

Background: The biogenesis of melanosomes and other lysosome-related organelles requires a pair of Rab GTPases, Rab32 and Rab38.
Results: Myosin Vc is a novel binding partner of these Rabs. Myosin Vc functions in the trafficking of integral membrane proteins to melanosomes.
Conclusion: Myosin Vc works in transport to and secretion of melanosomes.
Significance: These results advance understanding of melanosome biology.
Keywords: Melanogenesis, Membrane Protein, Membrane Trafficking, Myosin, Organelle, Protein Sorting, Rab
Abstract
Class V myosins are actin-based motors with conserved functions in vesicle and organelle trafficking. Herein we report the discovery of a function for Myosin Vc in melanosome biogenesis as an effector of melanosome-associated Rab GTPases. We isolated Myosin Vc in a yeast two-hybrid screening for proteins that interact with Rab38, a Rab protein involved in the biogenesis of melanosomes and other lysosome-related organelles. Rab38 and its close homolog Rab32 bind to Myosin Vc but not to Myosin Va or Myosin Vb. Binding depends on residues in the switch II region of Rab32 and Rab38 and regions of the Myosin Vc coiled-coil tail domain. Myosin Vc also interacts with Rab7a and Rab8a but not with Rab11, Rab17, and Rab27. Although Myosin Vc is not particularly abundant on pigmented melanosomes, its knockdown in MNT-1 melanocytes caused defects in the trafficking of integral membrane proteins to melanosomes with substantially increased surface expression of Tyrp1, nearly complete loss of Tyrp2, and significant Vamp7 mislocalization. Knockdown of Myosin Vc in MNT-1 cells more than doubled the abundance of pigmented melanosomes but did not change the number of unpigmented melanosomes. Together the data demonstrate a novel role for Myosin Vc in melanosome biogenesis and secretion.
Introduction
Melanosomes are lysosome-related organelles (LROs)3 that synthesize and store melanin pigments in skin melanocytes and retinal pigmented epithelial cells (1–3). The development or maturation of melanosomes depends on discrete organelle remodeling and trafficking events and can be classified into four distinct morphological stages (1, 4). The least mature melanosomes, stage I, are formed from vacuolar early endosomes. Stage I melanosomes are characterized by the presence of the structural protein Pmel17 (also known as PMEL) on the organelle limiting membrane and on intraluminal vesicles within the organelle and by a distinctive clathrin lattice on the limiting membrane (1, 4). Within the environment of stage I melanosomes, Pmel17 is cleaved by a furin-type proprotein convertase and then by a metalloproteinase (1, 5, 6). In stage II melanosomes, Pmel17 fragments oligomerize and form amyloid-like fibrils that span the length of the melanosome and are clearly evident in electron micrographs (4, 6–8). The subsequent delivery of integral membrane proteins such as the enzymes tyrosinase, tyrosinase-related protein 1 (Tyrp1), and Tyrp2 initiates the synthesis of melanin pigments that coat the Pmel17-fibrils to form partially pigmented stage III melanosomes (1). Tyrosinase is the key enzyme for melanin synthesis, with Tyrp1 and Tyrp2 serving modulatory roles (1, 3, 9). Additional melanin synthesis generates fully pigmented stage IV melanosomes (1).
Newly synthesized tyrosinase, Tyrp1, and other cargo required for melanosome maturation beyond stage II are delivered from early endosomal tubules and buds (1, 10–13). This traffic is complex and involves at least two routes that depend on adaptor protein-1 (AP-1), AP-3, biogenesis of lysosome-related organelle complex-1 (BLOC-1), BLOC-2, and BLOC-3 (1, 10–13). These components of the trafficking machinery are expressed in all cell types, reflecting their function in ubiquitous trafficking such as transport of lysosome-associated membrane proteins (LAMPs) to the lysosome (14–20). However, with the exception of AP-1, whose deficiency causes embryonic lethality, deficiency in any of these proteins manifests more noticeably in LRO-producing cell types and underlies various forms of Hermansky-Pudlak syndrome (14, 21–23). Hermansky-Pudlak syndrome is a disorder characterized by hypopigmentation due to defective melanosome biogenesis and additional manifestations caused by deficiencies in other LROs. Importantly, two closely related members of the Rab family, Rab32 and Rab38, interact physically and functionally with AP-1, AP-3, and the BLOCs to mediate transport of cargo from early endosomal domains to maturing melanosomes (24–26). Rab32 and Rab38 are expressed in a tissue-specific manner, are chiefly present in LRO-producing cell types, and provide an elegant mechanistic explanation to the repurposing of the ubiquitous trafficking machinery for LRO biogenesis in specialized cell types (25, 27–32). Consistently, rodents with mutations in Rab38 manifest with typical LRO deficiencies such as hypopigmentation due to impaired melanosome biogenesis (29, 31–35).
In skin, the stage IV melanosomes are secreted and transferred from melanocytes to keratinocytes where they remain and form a pigmented cap around the keratinocyte nucleus (2, 3). Several mechanisms have been proposed to explain the intercellular melanosome transfer including (i) the Rab11-dependent exocytosis of naked melanin or “melanocore” by melanocytes and subsequent endocytosis by keratinocytes, (ii) the formation of Rab17-dependent filopodia by melanocytes as conduits for melanosome transfer, (iii) the release of “globules” containing several melanosomes that are then taken up by keratinocytes, and (iv) the shedding of melanosome-rich packages subsequently phagocytosed by keratinocytes (36–41). The melanosome secretion and/or transfer mechanism is currently debated at a morphological level, and with the possible exception of Rab11 and Rab17, the molecular machinery involved is unknown. However, it is clear that before secretion and intercellular transfer can occur, stage IV melanosomes must move on microtubules to the cell periphery and be captured by the cortical actin cytoskeleton. Rab27a and the actin-based motor Myosin Va localize to and interact on stage IV melanosomes to mediate their association with the cortical actin close to the plasma membrane (42–45). The interaction between Rab27a and Myosin Va is mediated by melanophilin rather than direct (46, 47). Deficiency in Rab27a, Myosin Va, or melanophilin does not affect melanosome biogenesis but results in perinuclear clustering of mature melanosomes and diminished transfer to keratinocytes (42–51). Griscelli syndrome patients and the corresponding mice models have mutations in Rab27a, Myosin Va, or melanophilin and display hypopigmentation due to inefficient peripheral capture and subsequent transfer of melanosomes to keratinocytes (21). Myosin Va belongs to a family of well conserved actin-associated motors that function in vesicle and organelle trafficking events from yeast to humans (52). The other members of the mammalian class V Myosin family are Myosin Vb and Myosin Vc. Myosin Vb functions with Rab11 and Rab8 in the ubiquitous recycling of cargoes such as the transferrin receptor from early/recycling endosomal domains to the plasma membrane (53). Myosin Vc has been less well characterized but localizes to early/recycling endosomal domains in non-specialized cells and to secretory organelles in specialized cell types (53–56). To our knowledge no functional binding partner of Myosin Vc has been described to date.
Here we report that Myosin Vc interacts with Rab32, and Rab38 and functions in melanosome biogenesis and secretion. Myosin Vc functions in the trafficking of integral membrane proteins to melanosomes including Tyrp1, Tyrp2, and Vamp7. Myosin Vc deficiency also causes accumulation of pigmented melanosomes in melanocytes, but Myosin Vc is not abundant on pigmented melanosomes, suggesting an indirect role in melanosome secretion.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, and Yeast Two-hybrid Assay
The cDNA encoding for human Rab7a, Rab8a, Rab11a, Rab17, Rab27a, Rab32, Rab38, and Myosin Va full tail (encoding for aa 908–1855, gene ID 4644) were amplified from total RNA of MNT-1 cells by reverse transcriptase-PCR and subsequently cloned in-frame into pGBT9 and pGAD424 vectors (Clontech). The cDNA encoding for full-length human Myosin Vb was obtained from Promega Kazusa ORFs (pF1K-hMyo5b, gene ID 4645) and the corresponding full tail (encoding for aa 905–1848) amplified by PCR and cloned in-frame into pGAD424 vector. Plasmids for overexpression of human Myosin Vc-GFP full-length and full tail (encoding for aa 898–1742) were previously described (54, 55). The constructs encoding for Myosin Vc full-length and full tail utilized in this study correspond to the gene ID 55930 sequence and contained all exons that in other Myosin V family members are alternatively spliced (exons D, E, and F). The cDNA for Myosin Vc full tail was subcloned into the pGAD424 vector, and the cDNA for Myosin Vc globular tail (aa 1335–1742) was amplified by PCR and cloned into pGAD424. The cDNA for human tyrosinase and tyrp1 were amplified from total RNA of MNT-1 cells by reverse transcriptase-PCR and subsequently cloned in-frame into pmCherry. Site-directed mutagenesis of plasmids encoding for the Rab proteins was performed using the QuikChange system (Stratagene, La Jolla, CA). Deletion of Myosin Vc tail regions (exon D, E, and F) was achieved by the overlapping PCR method. Antibodies used were as follows: rabbit a-Myosin Va (57), chicken a-Myosin Vb (54), rabbit a-Myosin Vc (54), mouse a-tyrosinase (Santa Cruz, T311), mouse a-Tyrp1 (Santa Cruz, MEL5, clone Ta99), mouse a-Tyrp2 (Santa Cruz, C-9), mouse a-Pmel17 (Dako, HMB45), mouse a-Vamp7 (Abcam, SYBL1 clone 158.2), mouse a-Rab7a (Sigma, Rab7–117), mouse a-Rab8a (BD Biosciences, 610845), mouse a-Rab27a (Santa Cruz, E12A-1), rabbit a-Rab11a (Invitrogen, 71-5300), rabbit a-Rab32 and rabbit a-Rab38 (24), and mouse a-αtubulin (Sigma, DM1A). Alexa-488- Alexa-546-, and Alexa-647-conjugated secondary antibodies were from Invitrogen, and horseradish peroxidase-conjugated secondary antibodies were from GE Healthcare/Amersham Biosciences. Anti-mouse secondary antibodies complexed with 12- or 18-nm colloidal gold for transmission electron microscopy were from Jackson ImmunoResearch. Yeast two-hybrid experiments were performed using pGBT9 and pGAD424 vectors (Clontech) and AH109 Saccharomyces cerevisiae cells grown on Synthetic Dropout media lacking leucine and tryptophan as selection markers as previously described (58). 3-Amino-1,2,4-triazole was used to test for higher binding stringency.
Cell Culture
Human MNT-1 cells were cultured as described (59). Transfection for siRNA and plasmids for overexpression of GFP- and Cherry-fusion proteins was performed using the Nucleofector electroporation system (Lonza) and the NHEM-Neo kit with MNT-1 cells subcultured 2–3 days before transfection. Briefly, 1.5 × 106 cells were subjected to two sequential siRNAs treatments on days 1 and 4, and cells were analyzed on day 7. Cells transfected with plasmids for GFP-fusion protein overexpression (1.5 × 106 cells per transfection) were analyzed after 48 h. Oligonucleotides used for siRNA are as follows: Myosin Vc (Sigma; SASI_Hs01_00184026), Rab32 (Sigma; SASI_Hs02_00342400), Rab38 (Sigma; SASI_Hs01_00247037), δ subunit of AP-3 (12), and universal negative control siRNA (Sigma; SIC001). Transfection efficiency of plasmids was 70% or higher as judged by the percentage of cells displaying fluorescence upon microscopy observation.
Biochemical Procedures
Whole cell extracts were prepared as previously described (60). For immunoblotting analysis, proteins were fractionated on pre-cast 4–20% gradient SDS/polyacrylamide gels (Invitrogen) and transferred by electroblotting to PVDF membranes. Membranes were incubated sequentially with blocking buffer, primary antibody, and horseradish peroxidase-conjugated secondary antibody as described (61). Bound antibodies were detected by using ECL Prime Western blotting reagent (GE Healthcare/Amersham Biosciences). For subcellular fractionation, a post-nuclear supernatant was prepared by homogenizing MNT-1 cells with a Dounce homogenizer in buffer H (20 mm Hepes, pH 7.4) containing 0.32 m sucrose and protease inhibitors followed by centrifugation for 20 min at 800 × g at 4 °C. The post-nuclear supernatant (250 μl) was loaded onto a 12-ml linear sucrose gradient (20–55%) in buffer H. The sample was centrifuged at 113,000 × g for 6 h in a SW41Ti rotor in a Beckman L8–70 M ultracentrifuge at 4 °C. Fractions of 1 ml were collected and used for immunoblotting.
Melanin Content
MNT-1 cells were centrifuged at 90 × g for 10 min to pellet cells. Cell pellets were solubilized with Soulene 350 (PerkinElmer Life Sciences) and treated and analyzed as described (62) by a spectrophotometric method at 500 nm using purified Sepia officinalis melanin (Sigma) as a standard. Melanin secreted by MNT-1 cells to the culture media was determined as follows. After siRNA or plasmid transfection, the monolayer of MNT-1 cells was carefully washed with media to eliminate cells loosely attached to the culture dish, and fresh medium was added for a final 24-h incubation period. Culture media was collected and subjected to ultracentrifugation at 400,000 × g for 15 min at 4 °C, and the resulting pellets were treated as described for solubilization and melanin determination (62). The melanin content was normalized to the protein content in the cell monolayer, which was established in a Triton X-100 lysate by the Bradford method using a commercial kit (Bio-Rad) and BSA as standard.
Electron Microscopy
High pressure freezing was performed as previously described (63, 64). Briefly, MNT-1 cells treated with control or Myosin Vc siRNA were subjected to high pressure freezing in 15% dextran (9–11 kDa) in culture media, freeze-substitution in 0.25% glutaraldehyde, 0.1% uranyl acetate, and embedding in Lowicryl HM20 resin and processed for thin-section transmission electron microscopy. For immunogold labeling, 80-nm sections were collected on nickel slot grids, blocked with 20% goat serum in PBS, blotted, and then incubated in the primary antibodies overnight at 4 °C. Grids were washed in PBS-Tween and incubated in the secondary antibodies for 1.5 h. Anti-mouse secondary antibody complexed with 12-nm colloidal gold was used for detection of Pmel17. Anti-mouse secondary antibody complexed with 18-nm colloidal gold was used for detection of Vamp7. Grids were washed first in PBS-Tween and then in PBS, fixed in 0.5% glutaraldehyde in PBS, washed in distilled H2O, and dried. The grids were stained with 2% uranyl acetate in 70% methanol, 30% water for 7 min, rinsed in 70% methanol, 30% water, dried, and then post-stained in Reynold's lead stain, rinsed in distilled H2O, and dried. Images were taken on a JEOL 2000 TEM.
Live Cell Microscopy, Immunofluorescence Microscopy, and Antibody Internalization Assay
For live cell imaging experiments cells were plated on Matrigel-coated glass-bottom 35-mm dishes. The samples were imaged using a temperature-controlled chamber at 37 °C and 5% CO2 on an Olympus IX81 spinning-disk confocal fluorescence microscope with Photometrics Cascade II camera using a 100×/1.40NA objective. For immunofluorescence microscopy cells were grown on Matrigel-coated coverslips and fixed in a 50/50 mixture of cold (−20 °C) methanol/acetone at room temperature for 10 min, allowed to dry, and stored in PBS containing 0.1% sodium azide at 4 °C. Cells were permeabilized and blocked, incubated with primary antibody for 1 h, and incubated with species-specific secondary antibodies conjugated to Alexa-488, Alexa-546, or Alexa-647 (Invitrogen). Internalization of antibodies was performed as described (12, 17). Briefly, MNT1 cells transfected with siRNA or plasmids were grown on glass coverslips and incubated for 20 min at 37 °C in the presence of mouse a-Tyrp1 primary antibody diluted in DMEM, 1% (w/v) BSA, 25 mm HEPES buffer, pH 7.4. Subsequently cells were washed for 5 min in ice-cold PBS, fixed in 2% formaldehyde for 10 min, incubated for 1 h at room temperature in the presence of Alexa-488- or Alexa-647-labeled a-mouse secondary antibody (diluted in PBS, 0.1% (w/v) BSA, 0.1% (w/v) saponin), rinsed with PBS, and mounted onto glass slides with Fluoromount G. Tyrp1 surface expression represents the relative fluorescence intensity of each siRNA treatment versus control (irrelevant siRNA; see Fig. 7B) or each GFP-fusion protein versus control (GFP; see Fig. 7C) corrected for the corresponding overall level of Tyrp1 expression as determined by immunoblotting of total cell extracts. For live cell imaging of transferrin, MNT-1 cells previously transfected with Myosin Vc-GFP and Myosin Vb-GFP were incubated for 20 min at 37 °C in the presence of 0.16 mg/ml transferrin-Alexa-647 (Invitrogen) in culture media. Subsequently, cells were washed in culture media and immediately observed by spinning disk confocal fluorescence microscopy. Immunofluorescence microscopy samples were examined using the same microscope described above. Fixed and live cell sample images were acquired and analyzed in Slidebook 5.0 software (3i). For determination of the percent of colocalization, each channel was subjected to a Laplacian 2D filter with a 3 × 3 kernel (−1, 8, −1), and a binary mask was generated using a Ridler-Calvard automated threshold method both on each channel and on the overlap between the two individual channels in Slidebook software. Pixel area overlap from the overlap mask and each individual mask was used to calculate percent colocalization. Antibody internalization images were acquired at room temperature using a Nikon Diaphot 300 microscope with Photometrics Cool SNAP camera using Metamorph software under conditions optimized to prevent signal saturation. Images were analyzed for total fluorescence intensity with ImageJ software (National Institutes of Health) as previously described (12, 17).
FIGURE 7.

Myosin Vc knockdown and overexpression of Myosin Vc-tail-GFP disrupt trafficking of specific melanosomal cargoes. A, immunoblotting analysis of total cell extracts from siRNA control- and Myosin Vc-treated MNT-1 cells. Deficiency of Myosin Vc results in an accumulation of melanin synthesizing enzymes tyrosinase and Tyrp1 but a severe reduction in Tyrp2. Pmel17 is highly enriched in Myosin Vc-depleted samples. The experiments were repeated at least three times with similar results. B, live MNT-1 control cells or cells deficient for AP-3, both Rab32 and Rab38, or Myosin Vc were incubated with culture media containing the MEL-5 (Ta99) mouse anti-Tyrp1 antibody for 20 min at 37 °C, subsequently fixed, permeabilized, and immunostained with an Alexa-488-conjugated anti-mouse antibody. Cells were imaged using an epifluorescent microscope, and the relative amounts of surface and internalized anti-Tyrp1 antibody were estimated as the average fluorescence intensity per cell determined with ImageJ and normalized to control cells (means ± S.D.). Results from at least three independent experiments were compared with control cells. *, p < 0.01. C, live MNT-1 cells overexpressing GFP (Control) or the following GFP fusion proteins: full-length Myosin Vc (Myo Vc-GFP), the full tail of Myosin Vc (aa 898–1742) (Myo Vc-tail-GFP), the full tail of Myosin Vc carrying a deletion of exon F (Myo Vc-tailΔF-GFP), and full-length Myosin Vb (Myo Vb-GFP) were subjected to the same anti-Tyrp1 antibody internalization assay described in B with the exception that cells were immunostained with an Alexa-647-conjugated anti-mouse antibody. Result from three independent experiments were compared with control cells. *, p < 0.05. D, immunoelectron microscopy analysis of control and Myosin Vc-deficient MNT-1 cells labeled with the SYBL1 (158.2) antibody to Vamp7 (arrows, 18 nm). Vamp7 largely localizes to pigmented melanosomes in control MNT-1 cells (57 ± 6% of the gold particles, 5 cells). Insets 1 and 2 show examples of gold particles associated with stage III/IV melanosomes in control cell. Myosin Vc deficiency decreases the localization of Vamp7 to pigmented melanosomes (29 ± 7% of the gold particles, 7 cells, p < 0.001) and increases its localization to the plasma membrane and vacuolar compartments with morphological characteristics of endosomes (electron-lucent interior with few or no intraluminal vesicles). Insets 3, 4, and 5 show examples of gold particles associated with the plasma membrane, electron lucent vacuoles, and stage IV melanosomes, respectively, in a Myosin Vc-deficient cell. Bars, 500 nm.
RESULTS
Myosin Vc Interacts with Rab GTPases Involved in Melanosome Biogenesis
Previous work identified the closely related Rab32 and Rab38 as key components in the biogenesis of melanosomes, platelet dense granules, and other LROs. To identify novel binding partners of Rab38, a human bone marrow expression library was screened with the constitutively active (GTP-locked) human Rab38-Q69L mutant using the yeast two-hybrid system. Six colonies that grew in the absence of histidine and presence of 5 mm 3-amino-1,2,4-triazole were isolated. One of them, displaying the strongest interaction, was further characterized by DNA sequencing. The clone contained a cDNA segment encoding amino acid residues 901–1350 of Myosin Vc, spanning most of the coiled-coil part of its tail domain (Fig. 1C, left side) (54). To validate this result with independent constructs, the full tail of Myosin Vc (residues 898–1742) was tested and found to interact with the constitutively active, GTP-locked mutants of Rab32 and Rab38 but not with the wild-type proteins (Fig. 1A) or the corresponding dominant-negative mutants Rab38-T23N and Rab32-T39N (not shown). These interactions were highly specific because constitutively active Rab32 and Rab38 did not interact with the full tail of the other human class V Myosins, Myosin Va and Myosin Vb (Fig. 1B). Conversely, specificity was also demonstrated by the lack of interaction between the full tail of human Myosin Vc and constitutively active human Rab11a or Rab27a, which are known binding partners of Myosin Vb and Myosin Va, respectively (Fig. 1A) (42, 65). Specificity was further supported by the lack of Myosin Vc interaction with constitutively active human Rab17 (Fig. 1D).
FIGURE 1.

Interaction of Myosin Vc with Rab proteins involved in melanosome biogenesis. Yeast two-hybrid assays with the full-tail of human Myosin Va (aa 908–1855), Myosin Vb (aa 905–1848), and Myosin Vc (aa 898–1742) binding to human Rab GTPases associated with melanosome biogenesis in the GTP-locked, constitutively active state (Q-L mutations) is shown. Yeast cells (AH109) were co-transformed with expression plasmids containing GAL4 activation (PGAD424) and DNA binding (PGBT9) domains fused in-frame to the indicated Myosin V fragments and Rab proteins, respectively. Double transformants were first selected on minimal media lacking leucine and tryptophan and containing histidine (+his) and then spotted onto plates containing the same medium (as control) or selective medium lacking histidine and containing various concentrations of 3-amino-1,2,4-triazole (3-AT). Cell growth in panels lacking histidine indicates physical interaction between Myosin V and Rabs. Growth in higher concentrations of 3-amino-1,2,4-triazole demonstrates stronger binding. A, Myosin Vc binds to constitutively active Rab7a, Rab8a, Rab32, and Rab38 but not with Rab11a or Rab27a. B, specificity of Rab interactions with class V Myosins. Rab32 and Rab38 bind only to Myosin Vc, but Rab7a and Rab8a bind other class V myosins. C, mapping binding of Rab proteins to Myosin Vc tail. Regions in the tail of Myosin Vc homologous to Myosin Va exons D, E, and F were removed from the full-length tail and tested in yeast two-hybrid assays with Rab7a, Rab8a, Rab32, and Rab38. The binding site for Rab32 and Rab38 includes exon F, Rab8a and Rab38 require exon E, and Rab8 is the only one whose binding site includes exon D. D, yeast two-hybrid assays with the full-tail (aa 898–1742) or the globular (Glob.) tail (aa 1335–1742) of human Myosin Vc binding to human Rab GTPases Rab8a, Rab17, Rab32, and Rab38 in the GTP-locked, constitutively active state (Gln-to-Leu mutations). E, Rab7a independently binds to the coiled-coil (aa 898–1350) and globular tail (aa 1335–1742) domains of Myosin Vc.
In melanocytes, Rab7a localizes to immature stage I-III melanosomes and Rab8a to more mature, pigmented melanosomes and may be involved in melanosome biogenesis (66, 67). Therefore, we tested for interaction between the constitutively active, GTP-locked mutant version of these Rabs and Myosin Vc as well as Myosin Va and Myosin Vb (Fig. 1, A and B). Constitutively active Rab7a and both wild-type and constitutively active Rab8a interacted with Myosin Vc full tail (Fig. 1A). Rab7a had the least specificity in binding and was found to bind to all three human class V Myosins (Fig. 1B). Rab8a interacted with the full tails of Myosin Vb and Myosin Vc, but not with Myosin Va (Fig. 1B), consistent with a previous report (53). In addition, Rab11a bound only the tail of Myosin Vb, as previously reported (53), but at variance with another study reporting interaction with Myosin Va and Myosin Vb (68), and Rab27a showed no direct binding to any of the Myosins tested, consistent with the known necessity of melanophilin for Rab27a-Myosin Va interaction (43, 47). Therefore, constitutively active Rab32 and Rab38 showed the most specific binding to Myosin Vc, suggesting that Myosin Vc may function as an effector protein for these Rabs. Myosin Vc could function as an effector of additional Rabs (Rab7a and Rab8a) in melanosome biogenesis.
Rabs are known to bind the tail of class V myosins at the globular region or the coiled-coil region (Fig. 1C) (69, 70). The Myosin Vc globular tail domain alone did not interact with Rab32, Rab38, or Rab8 (Fig. 1D). Tissue-specific alternative splicing of Myosin V-tail exons in the coiled-coil domain is prevalent and important for regulating specific Rab interactions. For example, in melanocyte cells Myosin Va exists as an alternatively spliced isoform containing exon F, which is required for localization to melanosomes. Furthermore, the interaction between Rab8a and Myosin Vc in HeLa cells appears to depend on exon D (43, 69). Consequently, homologous exon sequences in the tail of Myosin Vc (naming is based on alignment with Myosin Va and Myosin Vb (54, 71)) were removed from the full-length tail and tested in yeast two-hybrid assays with Rab7a, Rab8a, Rab32, and Rab38 (Fig. 1C). Rab32 and Rab38 were found to have partially overlapping binding sites requiring exon F (Fig. 1C). Rab8a and Rab38 were also found to have partially overlapping binding sites requiring exon E (Fig. 1C). Of the Rabs tested, Rab8a was the only one requiring exon D for binding (Fig. 1C). Binding of Rab7a could not be mapped to any of these exons, but Rab7a turned out to be capable of binding to both the coiled-coil and globular domains in the tail of Myosin Vc (Fig. 1, C and E). It is not known if Myosin Vc is alternatively spliced in other tissues, but we found that in MNT-1 melanocytes the only isoform of Myosin Vc detected contained all of the exons D, E, and F. Therefore, Myosin Vc may be able to bind simultaneously to specific Rabs with non-overlapping binding sites (Fig. 1, C and E).
Myosin Vc Binds to a Canonical Effector Binding Surface on Rab32 and Rab38
The interaction between Rabs and effector proteins depends on exposed hydrophobic residues in the Rab switch I and switch II regions and an adjacent hydrophobic triad (71, 72). Because the switch conformation serves as a readout for the nucleotide status, Rab effector proteins must bind to this region to discriminate between active (GTP-bound) and inactive (GDP-bound) forms of the GTPase. A previously known effector of Rab32 and Rab38, Varp, interacts with the switch II region of these Rabs (73). Site-directed mutagenesis was performed for amino acids in the switch I, switch II, and hydrophobic triad regions, and a sub-family specific loop, shared by Rab32, Rab38, and Rab9a was deleted (72). Fig. 2A shows a model of the predicted Rab32 three-dimensional structure highlighting residues subjected to mutagenesis (purple and yellow residues); the homologous residues were also mutated in Rab38. The ability of both sets of mutants to interact with the tail of Myosin Vc was tested using the yeast two-hybrid system (Fig. 2). Mutation of the tyrosine residue present in the switch II region of Rab32 (Y95A) and Rab38 (Y79A) disrupted the interaction of both Rabs with Myosin Vc (Fig. 2, A and B). The adjacent residue in the switch II region (Val-94 in Rab32 and Val-78 in Rab38) was shown to participate in the binding of Rab32 and Rab38 to Varp (Fig. 2A, green residue) (73). We, therefore, mutated the valine residue in both Rab32 and Rab38 and tested for its effect on the interaction with Myosin Vc (Fig. 2C). Mutation of the valine residue individually did not appear to decrease binding of Rab32 or Rab38 to Myosin Vc, but the combined mutation of adjacent valine and tyrosine residues resulted in a loss of interaction with Myosin Vc that was greater than the effect of the tyrosine residue alone (Fig. 2C). From these experiments we conclude that Myosin Vc and Varp utilize an overlapping binding site in the switch II region of Rab32 and Rab38.
FIGURE 2.

Mapping Myosin Vc binding sites on Rab32 and Rab38. A, homology modeling of Rab32 onto the crystal structure of Rab4a (PDB 2BME) with the residues mutated to alanine in B and C shown in stick representation. Point mutations causing loss of Myosin Vc binding are highlighted in yellow (tyrosine-95) and green (valine-94), and those with no effect in pink. Deletion of the leucine 131-glycine 134 loop (LPNG, pink) also did not have an effect. Rab32 residue valine 94 (highlighted in green) is required for the binding of the Rab32 and Rab38 effector Varp. Homologous mutations were introduced in Rab38. B, yeast two-hybrid assay of constitutively active Rab32 (Rab32Q85L) and Rab38 (Rab38Q69L) binding to Myosin Vc. Residues on the surface of Rab32 and Rab38 were mutated to alanine as indicated. Mutation of Rab32Q85L-Y95A and Rab38Q69L-Y79A specifically disrupt interaction with Myosin Vc tail. C, yeast two-hybrid assays with mutations that disrupt interactions with Rab32 and Rab38 effectors Varp and Myosin Vc. A longer growth period was used to detect subtle defects in binding of Rab single and double mutants.
Myosin Vc Deficiency Causes the Accumulation of Pigmented Melanosomes
The interaction between Myosin Vc and Rab32 and Rab38 suggested a role for Myosin Vc in melanosome biogenesis or function. To test this possibility, siRNA-mediated knockdown against Myosin Vc was performed in MNT-1 melanocytes. Importantly, although significant Myosin Vc deficiency was achieved, related proteins Myosin Va or Myosin Vb were not knocked down (Fig. 3A). Instead, knockdown of Myosin Vc resulted in an increase in the abundance of both Myosin Va and Myosin Vb (Fig. 3A). It was found that deficiency of Myosin Vc caused a significant increase in the amount of melanin accumulated inside MNT-1 melanocytes (Fig. 3B). Transmission-electron micrographs of control and Myosin Vc-deficient cells demonstrated that the increase in melanin was due to an increase in the abundance of pigmented stage III and IV melanosomes (Fig. 3, D and E). Quantification of the number of non-pigmented, stage II melanosomes identified by the presence of striations and Pmel17 immunogold labeling demonstrated no difference between control and Myosin Vc-deficient cells (Fig. 3, F and G). These results suggest Myosin Vc deficiency may cause a defect in melanosome secretion, a misregulation in melanosome biogenesis, or both. To test for a possible secretion defect, the melanin content in the media of cultured MNT-1 cells was quantified. Consistent with a secretion defect, the culture media of Myosin Vc-deficient cells contained less melanin than control cells (Fig. 3H).
FIGURE 3.

Myosin Vc knockdown causes accumulation of pigmented melanosomes and a defect in melanin secretion. A, immunoblotting analysis of total cell extracts from MNT-1 cells subjected to control or Myosin Vc siRNA knockdown. Myosin Vc is highly depleted upon siRNA treatment, but Myosin Va and Myosin Vb are enriched in extracts from Myosin Vc-deficient cells. B, melanin was extracted from control MNT-1 cells or cells deficient for Myosin Vc and quantified by a spectrophotometric method. Error bars represent S.E. *, p < 0.05, n = 4 experiments. C, melanin was extracted from MNT-1 cells overexpressing GFP (control) or the following GFP fusion proteins: full-length Myosin Vc (Myo Vc-GFP), the full tail of Myosin Vc (aa 898–1742) (Myo Vc-tail-GFP), the full tail of Myosin Vc carrying a deletion of exon F (Myo Vc-tailΔF-GFP), and full-length Myosin Vb (Myo Vb-GFP). Melanin was quantified by a spectrophotometric method. Error bars represent S.E. *, p < 0.05, n = 3 experiments. D and E, control MNT-1 cells or cells deficient for Myosin Vc were subjected to high pressure freezing in 15% dextran (9–11 kDa) in culture media, freeze-substitution in 0.25% glutaraldehyde, 0.1% uranyl acetate, and embedding in Lowicryl HM20 resin and processed for thin-section transmission electron microscopy. Myosin Vc knockdown resulted in accumulation of pigmented (stage III and IV) melanosomes in MNT-1 cells. Error bars represent S.E. *, p < 0.0001, n > 10 cells. Bars, 1 μm. F and G, immunoelectron microscopy analysis of control MNT-1 cells or cells deficient for Myosin Vc labeled with the HMB45 antibody to Pmel17 (arrows, 12-nm colloidal gold). Pigmented melanosomes comprise melanosomes at stages III and IV, and Striated melanosomes reflects any organelle labeled with Pmel17 that has visible striations. Error bars represent S.E. *, p < 0.0001, n > 10 cells. Bars, 200 nm. H, quantification of melanin secreted to the culture media by control MNT-1 cells or cells deficient for Myosin Vc. Error bars represent S.E. *, p < 0.05, n = 5 experiments. I, quantification of melanin secreted to the culture media by MNT-1 cells overexpressing GFP (Control) or the following GFP fusion proteins: full-length Myosin Vc (Myo Vc-GFP), the full tail of Myosin Vc (aa 898–1742) (Myo Vc-tail-GFP), the full tail of Myosin Vc carrying a deletion of exon F (Myo Vc-tailΔF-GFP), and full-length Myosin Vb (Myo Vb-GFP). Error bars represent S.E. *, p < 0.05, n = 4 experiments.
An alternative approach to interfere with the function of class V Myosins has been the overexpression of their tail domain. This dominant negative strategy has been successfully used to investigate Myosin Va, Myosin Vb, and Myosin Vc (54). We thus utilized the same Myosin Vc tail-GFP construct previously used as a dominant negative (54), transfected MNT-1 melanocytes, and determined the amount of melanin accumulated inside the cells (Fig. 3C). For comparison, parallel experiments were carried out with MNT-1 cells overexpressing GFP (control), full-length Myosin Vc-GFP, full-length Myosin Vb-GFP, and Myosin Vc tail-GFP harboring a deletion of exon F, the binding site for Rab32 and Rab38. Overexpression of Myosin Vc-tail-GFP caused a significant increase in the melanin accumulated by MNT-1 cells, whereas full-length Myosin Vc or Myosin Vb did not (Fig. 3C). This result is consistent with the Myosin Vc siRNA data described above (Fig. 3B) and further shows that the phenotype is not due to up-regulation of Myosin Vb. Importantly, the increase in intracellular melanin elicited by overexpression of the Myosin Vc tail requires exon F, indicating that this phenotype depends on the Myosin Vc interaction with Rab32 and Rab38 (Fig. 3C). The dominant negative strategy was also utilized as an alternative approach to test for melanin secretion defect (Fig. 3I). The amount of melanin in the culture media of MNT-1 cells overexpressing Myosin Vc-tail-GFP was lower than cells overexpressing GFP (control), Myosin Vc-GFP, and Myosin Vb-GFP. This result supports the secretion defect observed upon Myosin Vc depletion by siRNA described above (Fig. 3H) and indicates the phenotype is not due to up-regulation of Myosin Vb. Furthermore, the decrease in secreted melanin caused by overexpression of the Myosin Vc tail depends on exon F, demonstrating that interference of Myosin Vc function requires its interaction with Rab32 and Rab38 (Fig. 3I).
Interestingly, the phenotype elicited by Myosin Vc deficiency is different from that of Myosin Va deficiency, which results in perinuclear clustering of melanosomes without significantly affecting melanosome number (44, 51). Instead, Myosin Vc deficient-MNT-1 cells showed no obvious defects in melanosome localization but significantly increased the number of pigmented melanosomes (Fig. 3, D and E). The distinct phenotypes of Myosin Va and Myosin Vc deficiency suggest that Myosin Vc functions independently from Myosin Va in melanocytes. Moreover, although Myosin Va localizes tightly to mature melanosomes (45), subcellular fractionation of MNT-1 extracts using sucrose density gradients and confocal fluorescence microscopy studies showed the bulk of Myosin Vc does not localize to melanosomes (Fig. 4, 5, and 6). Based on the fractionation patterns of melanosome resident proteins such as tyrosinase, Tyrp1, Tyrp2, Pmel17, and Myosin Va, melanosomes are concentrated in fractions 5–10 of the sucrose density gradient, but Myosin Vc is largely found in fractions 1–3 and to a lesser extent in fractions 4–6 (Fig. 4). Thus a small proportion of Myosin Vc is associated with melanosomes at steady state or the association is transient and does not withstand the lengthy fractionation procedure. Myosin Vc largely co-fractionates with cytosolic proteins, vesicles, and light organelles that are obtained in the first fractions (64). Notice that in addition to a cohort of Rab32 and Rab38 (and other Rabs) that co-fractionate with melanosomes (fractions 5–10), a proportion of these Rabs is obtained in lighter fractions, consistent with their known presence in small transport vesicles and early endosomal domains (24, 25, 64). Consistently, live cell confocal fluorescence microscopy of MNT-1 cells expressing full-length Myosin Vc-GFP and tyrosinase-Cherry showed a limited number of structures where both proteins colocalized over time (Fig. 5A). The overall colocalization between Myosin Vc-GFP and tyrosinase-Cherry was 21 ± 4% (Fig. 5A). Furthermore, the average diameter of the Myosin Vc-GFP structures (371 ± 139 nm; 682 structures from 8 cells) was smaller than the average diameter of tyrosinase-Cherry structures (613 ± 245 nm; 1198 structures from 8 cells; significance at p < 0.0001). At steady state tyrosinase mostly labels mature melanosomes that are 400–500 nm in size as determined by electron microscopy (1, 4). The smaller Myosin Vc-GFP structures are more consistent with vesicles and early endosome organelle domains. Similar results were obtained with MNT-1 cells expressing Myosin Vc-GFP and Tyrp1-Cherry (Fig. 5B). Additionally, a low degree of colocalization was observed between endogenous melanosome markers Pmel17 (25 ± 3%), Tyrp1 (15 ± 2%), or Rab27a (22 ± 2%) and endogenous Myosin Vc by confocal fluorescence microscopy of fixed and immunostained MNT-1 cells (Fig. 6). Although the quality of the antibody staining on fixed samples is not as good as the live cell imaging of overexpressed GFP/Cherry-tagged proteins, both approaches suggest a low level of Myosin Vc localization to melanosomes at steady state. Together, these results do not support a direct role of Myosin Vc in secretion of mature melanosomes. Rather, these results suggest the accumulation of mature melanosomes is an indirect effect of Myosin Vc deficiency in melanocytes. For example, during melanosome biogenesis Myosin Vc could be involved in vesicular transport of a cargo that is then required for melanosome secretion.
FIGURE 4.
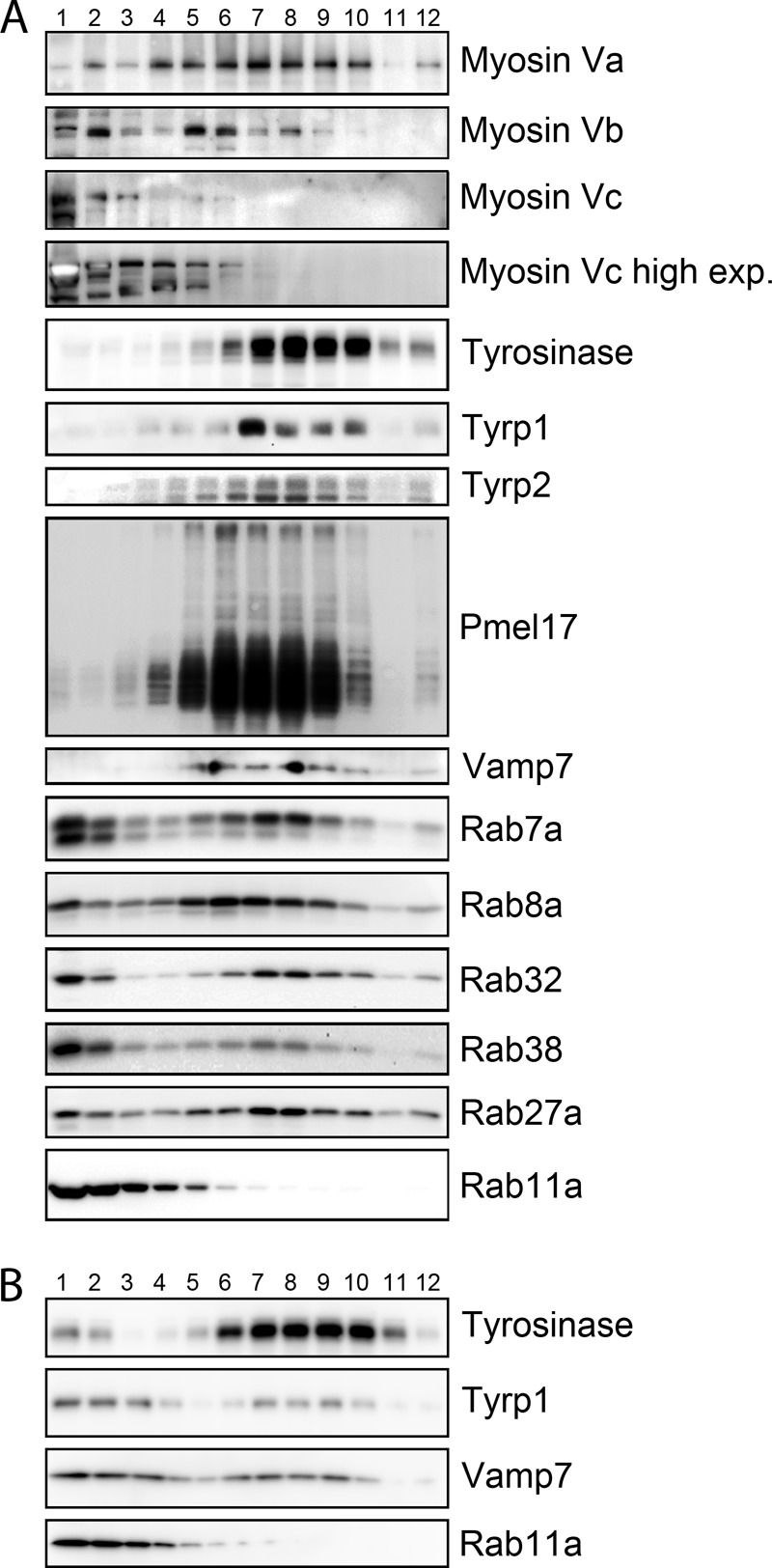
Myosin Vc is not abundant in melanosome-enriched subcellular fractions, but Myosin Vc knockdown alters the distribution of a subset of melanosomal proteins. Immunoblotting analysis of fractions obtained from control (A) and Myosin Vc-deficient MNT-1 post nuclear supernatants (B) subjected to subcellular fractionation with a 20–55% sucrose gradient. Fraction 1 corresponds to 20% sucrose and fraction 12–55% to sucrose. In control cells, melanosome markers tyrosinase, Tyrp1, Tyrp2, and Pme17 are enriched in fractions 5–10. A significant proportion of melanosome-associated Rabs, Rab7a, Rab8a, Rab32, Rab38, and Rab27a is also recovered in fractions 5–10. Myosin Va is highly enriched in fractions 5–10, consistent with its known localization to mature melanosomes. Myosin Vc is present primarily in fractions 1–5 and most enriched in fractions 1 and 2, similar to Rab11a. Notice the significant change in Tyrp1 and Vamp7 fractionation profile in Myosin Vc-deficient cells (B) compared with control cells (A). In contrast, tyrosinase and Rab11a are largely unchanged.
FIGURE 5.

Myosin Vc shows limited colocalization with the melanin-synthesizing enzymes tyrosinase and Tyrp1. Shown are spinning-disk confocal fluorescence microscopy images of a live MNT-1 cell expressing full-length Myosin Vc-GFP (5c) and the melanosome markers tyrosinase-cherry (A) or Tyrp1-cherry (T; B). A, the main picture shows the first frame of a movie, and the inset on the lower left corner shows examples of structures where the two proteins colocalize (yellow arrows). The same region of the cell is shown in the small panels on the right as a time series depicting five frames of the movie. Images were acquired at a rate of 1 frame/s. B, the picture corresponds to the first frame of a movie, and the panels on the upper right corner show examples of structures were the two proteins colocalize (yellow arrows). The same region of the cell is shown in the panels on the lower right corner as a time series depicting six frames of the movie. Images were acquired at a rate of 1 frame/s.
FIGURE 6.

Myosin Vc and Myosin Vb display limited colocalization with melanosome markers. Confocal fluorescent microscopy images of fixed/permeabilized MNT-1 cells immunolabeled for endogenous Myosin Vc (Alexa- 488) (A, C, and E) or Myosin Vb (Alexa-488) (B, D, and F) and endogenous melanosome markers Pmel17, Tyrp1, and Rab27a (Alexa-546). Both Myosin Vc (A, C, and E) and Myosin Vb (B, D, and F) show partial colocalization with early, Pmel17-labeled, melanosomes (A and B) and with more mature, pigmented melanosomes with Tyrp1 and Rab27a labeling (C–F). G, quantification of colocalization as determined with Slidebook software. Error bars represent S.E., n ≥ 10 cells.
Myosin Vc Functions in Cargo Trafficking to Melanosomes
In melanocytes deficient in AP-1, AP-3, BLOC-1, or BLOC-2, newly synthesized integral membrane protein cargo such as tyrosinase and Tyrp1 are not properly sorted and accumulate in early endosomal domains (10–13, 74). This cargo can subsequently a leak into the recycling pathway to the plasma membrane or enter into the degradative multivesicular body pathway (see Fig. 10). Thus, as a consequence of disrupting the normal transport from early endosomal domains to melanosomes, cargo proteins can exhibit increased traffic via the plasma membrane and/or are subjected to degradation. Melanocytes isolated from AP-3-, BLOC-1-, or BLOC-2-deficient mice showed both phenotypes (recycling through the plasma membrane and degradation) when Tyrp1 was analyzed (12, 13). Similarly, MNT-1 cells deficient for AP-1 accumulate Tyrp1 in early endosomes (10). MNT-1 cells deficient in Rab32 and Rab38 showed a modest but epistatic increase in recycling of Tyrp1 and degradation of tyrosinase and a drastic Rab32-specific loss of Tyrp2 (24). Here, we sought to determine if Myosin Vc deficiency also produces similar cargo trafficking phenotypes.
FIGURE 10.

Model for Myosin Vc-dependent and Myosin Vc-independent transport pathways to maturing melanosomes. Myosin Vc interacts with components of the trafficking machinery such as Rab32 and Rab38 at early/recycling endosome membrane domains where a subset of cargoes including Tyrp1 and Vamp7 (and possibly Tyrp2) is concentrated and packaged into vesicles for transport to maturing melanosomes. Myosin Vc does not participate, however, in pathways that deliver other melanosomal cargo such as tyrosinase. Unlike adaptor proteins (AP-1, AP-3) and clathrin that typically dissociate from the vesicle, Myosin Vc may remain bound to mediate vesicle transport and/or tethering to melanosomes. Myosin Vc deficiency elicits accumulation of cargo (such as Tyrp1, Vamp7, and possibly Tyrp2) in early/recycling endosomes that eventually leak into the recycling pathway to the plasma membrane or the late endosome/multivesicular body (MVB) degradative pathway. A component of the melanosome secretion machinery such as Vamp7 is not adequately incorporated into melanosomes in Myosin Vc-deficient cells causing accumulation of pigmented melanosomes within melanocytes.
First, immunoblotting analysis of total extracts from Myosin Vc-deficient MNT-1 cells showed increased levels of tyrosinase, Tyrp1, and Pmel17 and drastically decreased levels of Tyrp2 compared with control cells (Fig. 7A). The increased level of various melanosome resident proteins in Myosin Vc-deficient cells likely reflects the substantial accumulation of pigmented melanosomes. Hence, based on this assay it is unclear if a trafficking defect exists for cargo such as tyrosinase. However, the strong Tyrp2 phenotype resembles the result previously reported for Rab32-deficient MNT-1 cells and supports a role for Myosin Vc in transport to melanosomes.
Second, cell surface expression and recycling of Tyrp1 was determined with MNT-1 cells deficient in Myosin Vc and control cells. Live cells were incubated with antibodies to the luminal domain of Tyrp1 for 20 min and then fixed/permeabilized and processed for fluorescence microscopy (Fig. 7B). The total fluorescence intensity signal per cell corresponds to the sum of surface and internalized antibody and thus represents Tyrp1 undergoing recycling. This value is then corrected for the overall amount of Tyrp1 as determined by immunoblotting of total cell extracts to account for differences in expression levels. Myosin Vc deficiency produced a significant increase in Tyrp1 recycling suggesting Myosin Vc is involved in transport from early endosomes to maturing melanosomes. To compare the relative severity of the Tyrp1 recycling phenotype, similar experiments were carried out in parallel with MNT-1 cells deficient in AP-3 or simultaneously deficient in both Rab32 and Rab38 (Fig. 7B). The Tyrp1 recycling phenotype of Myosin Vc-deficient cells was more severe than the one observed in Rab32/Rab38 double-deficient cells but less severe than in AP-3-deficient cells assayed under the same conditions (Fig. 7B). These differences may be secondary to different levels of depletion obtained by the corresponding siRNA treatments or represent true differences in the dependence of Tyrp1 traffic with each of these proteins. Regardless, the data support a model in which Myosin Vc functions in transport from early/recycling endosomes to maturing melanosomes (see Fig. 10). We also used the dominant negative strategy of overexpressing the Myosin Vc tail as another approach to test for Tyrp1 mistrafficking (Fig. 7C). Indeed, MNT-1 cells overexpressing Myosin Vc-tail-GFP displayed significant Tyrp1 recycling (Fig. 7C). However, overexpression of Myosin Vc-GFP, Myosin Vb-GFP, or Myosin Vc-tail-GFP carrying a deletion of exon F did not show increased Tyrp1 recycling relative to control (GFP) (Fig. 7C). The lack of a recycling phenotype by overexpression of the later construct indicates Myosin Vc function in Tyrp1 transport depends on its ability to interact with Rab32 and Rab38.
Third, we investigated the SNARE protein Vamp7, considered to be both cargo and machinery of the pathway to melanosomes and lysosomes (73, 75, 76). In MNT-1 cells, normal steady state levels of Vamp7 depend on the BLOC-1 pathway, and blockage of this pathway results in increased surface recycling of Vamp7 (76). Furthermore, Vamp7 interacts physically with AP-3 and with the Rab32 and Rab38 effector Varp, and Vamp7 knockdown causes mistrafficking of Tyrp1 in melanocytes (73, 75, 77). Immunoblotting analysis showed increased steady state levels of Vamp7 protein in whole-cell extracts from Myosin Vc-deficient MNT-1 cells compared with control cells (Fig. 7A). This result suggests a significant proportion of Vamp7 may localize to pigmented melanosomes, which are highly abundant in Myosin Vc-deficient cells. Furthermore, Vamp7 distribution in sucrose gradient subcellular fractionation assays (fractions 5–10) correlated tightly with the melanosome resident proteins Pmel17, tyrosinase, Tyrp1, and Tyrp2 (Fig. 4). Indeed, localization of Vamp7 by immunogold electron microscopy of control MNT-1 cells revealed that 57 ± 6% of the label was associated with the limiting membrane of pigmented melanosomes, whereas 40 ± 5% of the label was associated with other intracellular membranes (69 gold particles, 5 cells) (Fig. 7D). Based on the lack of staining of the nucleus and mitochondria the nonspecific labeling was considered negligible, and only one gold particle was detected at the plasma membrane. Despite the increased number of pigmented melanosomes in Myosin Vc-deficient cells, only 29 ± 7% of the gold particles was associated with the limiting membrane of pigmented melanosomes (245 gold particles, 7 cells; p < 0.001, Myosin Vc knockdown versus siRNA control). Moreover, gold particles were abundant at the plasma membrane and vacuolar compartments with morphological characteristics of endosomes (electron-lucent interior with few or no intraluminal vesicles), likely reflecting increased recycling (Fig. 7D). From these experiments we conclude that in melanocytes at steady state a significant proportion of Vamp7 localizes to pigmented melanosomes and that its traffic to the melanosome depends on Myosin Vc (see Fig. 10).
Fourth, to test for Myosin Vc function in trafficking of Tyrp1 and Vamp7 to the melanosome with an independent approach, we fractionated extracts of MNT-1 cells deficient in Myosin Vc using sucrose density gradients (Fig. 4B). Although a cohort of Tyrp1 and Vamp7 was detected in fractions 5–10 corresponding to melanosomes, a significant proportion was found in lighter membrane fractions demonstrating significant mislocalization. Interestingly, the bulk of tyrosinase fractionated as in wild-type cells thus showing minimal mistargeting caused by Myosin Vc deficiency (Fig. 4B). Defects in the trafficking pathways used by Tyrp1 and Vamp7, but not tyrosinase, suggest that Myosin Vc functions in trafficking of a subset of cargoes from early endosomal domains to maturing melanosomes (see Fig. 10). In further support of this idea we found that a proportion of Myosin Vc-GFP localizes to early/recycling compartments in MNT-1 cells as labeled with internalized transferrin (Fig. 8). For this experiment, MNT-1 cells transfected with Myosin-Vc (and Myosin Vb) were incubated for 20 min with transferrin-Alexa 647, washed, and immediately observed by confocal fluorescence microscopy. The overall colocalization between Myosin Vc-GFP and transferrin-Alexa 647 was 28 ± 7%. In parallel experiments the well known component of the recycling pathway, Myosin Vb, showed 62 ± 9% colocalization with transferrin-Alexa 647. Furthermore, Rab32 and Rab38 have been shown to localize to early endosomal domains, transport vesicles, and melanosomes (see Fig. 10). We found that Myosin Vc-tail-GFP colocalized with Rab32-Cherry and Rab38-Cherry to a significant degree in transfected MNT-1 cells (48 ± 10% and 51 ± 8%, respectively) (Fig. 9). However, Myosin Vc-tailΔF-GFP harboring a deletion of exon F only marginally colocalized with Rab32 and Rab38 (17 ± 6% and 14 ± 5%, respectively) and showed more cytosolic background staining (Fig. 9). From these experiments we conclude that Myosin Vc functions in transport of a subset of melanosomal integral membrane proteins including Tyrp1 and Vamp7 from early endosomal domains to maturing melanosomes. Fig. 10 shows a model summarizing our interpretation of these results in the context of previous knowledge of the transport routes to melanosomes.
FIGURE 8.
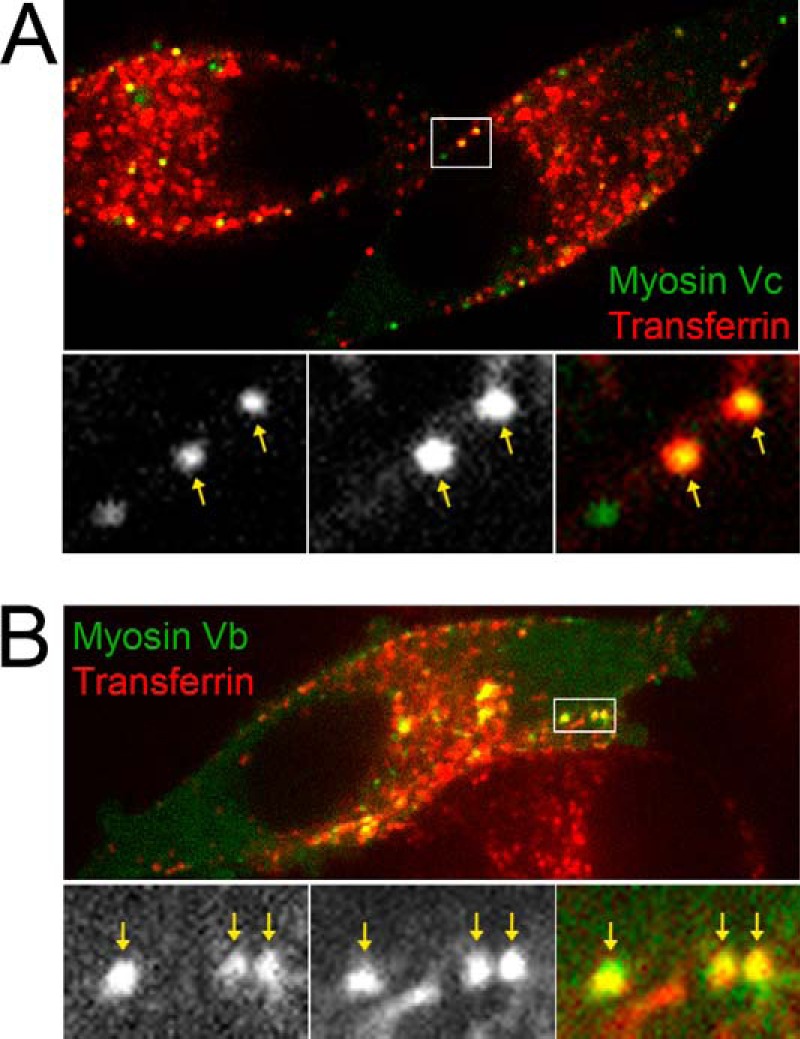
Myosin Vc-GFP shows partial colocalization with internalized transferrin-Alexa-647. Shown are spinning-disk confocal fluorescence microscopy images of live MNT-1 cells expressing full-length Myosin Vc-GFP (A) or full-length Myosin Vb-GFP (B) after 20 min of incubation with transferrin-Alexa-647 at 37 °C. Boxed areas are shown in the magnified bottom panels and contain examples of structures were the two proteins colocalize (yellow arrows).
FIGURE 9.

Myosin Vc-tail-GFP shows partial colocalization with Rab32-Cherry and Rab38-Cherry. Spinning-disk confocal fluorescence microscopy images of live MNT-1 cells expressing the full tail of Myosin Vc (aa 898–1742) tagged with GFP (Myosin Vc tail) and Rab32-Cherry (Rab32) (A), the full tail of Myosin Vc carrying a deletion of exon F tagged with GFP (Myosin Vc tail ΔF) and Rab32-Cherry (Rab32) (B), the full tail of Myosin Vc (aa 898–1742) tagged with GFP (Myosin Vc tail) and Rab38-Cherry (Rab38) (C), or the full tail of Myosin Vc carrying a deletion of exon F tagged with GFP (Myosin Vc tail ΔF) and Rab38-Cherry (D). Boxed areas in A and C are shown in the magnified panels and contain examples of structures were the two proteins colocalize (yellow arrows).
DISCUSSION
Several studies have demonstrated that the biogenesis of LROs in general and melanosomes in particular depends on a combination of ubiquitous and cell type-specific trafficking machinery (1, 14, 21–25, 27, 78). AP-1, AP-3, and the BLOCs are well known examples of the ubiquitous machinery, and Rab32 and Rab38 are key cell type-specific components. This machinery defines multiple vesicular transport pathways from specialized tubules and buds of the early endosomal system to melanosomes for delivery of integral membrane proteins required for organelle maturation and function (Fig. 10) (1, 27). In this study we show that Myosin Vc is a new component of the ubiquitous machinery that interacts physically and functionally with cell type-specific Rab32 and Rab38 to mediate transport of a subset of cargoes to maturing melanosomes and ultimately facilitate organelle secretion.
We show that Rab32 and Rab38 interact with Myosin Vc specifically when they are in the GTP-bound conformation and do not bind the related Myosin Va and Myosin Vb. Our results show that the interaction depends on the canonical switch II surface region used by Rabs to interact with effector proteins (71). These findings indicate Myosin Vc is likely one of the effectors of Rab32 and Rab38. The interaction also depends on regions of the coiled-coil domain of the Myosin Vc tail that have been shown to be important for the function of other class V Myosins (43, 69). Notably, Rab32 and Rab38 interaction with Myosin Vc depends on its exon F region similar to the melanocyte splice form of Myosin Va that depends on exon F to bind the Rab27/melanophilin complex and localize to melanosomes (43). In support of the idea that the interaction between Rab32 and Rab38 with Myosin Vc is biologically relevant and that Myosin Vc works in melanosome biogenesis, we show that a deficiency of Myosin Vc in melanocytes causes similar mistrafficking of a subset of melanosomal proteins, Tyrp1 and Tyrp2, as a deficiency of Rab32 and Rab38 (24). First, knockdown of Myosin Vc causes increased recycling of Tyrp1 through the plasma membrane, a defect previously reported for Rab32- and Rab38-deficient cells and reproduced here. Second, Myosin Vc deficiency causes reduced overall Tyrp2 levels paralleling the results previously obtained for Rab32-deficient melanocytes. Third, increased accumulation of intracellular melanin, decreased secretion, and abnormal Tyrp1 recycling phenotypes occurred only with the full Myosin Vc tail construct but not with the same Myosin Vc fragment lacking the binding site for Rab32 and Rab38, exon F. Finally, Myosin Vc-tail-GFP colocalization with Rab32 and Rab38 was dependent on the Myosin Vc region encoded by exon F. These results imply that at least some of Myosin Vc functions depend on Rab32 and Rab38. We also show that in control melanocytes a significant proportion of Vamp7 localizes to pigmented, stage III and IV melanosomes, but in Myosin Vc-deficient cells Vamp7 localization is abnormal and consistent with increased recycling. This is in line with previous reports showing Vamp7 trafficking phenotypes in non-specialized cells from AP-3 knock-out mice and in MNT-1 cells with a defective BLOC-1 pathway (76). However, it is not clear if Rab32 and Rab38 are needed for Vamp7 transport in melanocytes. Despite the abnormal traffic of some cargoes to melanosomes, Myosin Vc-deficient cells produce pigmented melanosomes. This is also the case with melanocytes from mice and patients deficient for AP-3, Rab38, and other components of the biogenesis machinery as well as MNT-1 cells depleted for AP-1 (10, 21, 29). The existence of multiple parallel trafficking pathways that deliver cargo to maturing melanosomes allows for enough traffic to produce pigmented melanosomes even when some of the pathways are defective. However, cargoes display different levels of dependence on the various parallel pathways and tyrosinase and Tyrp1 show little overlap (1) (Fig. 10). Notably, the trafficking of tyrosinase is largely unaffected by Myosin Vc deficiency (Fig. 4). This result explains the production of pigmented melanosomes in Myosin Vc-deficient MNT-1 cells because tyrosinase is the key enzyme in melanin synthesis, whereas Tyrp1 and Tyrp2 serve modulatory roles (1, 3, 9). In contrast Rab32 and Rab38 (and AP-3) are involved in the transport of both tyrosinase and Trp1 (11, 25, 27, 31), suggesting that Rab32 and Rab38 serve both MyosinVc-dependent and Myosin Vc-independent functions in melanosome biogenesis. Conversely, Myosin Vc may have Rab32/Rab38-independent functions, thus causing only partially overlapping phenotypes when either the Rabs or Myosin Vc are deficient. For example the interaction between Myosin Vc with Rab7 and Rab8 may mediate some Myosin Vc functions that are Rab32/Rab38-independent. The trafficking route followed by Tyrp2 is not nearly as well understood as those of tyrosinase and Tyrp1. For example, it has not been shown if Tyrp2 also traffics through the early/recycling endosomal system like tyrosinase and Tyrp1. In the future it will be important to clarify the transport route for Tyrp2, which appears to differ from Tyrp1 and tyrosinase based on this work and published data (24, 27). Nevertheless, the drastic reduction of Tyrp2 steady state level caused by Myosin Vc deficiency (Fig. 7) is remarkably similar to that elicited by Rab32 deficiency (24, 27). In contrast, Tyrp2 is unaffected by Rab38 deficiency, suggesting a closer functional relationship between Myosin Vc and Rab32 than Rab38 (24).
These cargo trafficking data together with the physical interactions and colocalization results are most consistent with a model in which Myosin Vc functions in pathways from early/recycling endosome-associated tubules to maturing melanosomes (Fig. 10). Such a location fits well with the established model for transport of Tyrp1 from early endosomes to maturing melanosomes. This model also explains the data on Vamp7 transport to melanosomes and places Myosin Vc as acting from early/recycling endosomes with potential roles in vesicle scission, motility, and docking events. This model also provides a satisfactory explanation to the observed cargo phenotypes upon interfering with Myosin Vc function by siRNA or dominant negative approaches. Disruption of transport from early/recycling endosomes toward melanosomes causes accumulation of the cargo in early endosomes and leakage into the recycling pathway to the plasma membrane and/or into the degradative multivesicular body/lysosomal pathway. The model is compatible with only partial Myosin Vc colocalization with the early/recycling endosome marker, transferrin, and mature melanosome markers, tyrosinase and Tyrp1, as these would represent the start and end point of the Myosin Vc transport pathway (Fig. 10). The presence of Myosin Vc in transport vesicles moving from early/recycling endosomes to melanosomes would fit the small average size of structures decorated by Myosin Vc-GFP as well as the presence of endogenous Myosin Vc in fractions of low sucrose density.
Interestingly, Myosin Vc deficiency elicits abnormal accumulation of pigmented melanosomes within melanocytes. This is evidenced by the increase in total melanin, the number of pigmented melanosomes (but not of their unpigmented precursors), and the steady state levels of several melanosome resident proteins. The accumulation of pigmented melanosomes in Myosin Vc-deficient cells indicates Myosin Vc may function in melanosome secretion in addition to melanosome biogenesis. Accordingly, the amount of melanin secreted by Myosin Vc-deficient MNT-1 cells was lower than in control cells (Fig. 3). Importantly, Myosin Vc is not abundant on melanosomes, arguing against a direct function of Myosin Vc in secretion. Rather, the data suggest a role for Myosin Vc in the traffic of a melanosome component that may be required for proper secretion. Vamp7 could be such component as it was previously shown to mediate exocytosis of lysosomes and LROs in several cell types, and we found it to be abundant on mature melanosomes in control cells but mislocalized in Myosin Vc-deficient cells (80). In the skin, melanocytes transfer mature melanosomes to keratinocytes by a poorly understood mechanism. Several different possibilities have been proposed ranging from the exocytosis of naked melanin by melanocytes and subsequent endocytosis by neighboring keratinocytes to the shedding of melanosome-rich packages by melanocytes coupled to phagocytosis by adjacent keratinocytes (36–41). A role for Vamp7 (or other vSNAREs (vesicle SNAREs)) could be easily envisioned in a scenario where fusion of the melanosome membrane with another membrane, such as the melanocyte plasma membrane, is involved in the secretion or transfer mechanism. We thus reason that an indirect function on Myosin Vc in melanosome secretion is more likely but do not rule out the possibility of a direct role.
Several other Rab proteins have been localized to melanosomes or implicated in their biology. Rab11 was reported to participate in the exocytosis of melanosomes via the naked melanin mechanism (36). Rab17 was shown to operate in the formation of melanocyte filopodia as conduits for melanosome secretion (37). The lack of interaction of Myosin Vc with Rab11 or Rab17 suggests the function ascribed to these Rabs in melanosome secretion is independent of Myosin Vc. Another pair of relevant Rabs is that of Rab7a and Rab8a, which in melanocytes localize to immature and mature melanosomes, respectively (66, 67). In this context the interaction between these Rabs and Myosin Vc (Fig. 1) could be important for melanosome biogenesis. Therefore, in addition to tissue-specific Rab32 and Rab38, ubiquitous Rab7a and Rab8a could cooperate with Myosin Vc during melanosome biogenesis. Our mapping experiments indicate that Rab7a binds to regions of the Myosin Vc tail not overlapping with Rab8a, Rab32, and Rab38 and that Rab8a overlaps only with Rab38. Therefore, several of these Rabs could simultaneously bind Myosin Vc.
An interesting observation is that the levels of Myosin Va and Myosin Vb are higher in Myosin Vc-deficient MNT-1 melanocytes (Fig. 3A). As Myosin Va is found on pigmented melanosomes, the increase in Myosin Va observed in Myosin Vc-deficient cells may be caused by the increase in pigmented melanosomes. Alternatively, the higher Myosin Va level might reflect compensation for the loss of Myosin Vc function by up-regulation of Myosin Va. The latter possibility would suggest that some degree of redundancy exists in the functions of Myosin Vc with Myosin Va. However, Myosin Va deficiency results in perinuclear clustering of melanosomes without affecting melanosome number (44, 51). Instead, Myosin Vc-deficient MNT-1 cells show no obvious defects in melanosome localization but significantly increased the number of pigmented melanosomes. Furthermore, although Myosin Va does not operate in the biogenesis of melanosomes, our data revealed a function for Myosin Vc in transport of several cargoes to melanosomes. Thus, the distinct phenotypes observed for melanocytes deficient in Myosin Va and Myosin Vc suggest that Myosin Vc functions independently from Myosin Va in melanocytes. It is unclear if the higher level of Myosin Vb in cells deficient for Myosin Vc is due to Myosin Vb localization and function on mature melanosomes or if Myosin Vb is up-regulated to functionally compensate for Myosin Vc deficiency. Our results show only low levels of colocalization between Myosin Vb and melanosome markers Pmel17, Tyrp1, and Rab27 in fixed and immunostained MNT-1 cells (Fig. 6). Importantly, overexpression of Myosin Vb-GFP did not cause the increased melanin accumulation within MNT-1 cells, the decreased melanin secretion, or Trp1 recycling observed upon Myosin Vc depletion, thus ruling out up-regulation of Myosin Vb as a cause for these phenotypes (Figs. 3 and 7).
Interestingly, another effector of Rab32 and Rab38, Varp, was shown to participate in Tyrp1 traffic to melanosomes (73, 81). Furthermore, Varp binds to Vamp7 and traps it in a closed, fusogenically inactive conformation (75). Therefore, both Rab32 and Rab38 effectors, Myosin Vc and Varp, may function in the same trafficking pathway from early endosome tubules and buds to melanosomes. Myosin Vc and Varp bind to an overlapping region on the surface of Rab32 and Rab38, suggesting mutually exclusive binding and thus sequential function during transport to melanosomes (Fig. 2) (73). If Myosin Vc and Varp interact with Rab32 and Rab38 during discrete steps, it is not obvious which effector would act upstream. Myosin Vc could function during budding of a transport intermediate from the early endosomal system, perhaps upstream Varp. Alternatively, Myosin Vc could compete out Varp, thus releasing Vamp7 from its closed conformation, to foster tethering/fusion of a transport intermediate with the maturing melanosome. Interestingly, Myosin Va was shown to tether synaptic vesicles to the plasma membrane such that vesicles are poised for fusion upon rise in Ca2+ concentration (79, 82). Myosin Vc could have an analogous role in tethering transport vesicles to melanosomes. The link between Myosin Vc, Varp, and the Rabs in the biogenesis of melanosomes and other LROs warrants future investigation.
Acknowledgments
We thank Thomas Giddings (University of Colorado, Boulder, CO) for high pressure freezing and Andrea Ambrosio for invaluable help with figure preparation. Microscopes used in this work were supported in part by the Microscope Imaging Network core infrastructure grant from Colorado State University.
This work was supported, in whole or in part, by National Institutes of Health Grant HL106186 (to S. M. D.). This work was also supported by American Heart Association Award 09SDG2280525 (to S. M. D.).
- LRO
- lysosome-related organelle
- Tyrp1
- tyrosinase-related protein 1
- AP-1
- adaptor protein-1
- BLOC-1
- biogenesis of lysosome-related organelle complex-1
- aa
- amino acids.
REFERENCES
- 1. Raposo G., Marks M. S. (2007) Melanosomes–dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 8, 786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wasmeier C., Hume A. N., Bolasco G., Seabra M. C. (2008) Melanosomes at a glance. J. Cell Sci. 121, 3995–3999 [DOI] [PubMed] [Google Scholar]
- 3. Kondo T., Hearing V. J. (2011) Update on the regulation of mammalian melanocyte function and skin pigmentation. Expert. Rev. Dermatol. 6, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hurbain I., Geerts W. J., Boudier T., Marco S., Verkleij A. J., Marks M. S., Raposo G. (2008) Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc. Natl. Acad. Sci. U.S.A. 105, 19726–19731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berson J. F., Harper D. C., Tenza D., Raposo G., Marks M. S. (2001) Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 12, 3451–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berson J. F., Theos A. C., Harper D. C., Tenza D., Raposo G., Marks M. S. (2003) Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 161, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harper D. C., Theos A. C., Herman K. E., Tenza D., Raposo G., Marks M. S. (2008) Premelanosome amyloid-like fibrils are composed of only golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J. Biol. Chem. 283, 2307–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Watt B., van Niel G., Fowler D. M., Hurbain I., Luk K. C., Stayrook S. E., Lemmon M. A., Raposo G., Shorter J., Kelly J. W., Marks M. S. (2009) N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 284, 35543–35555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ito S., Wakamatsu K. (2008) Chemistry of mixed melanogenesis: pivotal roles of dopaquinone. Photochem. Photobiol. 84, 582–592 [DOI] [PubMed] [Google Scholar]
- 10. Delevoye C., Hurbain I., Tenza D., Sibarita J. B., Uzan-Gafsou S., Ohno H., Geerts W. J., Verkleij A. J., Salamero J., Marks M. S., Raposo G. (2009) AP-1 and KIF13A coordinate endosomal sorting and positioning during melanosome biogenesis. J. Cell Biol. 187, 247–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Theos A. C., Tenza D., Martina J. A., Hurbain I., Peden A. A., Sviderskaya E. V., Stewart A., Robinson M. S., Bennett D. C., Cutler D. F., Bonifacino J. S., Marks M. S., Raposo G. (2005) Functions of adaptor protein-3 (AP-3) and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol. Biol. Cell 16, 5356–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Pietro S. M., Falcón-Pérez J. M., Tenza D., Setty S. R., Marks M. S., Raposo G., Dell'Angelica E. C. (2006) BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol. Biol. Cell 17, 4027–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Setty S. R., Tenza D., Truschel S. T., Chou E., Sviderskaya E. V., Theos A. C., Lamoreux M. L., Di Pietro S. M., Starcevic M., Bennett D. C., Dell'Angelica E. C., Raposo G., Marks M. S. (2007) BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol. Biol. Cell 18, 768–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dell'Angelica E. C. (2004) The building BLOC(k)s of lysosomes and related organelles. Curr. Opin. Cell Biol. 16, 458–464 [DOI] [PubMed] [Google Scholar]
- 15. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 16. Dell'Angelica E. C., Klumperman J., Stoorvogel W., Bonifacino J. S. (1998) Association of the AP-3 adaptor complex with clathrin. Science 280, 431–434 [DOI] [PubMed] [Google Scholar]
- 17. Dell'Angelica E. C., Shotelersuk V., Aguilar R. C., Gahl W. A., Bonifacino J. S. (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol. Cell 3, 11–21 [DOI] [PubMed] [Google Scholar]
- 18. Peden A. A., Oorschot V., Hesser B. A., Austin C. D., Scheller R. H., Klumperman J. (2004) Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 164, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saftig P., Klumperman J. (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 [DOI] [PubMed] [Google Scholar]
- 20. Salazar G., Craige B., Styers M. L., Newell-Litwa K. A., Doucette M. M., Wainer B. H., Falcon-Perez J. M., Dell'Angelica E. C., Peden A. A., Werner E., Faundez V. (2006) BLOC-1 complex deficiency alters the targeting of adaptor protein complex-3 cargoes. Mol. Biol. Cell 17, 4014–4026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huizing M., Helip-Wooley A., Westbroek W., Gunay-Aygun M., Gahl W. A. (2008) Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu. Rev. Genomics Hum. Genet. 9, 359–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei M. L. (2006) Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 19, 19–42 [DOI] [PubMed] [Google Scholar]
- 23. Di Pietro S. M., Dell'Angelica E. C. (2005) The cell biology of Hermansky-Pudlak syndrome: recent advances. Traffic 6, 525–533 [DOI] [PubMed] [Google Scholar]
- 24. Bultema J. J., Ambrosio A. L., Burek C. L., Di Pietro S. M. (2012) BLOC-2, AP-3, and AP-1 proteins function in concert with Rab38 and Rab32 proteins to mediate protein trafficking to lysosome-related organelles. J. Biol. Chem. 287, 19550–19563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wasmeier C., Romao M., Plowright L., Bennett D. C., Raposo G., Seabra M. C. (2006) Rab38 and Rab32 control post-Golgi trafficking of melanogenic enzymes. J. Cell Biol. 175, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerondopoulos A., Langemeyer L., Liang J. R., Linford A., Barr F. A. (2012) BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr. Biol. 22, 2135–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bultema J. J., Di Pietro S. M. (2013) Cell type-specific Rab32 and Rab38 cooperate with the ubiquitous lysosome biogenesis machinery to synthesize specialized lysosome-related organelles. Small GTPases 4, 16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hutagalung A. H., Novick P. J. (2011) Role of rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91, 119–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loftus S. K., Larson D. M., Baxter L. L., Antonellis A., Chen Y., Wu X., Jiang Y., Bittner M., Hammer J. A., 3rd, Pavan W. J. (2002) Mutation of melanosome protein RAB38 in chocolate mice. Proc. Natl. Acad. Sci. U.S.A. 99, 4471–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohen-Solal K. A., Sood R., Marin Y., Crespo-Carbone S. M., Sinsimer D., Martino J. J., Robbins C., Makalowska I., Trent J., Chen S. (2003) Identification and characterization of mouse Rab32 by mRNA and protein expression analysis. Biochim. Biophys. Acta 1651, 68–75 [DOI] [PubMed] [Google Scholar]
- 31. Brooks B. P., Larson D. M., Chan C. C., Kjellstrom S., Smith R. S., Crawford M. A., Lamoreux L., Huizing M., Hess R., Jiao X., Hejtmancik J. F., Maminishkis A., John S. W., Bush R., Pavan W. J. (2007) Analysis of ocular hypopigmentation in Rab38cht/cht mice. Invest. Ophthalmol. Vis. Sci. 48, 3905–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Osanai K., Oikawa R., Higuchi J., Kobayashi M., Tsuchihara K., Iguchi M., Jongsu H., Toga H., Voelker D. R. (2008) A mutation in Rab38 small GTPase causes abnormal lung surfactant homeostasis and aberrant alveolar structure in mice. Am. J. Pathol. 173, 1265–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oiso N., Riddle S. R., Serikawa T., Kuramoto T., Spritz R. A. (2004) The rat Ruby (R) locus is Rab38: identical mutations in Fawn-hooded and Tester-Moriyama rats derived from an ancestral Long Evans rat sub-strain. Mamm. Genome 15, 307–314 [DOI] [PubMed] [Google Scholar]
- 34. Ninkovic I., White J. G., Rangel-Filho A., Datta Y. H. (2008) The role of Rab38 in platelet dense granule defects. J. Thromb. Haemost. 6, 2143–2151 [DOI] [PubMed] [Google Scholar]
- 35. Lopes V. S., Wasmeier C., Seabra M. C., Futter C. E. (2007) Melanosome maturation defect in Rab38-deficient retinal pigment epithelium results in instability of immature melanosomes during transient melanogenesis. Mol. Biol. Cell 18, 3914–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tarafder A. K., Bolasco G., Correia M. S., Pereira F. J., Iannone L., Hume A. N., Kirkpatrick N., Picardo M., Torrisi M. R., Rodrigues I. P., Ramalho J. S., Futter C. E., Barral D. C., Seabra M. C. (2014) Rab11b mediates melanin transfer between donor melanocytes and acceptor keratinocytes via coupled exo/endocytosis. J. Invest. Dermatol. 134, 1056–1066 [DOI] [PubMed] [Google Scholar]
- 37. Beaumont K. A., Hamilton N. A., Moores M. T., Brown D. L., Ohbayashi N., Cairncross O., Cook A. L., Smith A. G., Misaki R., Fukuda M., Taguchi T., Sturm R. A., Stow J. L. (2011) The recycling endosome protein Rab17 regulates melanocytic filopodia formation and melanosome trafficking. Traffic 12, 627–643 [DOI] [PubMed] [Google Scholar]
- 38. Ando H., Niki Y., Yoshida M., Ito M., Akiyama K., Kim J. H., Yoon T. J., Matsui M. S., Yarosh D. B., Ichihashi M. (2011) Involvement of pigment globules containing multiple melanosomes in the transfer of melanosomes from melanocytes to keratinocytes. Cell. Logist. 1, 12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu X. S., Masedunskas A., Weigert R., Copeland N. G., Jenkins N. A., Hammer J. A. (2012) Melanoregulin regulates a shedding mechanism that drives melanosome transfer from melanocytes to keratinocytes. Proc. Natl. Acad. Sci. U.S.A. 109, E2101–E2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ando H., Niki Y., Ito M., Akiyama K., Matsui M. S., Yarosh D. B., Ichihashi M. (2012) Melanosomes are transferred from melanocytes to keratinocytes through the processes of packaging, release, uptake, and dispersion. J. Invest. Dermatol. 132, 1222–1229 [DOI] [PubMed] [Google Scholar]
- 41. Singh S. K., Kurfurst R., Nizard C., Schnebert S., Perrier E., Tobin D. J. (2010) Melanin transfer in human skin cells is mediated by filopodia: a model for homotypic and heterotypic lysosome-related organelle transfer. FASEB J. 24, 3756–3769 [DOI] [PubMed] [Google Scholar]
- 42. Wu X., Rao K., Bowers M. B., Copeland N. G., Jenkins N. A., Hammer J. A., 3rd. (2001) Rab27a enables myosin Va-dependent melanosome capture by recruiting the myosin to the organelle. J. Cell Sci. 114, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 43. Wu X., Wang F., Rao K., Sellers J. R., Hammer J. A., 3rd. (2002) Rab27a is an essential component of melanosome receptor for myosin Va. Mol. Biol. Cell 13, 1735–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu X., Bowers B., Rao K., Wei Q., Hammer J. A., 3rd. (1998) Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function in vivo. J. Cell Biol. 143, 1899–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu X., Bowers B., Wei Q., Kocher B., Hammer J. A., 3rd. (1997) Myosin V associates with melanosomes in mouse melanocytes: evidence that myosin V is an organelle motor. J. Cell Sci. 110, 847–859 [DOI] [PubMed] [Google Scholar]
- 46. Wu X. S., Rao K., Zhang H., Wang F., Sellers J. R., Matesic L. E., Copeland N. G., Jenkins N. A., Hammer J. A., 3rd. (2002) Identification of an organelle receptor for myosin-Va. Nat. Cell Biol. 4, 271–278 [DOI] [PubMed] [Google Scholar]
- 47. Fukuda M., Kuroda T. S., Mikoshiba K. (2002) Slac2-a/melanophilin, the missing link between Rab27 and myosin Va: implications of a tripartite protein complex for melanosome transport. J. Biol. Chem. 277, 12432–12436 [DOI] [PubMed] [Google Scholar]
- 48. Jenkins N. A., Copeland N. G., Taylor B. A., Lee B. K. (1981) Dilute (d) coat colour mutation of DBA/2J mice is associated with the site of integration of an ecotropic MuLV genome. Nature 293, 370–374 [DOI] [PubMed] [Google Scholar]
- 49. Mercer J. A., Seperack P. K., Strobel M. C., Copeland N. G., Jenkins N. A. (1991) Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature 349, 709–713 [DOI] [PubMed] [Google Scholar]
- 50. Wilson S. M., Yip R., Swing D. A., O'Sullivan T. N., Zhang Y., Novak E. K., Swank R. T., Russell L. B., Copeland N. G., Jenkins N. A. (2000) A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc. Natl. Acad. Sci. U.S.A. 97, 7933–7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Provance D. W., Jr., Wei M., Ipe V., Mercer J. A. (1996) Cultured melanocytes from dilute mutant mice exhibit dendritic morphology and altered melanosome distribution. Proc. Natl. Acad. Sci. U.S.A. 93, 14554–14558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hammer J. A., 3rd, Sellers J. R. (2012) Walking to work: roles for class V myosins as cargo transporters. Nat. Rev. Mol. Cell Biol. 13, 13–26 [DOI] [PubMed] [Google Scholar]
- 53. Roland J. T., Kenworthy A. K., Peranen J., Caplan S., Goldenring J. R. (2007) Myosin Vb interacts with Rab8a on a tubular network containing EHD1 and EHD3. Mol. Biol. Cell 18, 2828–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rodriguez O. C., Cheney R. E. (2002) Human myosin-Vc is a novel class V myosin expressed in epithelial cells. J. Cell Sci. 115, 991–1004 [DOI] [PubMed] [Google Scholar]
- 55. Jacobs D. T., Weigert R., Grode K. D., Donaldson J. G., Cheney R. E. (2009) Myosin Vc is a molecular motor that functions in secretory granule trafficking. Mol. Biol. Cell 20, 4471–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marchelletta R. R., Jacobs D. T., Schechter J. E., Cheney R. E., Hamm-Alvarez S. F. (2008) The class V myosin motor, myosin 5c, localizes to mature secretory vesicles and facilitates exocytosis in lacrimal acini. Am. J. Physiol. Cell Physiol. 295, C13–C28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Espreafico E. M., Cheney R. E., Matteoli M., Nascimento A. A., De Camilli P. V., Larson R. E., Mooseker M. S. (1992) Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J. Cell Biol. 119, 1541–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Starcevic M., Dell'Angelica E. C. (2004) Identification of snapin and three novel proteins (BLOS1, BLOS2, and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1). J. Biol. Chem. 279, 28393–28401 [DOI] [PubMed] [Google Scholar]
- 59. Dell'Angelica E. C., Aguilar R. C., Wolins N., Hazelwood S., Gahl W. A., Bonifacino J. S. (2000) Molecular characterization of the protein encoded by the Hermansky-Pudlak syndrome type 1 gene. J. Biol. Chem. 275, 1300–1306 [DOI] [PubMed] [Google Scholar]
- 60. Di Pietro S. M., Falcón-Pérez J. M., Dell'Angelica E. C. (2004) Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3, HPS5 and HPS6. Traffic 5, 276–283 [DOI] [PubMed] [Google Scholar]
- 61. Nazarian R., Starcevic M., Spencer M. J., Dell'Angelica E. C. (2006) Reinvestigation of the dysbindin subunit of BLOC-1 (biogenesis of lysosome-related organelles complex-1) as a dystrobrevin-binding protein. Biochem. J. 395, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ozeki H., Ito S., Wakamatsu K., Hirobe T. (1995) Chemical characterization of hair melanins in various coat-color mutants of mice. J. Invest. Dermatol. 105, 361–366 [DOI] [PubMed] [Google Scholar]
- 63. Meehl J. B., Giddings T. H., Jr., Winey M. (2009) High pressure freezing, electron microscopy, and immuno-electron microscopy of Tetrahymena thermophila basal bodies. Methods Mol. Biol. 586, 227–241 [DOI] [PubMed] [Google Scholar]
- 64. Ambrosio A. L., Boyle J. A., Di Pietro S. M. (2012) Mechanism of platelet dense granule biogenesis: study of cargo transport and function of Rab32 and Rab38 in a model system. Blood 120, 4072–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lapierre L. A., Kumar R., Hales C. M., Navarre J., Bhartur S. G., Burnette J. O., Provance D. W., Jr., Mercer J. A., Bähler M., Goldenring J. R. (2001) Myosin vb is associated with plasma membrane recycling systems. Mol. Biol. Cell 12, 1843–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jordens I., Westbroek W., Marsman M., Rocha N., Mommaas M., Huizing M., Lambert J., Naeyaert J. M., Neefjes J. (2006) Rab7 and Rab27a control two motor protein activities involved in melanosomal transport. Pigment Cell Res. 19, 412–423 [DOI] [PubMed] [Google Scholar]
- 67. Chabrillat M. L., Wilhelm C., Wasmeier C., Sviderskaya E. V., Louvard D., Coudrier E. (2005) Rab8 regulates the actin-based movement of melanosomes. Mol. Biol. Cell 16, 1640–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lindsay A. J., Jollivet F., Horgan C. P., Khan A. R., Raposo G., McCaffrey M. W., Goud B. (2013) Identification and characterization of multiple novel Rab-myosin Va interactions. Mol. Biol. Cell 24, 3420–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Roland J. T., Lapierre L. A., Goldenring J. R. (2009) Alternative splicing in class V myosins determines association with Rab10. J. Biol. Chem. 284, 1213–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pashkova N., Jin Y., Ramaswamy S., Weisman L. S. (2006) Structural basis for myosin V discrimination between distinct cargoes. EMBO J. 25, 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee M. T., Mishra A., Lambright D. G. (2009) Structural mechanisms for regulation of membrane traffic by rab GTPases. Traffic 10, 1377–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kloer D. P., Rojas R., Ivan V., Moriyama K., van Vlijmen T., Murthy N., Ghirlando R., van der Sluijs P., Hurley J. H., Bonifacino J. S. (2010) Assembly of the biogenesis of lysosome-related organelles complex-3 (BLOC-3) and its interaction with Rab9. J. Biol. Chem. 285, 7794–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tamura K., Ohbayashi N., Ishibashi K., Fukuda M. (2011) Structure-function analysis of VPS9-ankyrin-repeat protein (Varp) in the trafficking of tyrosinase-related protein 1 in melanocytes. J. Biol. Chem. 286, 7507–7521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Setty S. R., Tenza D., Sviderskaya E. V., Bennett D. C., Raposo G., Marks M. S. (2008) Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 454, 1142–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schäfer I. B., Hesketh G. G., Bright N. A., Gray S. R., Pryor P. R., Evans P. R., Luzio J. P., Owen D. J. (2012) The binding of Varp to VAMP7 traps VAMP7 in a closed, fusogenically inactive conformation. Nat. Struct. Mol. Biol. 19, 1300–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ryder P. V., Vistein R., Gokhale A., Seaman M. N., Puthenveedu M. A., Faundez V. (2013) The WASH complex, an endosomal Arp2/3 activator, interacts with the Hermansky-Pudlak syndrome complex BLOC-1 and its cargo phosphatidylinositol-4-kinase type IIα. Mol. Biol. Cell 24, 2269–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kent H. M., Evans P. R., Schäfer I. B., Gray S. R., Sanderson C. M., Luzio J. P., Peden A. A., Owen D. J. (2012) Structural basis of the intracellular sorting of the SNARE VAMP7 by the AP3 adaptor complex. Dev. Cell 22, 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Raposo G., Marks M. S., Cutler D. F. (2007) Lysosome-related organelles: driving post-Golgi compartments into specialisation. Curr. Opin. Cell Biol. 19, 394–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Watanabe M., Nomura K., Ohyama A., Ishikawa R., Komiya Y., Hosaka K., Yamauchi E., Taniguchi H., Sasakawa N., Kumakura K., Ushiki T., Sato O., Ikebe M., Igarashi M. (2005) Myosin-Va regulates exocytosis through the submicromolar Ca2+-dependent binding of syntaxin-1A. Mol. Biol. Cell 16, 4519–4530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chaineau M., Danglot L., Galli T. (2009) Multiple roles of the vesicular-SNARE TI-VAMP in post-Golgi and endosomal trafficking. FEBS Lett. 583, 3817–3826 [DOI] [PubMed] [Google Scholar]
- 81. Tamura K., Ohbayashi N., Maruta Y., Kanno E., Itoh T., Fukuda M. (2009) Varp is a novel Rab32/38-binding protein that regulates Tyrp1 trafficking in melanocytes. Mol. Biol. Cell 20, 2900–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hartman M. A., Finan D., Sivaramakrishnan S., Spudich J. A. (2011) Principles of unconventional myosin function and targeting. Annu. Rev. Cell Dev. Biol. 27, 133–155 [DOI] [PMC free article] [PubMed] [Google Scholar]