ART-induced cell death.
Earlier studies have shown that both lysosomes and proteasomes are involved in ferritin degradation (21). The lysosomal inhibitor BAF has been shown to be able to inhibit the ferritin degradation (20). However, the effect of BAF on the iron release from lysosome is controversial. On the one hand, BAF treatment attenuates iron release from lysosomes in MEFs (20). On the other hand, it has been reported that BAF treatment promotes the release of lysosomal iron in hepatocytes (66). Moreover, such a mechanism has been attributed to the fact that BAF is capable of enhancing the cytotoxicity of phthalocyanine 4-PDT (photodynamic therapy) in A431 cancer cells and in human head and neck squamous carcinoma cells via increased mitochondrial iron uptake (67, 68). In our study, we found that BAF decreases intracellular iron level, indicated by the increase of the IRP2 protein level (Fig. 8F). Therefore, the protective effect of BAF against ART-mediated cell death is achieved via blockage of ferritin degradation and subsequent iron release. However, one intriguing finding is that overexpression of FTH cannot completely prevent ART-induced cell death (Fig. 9B). One possibility is that although FTH homopolymers are functional in sequestering iron, they are less efficient than the H/L heteropolymers, based on an earlier report that FTH overexpression only led to a 50% reduction of labile iron pool (chelatable and redox active iron) in HeLa cells (48). Therefore, there is still a proportion of labile iron for the toxicity of artesunate. In addition, in cells with FTH overexpression, iron still can be released from FTL degradation (Fig. 9A), which will activate artesunate and cause its cytotoxicity.
As we have discussed earlier, functional lysosomes have been shown to be required for ART-induced cell death, although the mechanism is unknown (15). Our results also showed that lysosomes play an important role in ART-induced cell death, based on the following observations: (i) ART accumulated in the lysosomes (Fig. 2); (ii) ART activated lysosomal functions, as discussed earlier (Fig. 3); and (iii) BAF, a lysosomal V-ATPase inhibitor, was able to effectively inhibit the cell death induced by ART (Fig. 6C). Moreover, ART showed little toxicity effect on NCOA4 knockdown cells (Fig. 9D), further confirming the notion that ferritin delivery and degradation in lysosomes is required for the toxicity of ART. Therefore, lysosomal inhibitors significantly protect the cells from ART-induced cell death via blockage of ferritin degradation (Fig. 8D). In fact, we also found that chloroquine inhibited the ferritin degradation and cell death induced by ART as efficiently as BAF (data not shown). Interestingly, we observed reduced cell viability in NCOA4 knockdown cells (Fig. 9D), indicating the importance of ferritin delivery and degradation in lysosomes in maintaining cell viability.
In summary, we demonstrate a novel mechanism underlying ART-induced cancer cell death. As shown in Fig. 9E, ART accumulates in lysosomes and reacts with lysosomal iron. The activated ART then promotes lysosomal function via enhancing V-ATPase assembly. One major source of iron for the cytotoxicity of ART is attained from the degradation of ferritin by lysosomes. In conclusion, we propose a new model of action on the mechanism of ART-induced cell death by focusing on lysosomal activation and ferritin degradation. At present, there is evidence demonstrating higher iron levels in cancer cells than in non-cancerous cells, and iron has been targeted for cancer therapy and prevention (69). Therefore, the results from our study support the development of this important anti-malaria drug as a cancer therapeutic agent.
Acknowledgments
We thank Dr. N. Mizushima (Tokyo Medical and Dental University) for providing the HeLa cells with stable expression of GFP-LC3, Dr. A Ballabio (Telethon Institute of Genetics and Medicine, Italy) for providing the TFEB-luciferase construct, Dr. D. J. Kwiatkowski (Harvard University) for providing the pair of Tsc2 WT and KO MEFs, and Dr. T. W. Soong (National University of Singapore) for providing FLAG-FTH plasmid. We also thank Dr. Y. K. Tai (National University of Singapore) for insightful discussions.
This work was supported in part by research grants from Singapore National Medical Research Council (NMRC/1260/2010) and the Singapore Biomedical Research Council (BMRC/08/1/21/19/554) (to H. M. S.). K. S. W. T. is founding director of BioLynx Technologies (Singapore), which provided the LynxTag-ARTTMASBlue used in this study.
- ART
- artesunate
- NCOA4
- nuclear receptor coactivator 4
- ROS
- reactive oxygen species
- BAF
- bafilomycin A1
- NAC
- N-acetylcysteine
- DFO
- deferoxamine mesylate
- FTH
- ferritin heavy chain
- FTL
- ferritin light chain
- V-ATPase
- vacuolar H+-ATPase
- DHA
- dihydroartemisinin
- MEF
- mouse embryonic fibroblast
- Z
- N-benzyloxycarbonyl
- fmk
- fluoromethylketone
- LTR
- LysoTracker Red
- LTG
- LysoTracker Green
- MTR
- MitoTracker Red
- MSR
- MitoSox Red
- TFEB
- transcription factor EB
- Ctrl
- control
- PI
- propidium iodide
- LC3
- light chain 3
- mTORC1
- mammalian or mechanistic target of rapamycin complex 1
- ATG
- autophagy-related gene
- IRP
- iron-responsive protein.
REFERENCES
- 1. China Cooperative Research Group on Qinghaosu and Its Derivatives as Antimalarials (1982) Chemical studies on qinghaosu (artemisinine). J. Tradit. Chin. Med. 2, 3–8 [PubMed] [Google Scholar]
- 2. Woerdenbag H. J., Moskal T. A., Pras N., Malingré T. M., el-Feraly F. S., Kampinga H. H., Konings A. W. (1993) Cytotoxicity of artemisinin-related endoperoxides to Ehrlich ascites tumor cells. J. Nat. Prod. 56, 849–856 [DOI] [PubMed] [Google Scholar]
- 3. Ho W. E., Peh H. Y., Chan T. K., Wong W. S. (2014) Artemisinins: pharmacological actions beyond anti-malarial. Pharmacol. Ther. 142, 126–139 [DOI] [PubMed] [Google Scholar]
- 4. Du J. H., Zhang H. D., Ma Z. J., Ji K. M. (2010) Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo. Cancer Chemother. Pharmacol. 65, 895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou C., Pan W., Wang X. P., Chen T. S. (2012) Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. J. Cell Physiol. 227, 3778–3786 [DOI] [PubMed] [Google Scholar]
- 6. Zhao Y., Jiang W., Li B., Yao Q., Dong J., Cen Y., Pan X., Li J., Zheng J., Pang X., Zhou H. (2011) Artesunate enhances radiosensitivity of human non-small cell lung cancer A549 cells via increasing NO production to induce cell cycle arrest at G2/M phase. Int. Immunopharmacol. 11, 2039–2046 [DOI] [PubMed] [Google Scholar]
- 7. Dell'Eva R., Pfeffer U., Vené R., Anfosso L., Forlani A., Albini A., Efferth T. (2004) Inhibition of angiogenesis in vivo and growth of Kaposi's sarcoma xenograft tumors by the anti-malarial artesunate. Biochem. Pharmacol. 68, 2359–2366 [DOI] [PubMed] [Google Scholar]
- 8. Rasheed S. A., Efferth T., Asangani I. A., Allgayer H. (2010) First evidence that the antimalarial drug artesunate inhibits invasion and in vivo metastasis in lung cancer by targeting essential extracellular proteases. Int. J. Cancer 127, 1475–1485 [DOI] [PubMed] [Google Scholar]
- 9. Hou J., Wang D., Zhang R., Wang H. (2008) Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin. Cancer Res. 14, 5519–5530 [DOI] [PubMed] [Google Scholar]
- 10. Eskelinen E. L., Tanaka Y., Saftig P. (2003) At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 13, 137–145 [DOI] [PubMed] [Google Scholar]
- 11. Saftig P., Klumperman J. (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 [DOI] [PubMed] [Google Scholar]
- 12. Mindell J. A. (2012) Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86 [DOI] [PubMed] [Google Scholar]
- 13. Mizushima N., Komatsu M. (2011) Autophagy: renovation of cells and tissues. Cell 147, 728–741 [DOI] [PubMed] [Google Scholar]
- 14. Settembre C., Fraldi A., Medina D. L., Ballabio A. (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamacher-Brady A., Stein H. A., Turschner S., Toegel I., Mora R., Jennewein N., Efferth T., Eils R., Brady N. R. (2011) Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. J. Biol. Chem. 286, 6587–6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen K., Shou L. M., Lin F., Duan W. M., Wu M. Y., Xie X., Xie Y. F., Li W., Tao M. (2014) Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs 25, 652–662 [DOI] [PubMed] [Google Scholar]
- 17. Rouault T. A. (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406–414 [DOI] [PubMed] [Google Scholar]
- 18. Ford G. C., Harrison P. M., Rice D. W., Smith J. M., Treffry A., White J. L., Yariv J. (1984) Ferritin: design and formation of an iron-storage molecule. Philos. Trans. R. Soc. Lond. B Biol. Sci. 304, 551–565 [DOI] [PubMed] [Google Scholar]
- 19. Harrison P. M., Arosio P. (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1275, 161–203 [DOI] [PubMed] [Google Scholar]
- 20. Asano T., Komatsu M., Yamaguchi-Iwai Y., Ishikawa F., Mizushima N., Iwai K. (2011) Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell Biol. 31, 2040–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Domenico I., Ward D. M., Kaplan J. (2009) Specific iron chelators determine the route of ferritin degradation. Blood 114, 4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mancias J. D., Wang X., Gygi S. P., Harper J. W., Kimmelman A. C. (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang H., Cicchetti G., Onda H., Koon H. B., Asrican K., Bajraszewski N., Vazquez F., Carpenter C. L., Kwiatkowski D. J. (2003) Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Invest. 112, 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarvius M., Paulsson J., Weibrecht I., Leuchowius K. J., Andersson A. C., Wählby C., Gullberg M., Botling J., Sjöblom T., Markova B., Ostman A., Landegren U., Söderberg O. (2007) In situ detection of phosphorylated platelet-derived growth factor receptor β using a generalized proximity ligation method. Mol. Cell Proteomics 6, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 25. Aon M. A., Cortassa S., Marbán E., O'Rourke B. (2003) Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744 [DOI] [PubMed] [Google Scholar]
- 26. Mukhopadhyay P., Rajesh M., Yoshihiro K., Haskó G., Pacher P. (2007) Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 358, 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S., Banfi S., Parenti G., Cattaneo E., Ballabio A. (2009) A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 [DOI] [PubMed] [Google Scholar]
- 28. Luo J., Zhu W., Tang Y., Cao H., Zhou Y., Ji R., Zhou X., Lu Z., Yang H., Zhang S., Cao J. (2014) Artemisinin derivative artesunate induces radiosensitivity in cervical cancer cells in vitro and in vivo. Radiat. Oncol. 9, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thanaketpaisarn O., Waiwut P., Sakurai H., Saiki I. (2011) Artesunate enhances TRAIL-induced apoptosis in human cervical carcinoma cells through inhibition of the NF-κB and PI3K/Akt signaling pathways. Int. J. Oncol. 39, 279–285 [DOI] [PubMed] [Google Scholar]
- 30. Chen H. H., Zhou H. J., Fang X. (2003) Inhibition of human cancer cell line growth and human umbilical vein endothelial cell angiogenesis by artemisinin derivatives in vitro. Pharmacol. Res. 48, 231–236 [DOI] [PubMed] [Google Scholar]
- 31. Yoshimori T., Yamamoto A., Moriyama Y., Futai M., Tashiro Y. (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266, 17707–17712 [PubMed] [Google Scholar]
- 32. Zhou J., Tan S. H., Nicolas V., Bauvy C., Yang N. D., Zhang J., Xue Y., Codogno P., Shen H. M. (2013) Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 23, 508–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng C., Ho W. E., Goh F. Y., Guan S. P., Kong L. R., Lai W. Q., Leung B. P., Wong W. S. (2011) Anti-malarial drug artesunate attenuates experimental allergic asthma via inhibition of the phosphoinositide 3-kinase/Akt pathway. PLoS One 6, e20932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakatogawa H., Ichimura Y., Ohsumi Y. (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130, 165–178 [DOI] [PubMed] [Google Scholar]
- 36. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Punnonen E. L., Reunanen H. (1990) Effects of vinblastine, leucine, and histidine, and 3-methyladenine on autophagy in Ehrlich ascites cells. Exp. Mol. Pathol. 52, 87–97 [DOI] [PubMed] [Google Scholar]
- 38. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mercer A. E., Copple I. M., Maggs J. L., O'Neill P. M., Park B. K. (2011) The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. J. Biol. Chem. 286, 987–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winterbourn C. C., Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 41. Turrens J. F. (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lai H. C., Singh N. P., Sasaki T. (2013) Development of artemisinin compounds for cancer treatment. Invest. New Drugs 31, 230–246 [DOI] [PubMed] [Google Scholar]
- 43. Terman A., Kurz T. (2013) Lysosomal iron, iron chelation, and cell death. Antioxid. Redox Signal. 18, 888–898 [DOI] [PubMed] [Google Scholar]
- 44. Cable H., Lloyd J. B. (1999) Cellular uptake and release of two contrasting iron chelators. J. Pharm. Pharmacol. 51, 131–134 [DOI] [PubMed] [Google Scholar]
- 45. Lloyd J. B., Cable H., Rice-Evans C. (1991) Evidence that desferrioxamine cannot enter cells by passive diffusion. Biochem. Pharmacol. 41, 1361–1363 [DOI] [PubMed] [Google Scholar]
- 46. Kurz T., Leake A., Von Zglinicki T., Brunk U. T. (2004) Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem. J. 378, 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pantopoulos K., Porwal S. K., Tartakoff A., Devireddy L. (2012) Mechanisms of mammalian iron homeostasis. Biochemistry 51, 5705–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cozzi A., Corsi B., Levi S., Santambrogio P., Albertini A., Arosio P. (2000) Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J. Biol. Chem. 275, 25122–25129 [DOI] [PubMed] [Google Scholar]
- 49. Pandey A. V., Tekwani B. L., Singh R. L., Chauhan V. S. (1999) Artemisinin, an endoperoxide antimalarial, disrupts the hemoglobin catabolism and heme detoxification systems in malarial parasite. J. Biol. Chem. 274, 19383–19388 [DOI] [PubMed] [Google Scholar]
- 50. Eckstein-Ludwig U., Webb R. J., Van Goethem I. D., East J. M., Lee A. G., Kimura M., O'Neill P. M., Bray P. G., Ward S. A., Krishna S. (2003) Artemisinins target the SERCA of Plasmodium falciparum. Nature 424, 957–961 [DOI] [PubMed] [Google Scholar]
- 51. Stocks P. A., Bray P. G., Barton V. E., Al-Helal M., Jones M., Araujo N. C., Gibbons P., Ward S. A., Hughes R. H., Biagini G. A., Davies J., Amewu R., Mercer A. E., Ellis G., O'Neill P. M. (2007) Evidence for a common non-heme chelatable-iron-dependent activation mechanism for semisynthetic and synthetic endoperoxide antimalarial drugs. Angew Chem. Int. Ed. Engl. 46, 6278–6283 [DOI] [PubMed] [Google Scholar]
- 52. Cipriano D. J., Wang Y., Bond S., Hinton A., Jefferies K. C., Qi J., Forgac M. (2008) Structure and regulation of the vacuolar ATPases. Biochim. Biophys. Acta 1777, 599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nishi T., Forgac M. (2002) The vacuolar (H+)-ATPases: nature's most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 3, 94–103 [DOI] [PubMed] [Google Scholar]
- 54. Li S. C., Kane P. M. (2009) The yeast lysosome-like vacuole: endpoint and crossroads. Biochim. Biophys. Acta 1793, 650–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen H. M., Mizushima N. (2014) At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem. Sci. 39, 61–71 [DOI] [PubMed] [Google Scholar]
- 56. Korolchuk V. I., Saiki S., Lichtenberg M., Siddiqi F. H., Roberts E. A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F. M., O'Kane C. J., Deretic V., Rubinsztein D. C. (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13, 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Z., Hu W., Zhang J. L., Wu X. H., Zhou H. J. (2012) Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio. 2, 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Du X. X., Li Y. J., Wu C. L., Zhou J. H., Han Y., Sui H., Wei X. L., Liu L., Huang P., Yuan H. H., Zhang T. T., Zhang W. J., Xie R., Lang X. H., Jia D. X., Bai Y. X. (2013) Initiation of apoptosis, cell cycle arrest and autophagy of esophageal cancer cells by dihydroartemisinin. Biomed. Pharmacother. 67, 417–424 [DOI] [PubMed] [Google Scholar]
- 59. Hu W., Chen S. S., Zhang J. L., Lou X. E., Zhou H. J. (2014) Dihydroartemisinin induces autophagy by suppressing NF-κB activation. Cancer Lett. 343, 239–248 [DOI] [PubMed] [Google Scholar]
- 60. Zhang S., Gerhard G. S. (2009) Heme mediates cytotoxicity from artemisinin and serves as a general anti-proliferation target. PLoS One 4, e7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cheng R., Li C., Li C., Wei L., Li L., Zhang Y., Yao Y., Gu X., Cai W., Yang Z., Ma J., Yang X., Gao G. (2013) The artemisinin derivative artesunate inhibits corneal neovascularization by inducing ROS-dependent apoptosis in vascular endothelial cells. Invest. Ophthalmol. Vis. Sci. 54, 3400–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Efferth T., Giaisi M., Merling A., Krammer P. H., Li-Weber M. (2007) Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS One 2, e693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ho W. E., Cheng C., Peh H. Y., Xu F., Tannenbaum S. R., Ong C. N., Wong W. S. (2012) Anti-malarial drug artesunate ameliorates oxidative lung damage in experimental allergic asthma. Free Radic. Biol. Med. 53, 498–507 [DOI] [PubMed] [Google Scholar]
- 64. Mercer A. E., Maggs J. L., Sun X. M., Cohen G. M., Chadwick J., O'Neill P. M., Park B. K. (2007) Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. J. Biol. Chem. 282, 9372–9382 [DOI] [PubMed] [Google Scholar]
- 65. Kidane T. Z., Sauble E., Linder M. C. (2006) Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 291, C445–C455 [DOI] [PubMed] [Google Scholar]
- 66. Uchiyama A., Kim J. S., Kon K., Jaeschke H., Ikejima K., Watanabe S., Lemasters J. J. (2008) Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology 48, 1644–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saggu S., Hung H. I., Quiogue G., Lemasters J. J., Nieminen A. L. (2012) Lysosomal signaling enhances mitochondria-mediated photodynamic therapy in A431 cancer cells: role of iron. Photochem. Photobiol. 88, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hung H. I., Schwartz J. M., Maldonado E. N., Lemasters J. J., Nieminen A. L. (2013) Mitoferrin-2-dependent mitochondrial iron uptake sensitizes human head and neck squamous carcinoma cells to photodynamic therapy. J. Biol. Chem. 288, 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Torti S. V., Torti F. M. (2013) Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13, 342–355 [DOI] [PMC free article] [PubMed] [Google Scholar]