

Background: Munc18-1 has multiple roles in neuronal exocytosis by regulating SNARE proteins.
Results: Mutations within domain-3a of Munc18-1 perturb syntaxin-1 chaperoning function and exocytosis.
Conclusion: Domain-3a plays a crucial role in syntaxin-1 chaperoning in addition to the priming function, and Pro-335 is pivotal in regulating the balance between these two functions.
Significance: This work provides mechanistic insights about how Munc18-1 controls exocytosis.
Keywords: Exocytosis, Membrane Fusion, Secretion, Soluble NSF Attachment Protein Receptor (SNARE), Vesicles, Munc18, Dense Core Vesicles
Abstract
Munc18-1 plays essential dual roles in exocytosis: (i) stabilizing and trafficking the central SNARE protein, syntaxin-1 (i.e. chaperoning function), by its domain-1; and (ii) priming/stimulating exocytosis by its domain-3a. Here, we examine whether or not domain-3a also plays a significant role in the chaperoning of syntaxin-1 and, if so, how these dual functions of domain-3a are regulated. We demonstrate that introduction of quintuple mutations (K332E/K333E/P335A/Q336A/Y337L) in domain-3a of Munc18-1 abolishes its ability to bind syntaxin-1 and fails to rescue the level and trafficking of syntaxin-1 as well as to restore exocytosis in Munc18-1/2 double knockdown cells. By contrast, a quadruple mutant (K332E/K333E/Q336A/Y337L) sparing the Pro-335 residue retains all of these capabilities. A single point mutant of P335A reduces the ability to bind syntaxin-1 and rescue syntaxin-1 levels. Nonetheless, it surprisingly outperforms the wild type in the rescue of exocytosis. However, when additional mutations in the neighboring residues are combined with P335A mutation (K332E/K333E/P335A, P335A/Q336A/Y337L), the ability of the Munc18-1 variants to chaperone syntaxin-1 and to rescue exocytosis is strongly impaired. Our results indicate that residues from Lys-332 to Tyr-337 of domain-3a are intimately tied to the chaperoning function of Munc18-1. We also propose that Pro-335 plays a pivotal role in regulating the balance between the dual functions of domain-3a. The hinged conformation of the α-helix containing Pro-335 promotes the syntaxin-1 chaperoning function, whereas the P335A mutation promotes its priming function by facilitating the α-helix to adopt an extended conformation.
Introduction
Munc18-1, a neuronal Sec1/Munc18 protein, and its orthologues, have been shown to be indispensable for neuronal exocytosis in mice (1, 2), Drosophila (3), and Caenorhabditis elegans (4). The precise modality of the contribution of Munc18-1 to exocytosis has been extensively studied in recent years through rescue assays with Munc18-1-deficient neurons (5, 6), chromaffin cells (7) and Munc18-1/2 double knockdown neuroendocrine PC12 cells (8–12), as well as through liposome fusion assays (13–17). At least two important functions of Munc18-1 have been proposed (18): (i) molecular chaperoning of syntaxin-1 allowing proper localization and expression of syntaxin-1 (8–10, 19–23); and (ii) priming or promoting SNARE complex-mediated membrane fusion (13, 14, 24, 25). The former function is mediated primarily by the binding between the domain-1 cleft of Munc18-1 and “closed” syntaxin-1. The K46E/E59K mutant was discovered as the key “chaperoning mutant” that essentially loses its abilities to bind to the closed conformation of syntaxin-1 and consequently becomes unable to restore syntaxin-1 expression, localization, dense core vesicle docking, and secretion in Munc18-1/2 double knockdown PC12 cells (8, 9).
The molecular mechanisms underlying priming function of Munc18-1 are not well characterized and are still under extensive investigation. Several groups have suggested that the direct binding of Munc18-1 to the SNARE complex is a key mechanism that underlies Munc18-1-dependent priming and/or stimulation of SNARE-mediated membrane fusion (13, 24, 26, 27). This direct interaction has been proposed to occur in two possible ways: (i) binding between the hydrophobic pocket of Munc18-1 and N-terminal peptide of syntaxin-1 (13, 26, 28); or (ii) interaction between domain-3a of Munc18-1 and synaptobrevin-2 within the SNARE complex. The first binding mode represents the interaction between the Munc18-1 hydrophobic pocket and syntaxin-1 N-peptide. However, mutating the residues that line the hydrophobic pocket region (F115E, E132K, F115E/E132K, L130K) of Munc18-1 showed limited or no phenotype in the rescue of exocytosis in Munc18-1 single knockdown (10) and Munc18-1/2 double knockdown PC12 cells (8) as well as Munc18-1-deficient neurons (5). In addition, syntaxin N-peptide binding to Munc18-1 was shown to be unselective (29). This suggests that Munc18-1-dependent exocytosis, which relies on the specific interaction between Munc18-1 and its cognate SNARE complex, involves another mode of direct interaction. Munc18-1 has also been suggested to directly interact with the assembled SNARE complex through its domain-3a. The cross-linking study has shown that the residues 333–339 of domain-3a (KMPQYQK) of Munc18-1 bind to the transmembrane proximal residues 87–91 (KYWWK) of synaptobrevin-2 (30). In addition, disrupting this region of the domain-3a of Munc18-1 by introducing deletion (Del 317–333) or insertion mutant (KE/5I)3 caused severe secretion defects without impairing the chaperoning activity of Munc18-1 (11, 12). Furthermore, the latter insertion mutant was found to interfere with the ability of Munc18-1 to bind to the preassembled SNARE complex (11). Moreover, another mutation, L348R, in domain-3a, which abolishes the Munc18-1-dependent stimulation of liposome fusion, was also found to impair the binding to synaptobrevin-2 (17). These studies collectively support the proposition that domain-3a of Munc18-1 mediates the Munc18-1-dependent priming of membrane fusion through its direct interaction with synaptobrevin-2 within the SNARE complex.
Despite the accumulating evidence that supports the importance of domain-3a of Munc18-1 in the priming of membrane fusion, whether domain-3a additionally contributes to the chaperone activity of Munc18-1 has not been assessed. Previous x-ray crystallography has revealed the tight binary interaction between closed syntaxin-1 and Munc18-1 cleft formed by domain-1 and domain-3a (see Fig. 1) (31). A more recent structural analysis revealed that domain-3a of Munc18-1 can undergo a conformational change, which serves to release Munc18-1 from the binary interaction with closed syntaxin-1, thus allowing the sequential interaction with assembled SNARE complex. This study has highlighted that Pro-335 residue of Munc18-1 acts as a hinge point that mediates the conformational change (29). This clearly indicates that domain-3a of Munc18-1 plays an essential role while it switches its partner from the closed syntaxin-1 to the assembled SNARE complex. However, the specific contribution of domain-3a during the binary interaction is unknown. Furthermore, how the Pro-335 residue acts as a critical hinge point that regulates this binary interaction requires further investigation.
FIGURE 1.
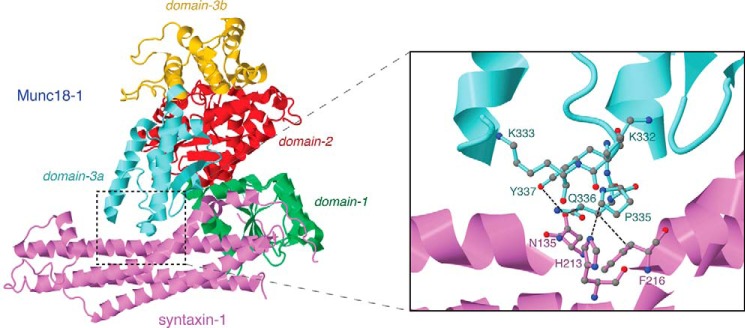
Ribbon representative of Munc18-1 structure bound to syntaxin-1. Each domain of Munc18-1 is represented in a different color. Domain-1, -2, -3a, and -3b are shown in green, red, yellow, and cyan, respectively. Syntaxin-1 is represented in purple. On the right is an enlarged ribbon representative highlighting the domain-3a residues that have been mutated in the study as well as the interacting residues of syntaxin-1.
Here, we uncover the additional function of the domain-3a during syntaxin-1 chaperoning activity by disclosing the first domain-3a chaperoning mutant, K332E/K333E/P335A/Q336A/Y337A. Our analyses of this mutant together with those of the P335A single mutant, as well as the combinations of the other various mutations in neighboring residues reveal that the Pro-335 residue plays a pivotal role in regulating the balance between the dual functions of domain-3a during regulated exocytosis.
EXPERIMENTAL PROCEDURES
General Materials
Parental pLVX-EmGFP-IRES-blast plasmid for lentivirus-mediated Munc18-1 expression was described previously (9). psPAX2 was purchased from Addgene (Cambridge, MA), and pCMV-VSVG was a kind gift from Dr. Herbert Gaisano (University of Toronto). We obtained monoclonal antibodies against syntaxin-1 (clone HPC-1) (32) from Sigma (Oakville, Ontario, Canada); Munc18-1 from BD Biosciences (Mississauga, Ontario, Canada); and GAPDH (clone 6C5) from Millipore (Billerica, MA).
Lentivirus-mediated Expression of Munc18-1 Variants in Munc18-1/2 Double Knockdown Cells
Two clonal lines of Munc18-1/2 double knockdown cells (D7, D16) were maintained as described previously (11). The constructs for lentivirus-mediated expression of various Munc18-1 mutants were generated so that these proteins could stably express in the Munc18-1/2 double knockdown cells. The Munc18-1 gene with silent nucleotide mutations (WT or its indicated mutant) was subcloned into the same site of the pLVX-EmGFP-IRES-blast plasmid. This Munc18-1 expression plasmid was cotransfected with psPAX2 and pCMV-VSVG into HEK-293FT cells to generate recombinant lentiviruses that express Munc18-1 WT or its variant fused with EmGFP. The Munc18-1/-2 double knockdown cells that were infected with lentiviruses expressing rescue proteins were selected with blasticidin (5 μg/ml).
[3H]Noradrenaline (NA) Release Assays from PC12 Cells
PC12 cells were plated in 24-well plates; 3–4 days after plating, the cells were labeled with 0.5 μCi of [3H]NA4 in the presence of 0.5 mm ascorbic acid for 12–16 h. The labeled PC12 cells were incubated with the fresh complete DMEM for 1–5 h to remove unincorporated [3H]NA. The cells were washed once with physiological saline solution (PSS) containing 145 mm NaCl, 5.6 mm KCl, 2.2 mm CaCl2, 0.5 mm MgCl2, 5.6 mm glucose, and 15 mm HEPES, pH 7.4, and NA secretion was stimulated with 200 μl of PSS or high K+-PSS (containing 81 mm NaCl and 70 mm KCl). Secretion was terminated after a 15-min incubation at 37 °C by chilling to 0 °C, and samples were centrifuged at 4 °C for 3 min. Supernatants were removed, and the pellets were solubilized in 0.1% Triton X-100 for liquid scintillation counting.
Cell Preparation for Confocal Immunofluorescence Microscopy
Cells were allowed to adhere to the coverslips prepared as described by Han et al. (11) overnight and then differentiated on the coverslips for 3–4 days in DMEM that contained 100 ng/ml NGF (Sigma), 1% horse serum, 1% calf serum, and penicillin/streptomycin. The cells were washed with PBS and fixed for 15 min with PBS containing 4% paraformaldehyde. Paraformaldehyde was then removed from each well, and cells were rinsed three times (10 min each time) with 1 ml of PBS per well. The fixed cells were then permeabilized with PBS containing 0.2% Triton X-100 and 0.3% BSA for 5 min followed by washing three times with PBS. Nonspecific sites were blocked for 1 h at room temperature in PBS containing 0.3% BSA. Primary antibodies against syntaxin-1 (HPC-1 diluted 1:1000) were applied to the cell for 1 h. After three washes in blocking buffer, Rhodamine Red-x-conjugated anti-mouse antibodies (diluted 1:1000) (Jackson ImmunoResearch Laboratories, West Grove, PA) were applied for 1 h. Samples were washed again three times in blocking buffer and mounted in Fluoromount-G reagent (Southern Biotechnology, Birmingham, AL). Immunofluorescence staining was recorded with a laser confocal scanning microscope (LSM510; Carl Zeiss, Jena, Germany) with an oil immersion objective lens (63×).
Yeast Two-hybrid Assays
Detailed procedure was previously described in Refs. 8, 9, and 11. Yeast strain L40 (33) was transfected with full-length WT Munc18-1 with silent nucleotide mutations or indicated mutant Munc18-1 (silent nucleotide mutations) and a cytoplasmic domain (residues 1–264) of rat syntaxin-1A (34). Transformants were plated on selection plates lacking uracil, tryptophan, and leucine. After 2 days of incubation at 30 °C, colonies were inoculated into supplemented minimal medium lacking uracil, tryptophan, and leucine and placed in a shaking incubator at 30 °C for 2 days.
β-Galactosidase assays were performed as follows. 80 μl of the yeast extract prepared as described previously (11) was added to 720 μl of Z buffer (60 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4, and 2.7 ml/liter β-mercaptoethanol, pH 7.0). The mixture was then incubated in a water bath at room temperature for 5 min. The reaction was initiated by adding 0.16 ml of stock solution (4 mg/ml o-nitrophenyl-β-d-galactoside in Z buffer; 4 °C), and the reaction mixture was incubated at room temperature. The reaction was precisely terminated at the end of a 7-min incubation by the addition of 0.4 ml of 1 m Na2CO3 stock solution in distilled water, and the optical density of the reaction mixture was measured at 420 nm by using a spectrophotometer. At the same time, the protein concentration in the extract was measured using Bradford dye binding assay. The specific activity of β-galactosidase in the extract was calculated according to the following formula: (A420 × 1.36)/(0.0045 × protein concentration (mg/ml) × extract volume (0.08 ml) × 7 min), where A420 is the optical density of the product o-nitrophenol at 420 nm. The unit of β-galactosidase-specific activity is expressed as nanomoles per minute per milligram of protein.
Purification of Bacterially Expressed Proteins
pGex-KG containing cytosolic syntaxin-1A (1–264) were described previously (35). pET21a plasmids containing full-length rat Munc18-1 variants (wild type, K332E/K333E, P335A, K332E/K333E/P335A) were generated by subcloning of a 1.8-kb fragment of Munc18-1 (without stop codon) into the EcoRI/HindIII site of pET21a (Novagen, Madison, WI). GST-syntaxin-1A was expressed and purified as described previously (35). pET21a-Munc18-1 constructs were transformed into BL21 (DE3) cells, and the bacteria were grown at 37 °C until confluent. Recombinant Munc18-1 expression was induced by adding 125 μm isopropylthio-β-d-galactoside at 15 °C overnight. The cultures were then centrifuged at 3000 rpm for 10 min at 4 °C using a JA-14 rotor (Pasadena, CA). Munc18-1 pellets were resuspended in a lysis buffer containing 20 mm Tris-HCl, 150 mm NaCl, 0.4% Nonidet P-40, 10 mm imidazole, 10 mm 2-mercaptoethanol, 1 mm PMSF, and 20 ng/ml DNase. Bacterial cell lysis was performed by applying a cell pressure of 1000 p.s.i. for 30 s three times using a French pressure cell press (Thermo Scientific). The lysate was then centrifuged at 15,000 rpm for 30 min at 4 °C using a JA-21 rotor. His6-Munc18-1 supernatant was mixed with HisPur nickel-nitrilotriacetic acid resin (Thermo Scientific). The binding to beads was performed for 2 h at 4 °C. Then, the washing was performed first by high salt buffer (20 mm Tris-HCl, 500 mm NaCl, 20 mm imidazole, 10 mm 2-mercaptoethanol, 10% glycerol) followed by mid-salt buffer (same contents as high salt buffer but with 300 mm NaCl instead). Then, Munc18-1 proteins were eluted by elution buffer (20 mm Tris-HCl, 150 mm NaCl, 250 mm imidazole, 10 mm 2-mercaptoethanol, 10% glycerol). Eluted His6-Munc18-1 and bead-immobilized GST-syntaxin-1A proteins were then saved for in vitro binding experiments.
GST Pulldown Assays between GST-Syntaxin-1A and His6-Munc18-1 Variants
∼12 μg of His6-Munc18-1 proteins was added to 500 μl of KGlu binding buffer (20 mm Hepes, pH 7.2, 20 mm potassium acetate, 120 mm potassium glutamate, 2 mm EGTA, 0.1% Triton X-100) containing 10 μl of agarose beads with ∼5 μg of immobilized GST-syntaxin-1A and incubated for 30 min at room temperature. The beads were washed five times with the binding buffer. After the washing, the total agarose beads were loaded onto SDS-PAGE gel and stained with Coomassie Brilliant Blue.
Binding of Recombinant Syntaxin-1A and Munc18-1 Expressed in HEK293FT Cells
Three 10-cm dishes of HEK293 cells expressing exogenous GFP-fused Munc18-1 wild-type and mutant forms after transient transfection were harvested and pelleted. The cells were washed with PBS containing 1 mm EDTA and protease inhibitors (10 μg/ml leupeptin and 10 μg/ml aprotinin), and then 1 ml of KGlu binding buffer containing 0.1% Triton X-100 was added to the pellet. After the cells were vortexed, they were homogenized using a 23.5-gauge needle. The soluble part separated by centrifugation was incubated overnight with the purified syntaxin-1A GST fusion proteins immobilized on the glutathione beads. After incubation, Munc18-1 bound GST-syntaxin-1 forms were washed five times with KGlu binding buffer and subjected to SDS-PAGE, transferred to nitrocellulose, and subsequently subjected to Ponceau S staining and immunoblotting.
RESULTS
Simultaneous Mutations of Lys-332, Lys-333, Pro-335, Gln-336, and Tyr-337 of Munc18-1 Prevents Binary Interaction between Munc18-1 and Syntaxin-1
The syntaxin-1 chaperoning function of Munc18-1 is primarily mediated by the tight binary interaction between closed syntaxin-1 and Munc18-1 cleft formed by domain-1 and domain-3a (31). We previously discovered a key double point mutant, K46E/E59K, in domain-1 of Munc18-1, that abolishes the binding with monomeric closed syntaxin-1 and consequently impairs the chaperoning ability of Munc18-1 (8, 9). In contrast, the key residues in domain-3a that are crucial for the binding to syntaxin-1 remain unknown. We previously investigated the phenotypes of K332E, K333E, K332E/K333E, Q336A/Y337L, Y337L/Q338A, and Q336A/Y337L/Q338A single, double, or triple mutants on the binary interaction with syntaxin-1 as well as their ability to rescue secretion of Munc18-1/2 double knockdown (DKD) cells (11). Among the tested residues, Gln-336 and Tyr-337 are shown to interact with Asn-135 and Phe-216 of syntaxin-1A based on x-ray crystallography (Fig. 1). Nonetheless, we found that all the mutants retain the intact binary interaction with monomeric syntaxin-1 and exhibit normal ability to rescue secretion defect of Munc18-1/2 DKD cells (11). Based on this observation, we speculated that the structure of the subregion we tested in domain-3a is highly flexible and thus its function may be resistant to point mutations.
However, a recent structural study revealed that Pro-335 functions as a flexible hinge point in the conserved α-helical region and that its extended conformation can interfere with the binding to closed syntaxin-1A (29). Therefore, the P335A mutation potentially reduces the interaction with closed syntaxin-1A by favoring the extended conformation. In addition, Pro-335 also directly interacts with the residue His-213 of closed syntaxin-1A (Fig. 1), suggesting that this interaction is likely to be impaired upon introducing the P335A mutation. Although the P335A single mutation alone has not shown a significant effect on Munc18-1 activity in previous work (12), we hypothesized that this residue would still play a significant role in binding with closed syntaxin-1A in a collaboration with its neighboring residues. To test our hypothesis, we simultaneously mutated the Pro-335 residue (P335A) and its flanking residues, Lys-332, Lys-333, Gln-336, and Tyr-337, generating a quintuple mutant, K332E/K333E/P335A/Q336A/Y337L (Fig. 2A). We found that this quintuple mutation completely abolishes the ability of Munc18-1 to interact with monomeric syntaxin-1 in yeast two-hybrid assays (Fig. 2B). Importantly, this mutant still retains binding to Mint-1, another Munc18-1-binding protein (36), as effectively as the wild type (Fig. 2B). This implies that its impaired binding to syntaxin-1A is specific and is not likely due to the loss of expression or inability to transport into the nucleus during the yeast two-hybrid assays. This novel mutant reveals, for the first time, the crucial contribution of domain-3a in the binary interaction of Munc18-1 and monomeric closed syntaxin-1A.
FIGURE 2.

The quintuple K332E/K333E/P335A/Q336A/Y337L (KKPQY) mutant impairs the ability of Munc18-1 to interact with syntaxin-1, to restore syntaxin-1, and to restore secretion. A, sequence alignment of the KKPQY mutant containing mutations at Lys-332, Lys-333, Pro-335, Gln-336, and Tyr-337. Mutated residues are shown in red. M18-1, Munc18-1. B, the binding between Munc18-1 domain-3a KKPQY mutant and syntaxin-1 or Mint-1 was analyzed by yeast two-hybrid assays. In this assay, β-galactosidase activities of the transformed yeast clones were quantified and normalized so that the activity of the yeast clones transformed with the wild-type Munc18-1 was set to 100%. Error bars indicate S.E. (n = 12–13 for syntaxin-1 interaction; n = 10–12 for Mint-1 interaction). C, stable re-expression of Munc18-1 KKPQY mutant in D7 or D16 clone fails to restore syntaxin-1 levels. EGFP, emerald green fluorescent protein. D, secretion defects are not rescued upon reintroduction of the KKPQY-EmGFP mutant in Munc18-1/-2 double knockdown cells (D7 clone). NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 9). E, secretion defects are not rescued upon reintroduction of the KKPQY-EmGFP mutant in Munc18-1/-2 double knockdown cells (D16 clone). NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 6).
The Quintuple K332E/K333E/P335A/Q336A/Y337L Mutant Does Not Rescue Syntaxin-1 Levels, Syntaxin-1 Localization, and Exocytosis
We then tested the ability of this quintuple mutant to rescue syntaxin-1 levels and exocytosis of our Munc18-1/2 double knockdown PC12 cells (8). The mutant expresses as efficiently as wild-type Munc18-1 upon re-expression in the double knockdown cells (D7 and D16) (Fig. 2C), suggesting the normal folding of the mutant. When the ability of this mutant to restore syntaxin-1 was assessed, we found that it fails to restore syntaxin-1 expression (Fig. 2C). This indicates that upon mutating the five residues, the ability of Munc18-1 to stabilize syntaxin-1 is disturbed due to the loss of binding to monomeric closed syntaxin-1. We also found that this quintuple mutant is unable to restore plasmalemmal localization of syntaxin-1 in contrast to the wild type (Fig. 3, A and B). These results indicate that domain-3a contributes to stabilizing and trafficking syntaxin-1A in concert with domain-1.
FIGURE 3.

Rescue of syntaxin-1 localization in Munc18-1/2 DKD (D16) clones upon reintroduction of wild-type Munc18-1-EmGFP (A), Munc18-1 quintuple K332E/K333E/P335A/Q336A/Y337L (KKPQY) mutant (B), or Munc18-1 quadruple K332E/K333E/Q336A/Y337L (KKQY) mutant (C). These cells were stained with anti-syntaxin-1 antibodies followed by Red-X-conjugated anti-mouse antibodies (middle panels). Left panels, intrinsic green fluorescence signals from Munc18-1 fused with emerald GFP. Right panels are merged pictures. Bar, 10 μm. M18-1, Munc18-1.
We previously showed that a Munc18-1 domain-1 mutant (K46E/E59K) that loses the ability to stabilize and traffic syntaxin-1 consequently results in defective secretion (8). When we examined the ability of the quintuple mutant to restore the secretion defect of DKD cells, we observed that this mutant failed to rescue the secretion defect (Fig. 2, D and E). This demonstrates that the domain-3a contributes to the chaperoning activity of Munc18-1.
The Quadruple K332E/K333E/Q336A/Y337A Can Rescue Syntaxin-1 Levels, Syntaxin-1 Localization, and Exocytosis
Previously analyzed single, double, and triple mutants that did not exhibit defective phenotypes were K332E, K333E, K333E/K333E, E278K/K332E/K333E, Q336A/Y337L, Y337L/Q338A, and Q336A/Y337L/Q338A (11). Therefore, we hypothesized that the severe phenotype observed for the quintuple mutant is due to the introduction of the P335A mutation. To validate the contribution of the P335A mutation to the observed phenotype of the quintuple mutant, we further generated a quadruple mutant, K332A/K333A/Q336A/Y337L, without modifying Pro-335 residue (Fig. 4A). We found that this quadruple mutant retained intact binary interaction with monomeric syntaxin-1 as assessed by yeast two-hybrid assays (Fig. 4B). This suggests that the impaired syntaxin-1 binding ability of the quintuple mutant (Fig. 2) is at least in part due to the P335A mutation.
FIGURE 4.

The quadruple K332E/K333E/Q336A/Y337A (KKQY) mutant retains the ability of Munc18-1 to interact with syntaxin-1, to restore syntaxin-1, and to restore secretion. A, sequence alignment of the KKQY mutant containing mutations at Lys-332, Lys-333, Gln-336, and Tyr-337. Mutated residues are shown in red. M18-1, Munc18-1. B, the binding between Munc18-1 domain-3a KKQY mutant and syntaxin-1 or Mint-1 was analyzed by yeast two-hybrid assays. In this assay, β-galactosidase activities of the transformed yeast clones were quantified and normalized so that the activity of the yeast clones transformed with the wild-type Munc18-1 was set to 100%. Error bars indicate S.E. (n = 16 for syntaxin-1 interaction; n = 10–12). C, stable re-expression of Munc18-1 KKQY mutant in D7 or D16 clone restores syntaxin-1 levels. EGFP, emerald green fluorescent protein. D, secretion defects are rescued upon reintroduction of the KKQY-EmGFP mutant in Munc18-1/-2 double knockdown cells (D7 clone). NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 7–13). E, secretion defects not rescued upon reintroduction of the KKQY-EmGFP mutant in Munc18-1/-2 double knockdown cells (D16 clone). NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 9–12).
We further examined the ability of the quadruple mutant to rescue syntaxin-1 levels and localization upon expression in DKD cells. We found that this mutant is able to restore the syntaxin-1A levels (Fig. 4C) and plasmalemmal localization (Fig. 3C). We also examined the ability of this mutant to rescue exocytosis to assess whether this mutant interferes with the priming of exocytosis. In our previous work, the insertion of 5 residues in addition to K332E/K333E double mutant (KE/5I) showed completely abolished secretory capability despite its intact ability to rescue syntaxin-1 levels and localization (11). Unlike the KE/5I mutant, however, the quadruple mutant retained the ability to rescue exocytosis (Fig. 4, D and E). Therefore, we conclude that this quadruple mutant retains both priming and chaperoning functions.
The P335A Mutation of Munc18-1 Destabilizes the Binding with Monomeric Closed Syntaxin-1A while Enhancing Exocytosis
The striking difference between the phenotypes of the quintuple mutant (Fig. 2) and those of the quadruple mutant (Fig. 4) strongly indicates the crucial role of Pro-335 in the binding of Munc18-1 to closed syntaxin-1A. Therefore, we next examined the ability of P335A mutants with or without mutations in the neighboring residues to bind syntaxin-1A through yeast two-hybrid assays (Fig. 5B). We found that P335A alone partially reduces the binary interaction with syntaxin-1A by ∼30% when compared with the wild type, which is statistically significant (p < 0.05) (Fig. 5B). When the additional mutations were introduced in the presence of P335A mutation, we observed very interesting phenomena; the domain-3a mutants such as K332E/K333E/P335A, P335A/Y337L, and P335A/Q336A/Y337L almost completely lost the ability to interact with syntaxin-1A, whereas a mutant like P335A/Q336A exhibited no defects in syntaxin-1 binding. In our previous study, we showed that K332E/K333E alone has no deteriorating effect on syntaxin-1A binding (supplemental Fig. 1 of Ref. 11). Thus, our results show that although the P335A mutation alone is not severe enough to completely disrupt the binary interaction, it destabilizes this interaction. Then, introducing the additional mutations in the neighboring residues (except for Gln-336) further exacerbates the destabilizing effect of the P335A mutant on syntaxin-1A binding. Importantly, all of these mutants retain the ability to interact with Mint-1 (Fig. 5C), implying that the observed effects are specific to the binding with syntaxin-1A.
FIGURE 5.

The P335A mutant destabilizes the binding with monomeric closed syntaxin-1A. A, sequence alignment of the K332E/K33E/P335A (KE/KE/PA), P335A, P335A/Q336A (PA/QA), P335A/Y337L (PA/YL), and P335A/Q336A/Y337L (PA/QA/YL) mutants. M18-1, Munc18-1; KE/KE, K332E/K33E. B and C, the binding between Munc18-1 domain-3a mutants and syntaxin-1 (B) or Mint-1 (C) was analyzed by yeast two-hybrid assays. In this assay, β-galactosidase activities of the transformed yeast clones were quantified and normalized so that the activity of the yeast clones transformed with the wild-type Munc18-1 was set to 100%. Error bars indicate S.E. (n = 12–14 for syntaxin-1 interaction; n = 10–12 for Mint-1 interaction). *, p < 0.05. D–F, GST pulldown experiments testing the interactions between Munc18-1 domain-3a mutants (recombinant His6-Munc18-1 (D) or HEK-293FT expressed Munc18-1 (E and F)) with GST-fused cytosolic syntaxin-1A (1–264). D, the GST-syntaxin-1A pulled down Munc18-1 variants were shown as Coomassie Brilliant Blue staining. E and F, Ponceau S staining and immunoblotting with anti-Munc18-1 antibody. KKP, K332E/K333E/P335A.
We also performed GST pulldown experiments to verify the effects of P335A mutation as well as mutations in the surrounding residues on binding of Munc18-1 and syntaxin-1A using more direct biochemical assays. We expressed cytosolic syntaxin-1A (residues 1–264) as a GST fusion protein. In one set of experiments, Munc18-1 variants (WT, K332E/K333E, P335A, K332E/K333E/P335A) were expressed in bacteria as recombinant His6 fusion proteins and purified with a nickel-agarose column (Fig. 5D). In another set of experiments, Munc18-1 variants were expressed in mammalian HEK-293FT cells (Fig. 5, E and F). In both sets of the GST pulldown experiments, we found that P335A single mutation partially reduces the binding to GST-syntaxin-1A. Although K332E/K333E mutations alone did not affect the binding, the triple mutations of K332E/K333E/P335A more severely impaired the binding (Fig. 5, D–F). Thus, the results of our biochemical binding experiments are consistent with those of the yeast two-hybrid assays (Fig. 5B).
We then tested the ability of these mutants to restore syntaxin-1A levels and exocytosis of the DKD cells (Figs. 6 and 7). In Fig. 6, A and B, we included the previously generated DKD cells that were rescued with the K332E/K333E mutant (11) for the purpose of side-by-side comparison with the other mutants. We found that the P335A mutation of Munc18-1 decreased the level of its expression upon re-expression in D7 and D16 cells when compared with other overexpressed Munc18-1 domain-3a variants. However, the expression level of P335A was still comparable with the endogenous Munc18-1 level (Fig. 6, A and B). This reduced expression may result from a possibility that the introduction of P335A point mutation alone induces the extension of the ordered α-helical structure even before encountering its SNARE complex partner (29), which may cause an instability in the overall structure of Munc18-1, yielding such reduced expression. Also, we found that the recovery of syntaxin-1A levels is reduced upon re-expression of the P335A mutant when compared with the cells rescued by the wild-type Munc18-1 (Fig. 6, A and B). This result is likely due to the combined effects of the reduced expression of the P335A mutant itself as well as its reduced ability to bind with syntaxin-1A (Fig. 5). Surprisingly, however, the P335A mutant restored exocytosis more efficiently than that of the wild type in both D7 and D16 cells, which is statistically significant (p < 0.05) (Fig. 6, C and D). This finding suggests that the P335A mutant seemed to exhibit a gain-of-function phenotype in secretion. This surprising result seems to be consistent with the recent finding that the identical P335A mutant outperforms the wild type in promoting SNARE-mediated liposome fusion (17). Thus, Pro-335 residue seems to regulate the balance between the dual functions of Munc18-1 such that the P335A mutation enhances the priming or fusogenic activity, whereas this mutation reduces the chaperoning function by loosening its binary interaction with monomeric closed syntaxin-1.
FIGURE 6.

The ability of the P335A single mutant and the K332E/K333E/P335A triple mutant to restore syntaxin-1 expression levels and to rescue secretion varies. A and B, the effect of stable re-expression of Munc18-1 K332E/K333E, P335A, or K332E/K333E/P335A mutant in Munc18-1/-2 double knockdown clones D7 (A) and D16 (B) on syntaxin-1 expression. A number on the left indicates the position of a molecular weight marker. KE/KE, K332E/K33E; KKP, K332E/K333E/P335A. C and D, the ability of the mutants to rescue the secretion defects upon reintroduction in D7 (C) and D16 (D) clones. NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 6 for K332E/K333E/P335A D7, n = 12 for all other D7; n = 14 for D16). *, p < 0.05. M18WT, Munc18-1 WT.
FIGURE 7.
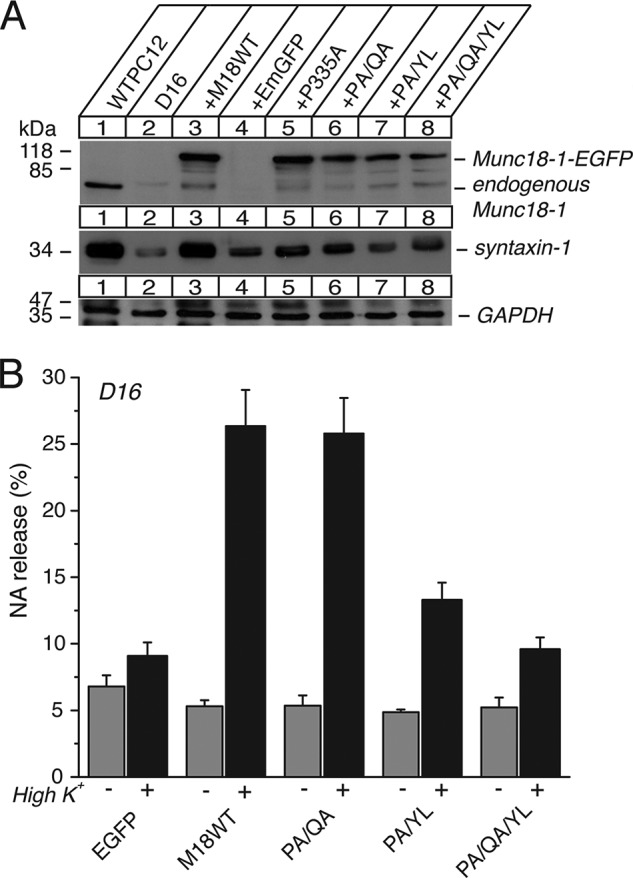
The effect of the P335A/Q336A, P335A/Y337L, and P335A/Q336A/Y337L mutants on syntaxin-1 expression levels and vesicle secretion. A, the effect of stable re-expression of Munc18-1 P335A/Q336A (PA/QA), P335A/Y337L (PA/YL), and P335A/Q336A/Y337L (PA/QA/YL) in Munc18-1/-2 double knockdown clone D16 on syntaxin-1 expression. A number on the left indicates the position of a molecular weight marker. EGFP, emerald green fluorescent protein. B, the ability of the mutants to rescue the secretion defects upon reintroduction in D16 clones. NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 6). M18WT, Munc18-1 WT.
Unlike the P335A mutant, the K332E/K333E mutant and the K332E/K333E/P335A mutant were expressed as efficiently as wild-type Munc18-1 (Fig. 6, A and B). Although the K332E/K333E mutation alone restored syntaxin-1A levels as effectively as WT, K332E/K333E/P335A could not restore syntaxin-1A levels. These results seem to be consistent with their differential ability to bind syntaxin-1A (Fig. 5). The ability of the K332E/K333E/P335A mutant to rescue exocytosis was significantly (p < 0.05) decreased when compared with the wild type (Fig. 6, C and D). On the other hand, our previous data indicated that K332E/K333E retains the ability to restore exocytosis as effectively as the wild type (11).
The other combinations of the domain-3a mutants, P335A/Q336A, P335A/Y337L, P335A/Q336A/Y337L, did not express well (Fig. 7), similar to P335A alone, which has challenged the interpretation of their phenotypes. Among these mutants, the P335A/Q336A was the only one that could rescue exocytosis as effectively as the wild type, whereas the ability of other mutants to rescue exocytosis seemed to be impaired (Fig. 7B). Because the P335A/Q336A has been shown to retain the ability to bind closed syntaxin-1 (Fig. 5), it is not surprising that this mutant is able to restore secretion. Although the other mutants reduced the ability to rescue exocytosis, they did not completely abolish it despite their strongly impaired ability to restore syntaxin-1 levels (Fig. 7, A and B). We suggest that some persistent rescuing activity of these mutants is likely due to the positive fusogenic effect of P335A on the priming activity of Munc18-1 as described above.
K332E/K333E Mutation with 4-Residue (EMPQ) Insertion, but Not with 3-Residue (MPQ) Insertion, Is Sufficient to Abolish the Priming Activity
By discovering the K332E/K333E/P335A/Q337A/Y337L quintuple mutant, we found that domain-3a of Munc18-1 also contributes to the Munc18-1-syntaxin-1a binary interaction (Fig. 2). This is very interesting as we have previously identified a mutant harboring K332E/K333E mutations with 5-residue (EEMPQ) insertion (KE/5I), which disrupts the priming function, whereas it retains the ability to bind closed syntaxin-1 and stabilize and traffic syntaxin-1 (11). Thus, introducing mutations to the similar region of domain-3a with the overlapping residues of Lys-332 and Lys-333 can cause strikingly different phenotypes. As the first step toward a better understanding of the structural determinant that causes such selective phenotype, we attempted to find the shortest insertion that is necessary to disrupt the priming function of Munc18-1. For this purpose, we reduced the insertion size from the previously identified 5 residues (EEMPQ) to 4 residues (EMPQ, KE/4I mutant) or 3 residues (MPQ, KE/3I mutant) and investigated their chaperoning and priming ability (Fig. 8A). We found that both of the mutants bind to monomeric syntaxin-1A (Fig. 8B) and restore the expression of syntaxin-1 (Fig. 8, C and D) as effectively as the wild type. Although these mutants contain insertions, the residues of Met-334, Pro-335, and Gln-336 are intact, which seems to enable the intact binding of these insertion mutants with syntaxin-1. Surprisingly, however, their ability to restore exocytosis varied; KE/4I abolishes the ability to restore exocytosis, whereas KE/3I restores exocytosis with the efficiency of 60% of the wild type (Fig. 8, E and F). Thus, the insertion of 4 residues, EMPQ, in presence of the K332E/K333E mutations in domain-3a is necessary to disrupt the priming action of Munc18-1.
FIGURE 8.

The K332E/K333E mutation with 4-residue (EMPQ) insertion (KE/4I), but not 3-residue (MPQ) insertion (KE/3I), is sufficient to abolish the priming activity. A, sequence alignment of the KE/4I and KE/3I mutants that contains insertion of 4 or 3 residues in the presence of K332E/K333E mutations. Mutated residues are shown in red. M18-1, Munc18-1. R., rat; D, Drosophila; C, C. elegans. B, the binding between Munc18-1 domain-3a insertion mutants (KE/4I and KE/3I) and syntaxin-1 was analyzed by yeast two-hybrid assays. In this assay, β-galactosidase activities of the transformed yeast clones were quantified and normalized so that the activity of the yeast clones transformed with the wild-type Munc18-1 (M18WT) was set to 100%. Error bars indicate S.E. (n = 10–14). Note that both mutants retain binding to syntaxin-1. C and D, stable re-expression of Munc18-1 KE/4I or KE/3I mutant in D16 (C) or D7 (D) clone restores syntaxin-1 levels. E and F, secretion defects are not rescued upon reintroduction of the KE/4I-EmGFP mutant, whereas they are restored to some degree upon re-expression of the KE/3I-EmGFP mutant in Munc18-1/-2 double knockdown clones D16 (E) or D7 (F). NA release was stimulated by 70 mm KCl for 15 min in the rescued cells. Error bars indicate S.E. (n = 3 for KE/3I D7, n = 9 for all other D7; n = 6–9 for D16).
DISCUSSION
In the present study, we discovered the first chaperoning mutant with mutations in domain-3a of Munc18-1. Our results clearly indicate that the binding between closed syntaxin-1A and Munc18-1 requires the concerted actions of domain-1 and domain-3a. The identified mutant harbors simultaneous quintuple mutations of K332E/K333E/P335A/Q336A/Y337L (Fig. 2). Domain-3a mediates the binding of Munc18-1 to syntaxin-1A and was found to be essential for the maintenance of syntaxin-1 levels, its trafficking, and its exocytosis (Figs. 2 and 3). In contrast to the quintuple mutant, the quadruple mutant sparing P335A does not impair the binary interaction and the chaperoning activity (Fig. 4), indicating the crucial role of Pro-335 residue in these processes. Although the P335A mutation alone does not strongly impair binding with closed syntaxin-1A, in combinations with the mutations in neighboring residues, it destabilizes the binary interaction, highlighting the contribution of the Pro-335 residue in this interaction (Figs. 5–7). This effect could be due to the loss of the direct contact with the residue His-213 of syntaxin-1 upon mutating Pro-335 (Fig. 1) (31). Alternatively, impairment in the structural flexibility of domain-3a, which may underlie the ability of Munc18-1 to adopt different binding modes, occurs upon the Pro-335 mutation (29). Therefore, further research needs to be conducted to elucidate the mechanisms that destabilize the interaction between the P335A mutant and syntaxin-1.
We also observed the unexpected increase in rescuing exocytosis upon expression of the P335A mutant despite its inefficient expression and impaired ability to rescue syntaxin-1 levels (Fig. 6). This strongly suggests that P335A mutation enhances the priming or exocytotic activity of Munc18-1. Indeed, very recent results using liposome fusion assays indicated that this mutant strikingly increases the ability of Munc18-1 to stimulate liposome fusion (17). Thus, Pro-335 seems to play a pivotal role in the transition of the dual functions of Munc18-1 from the chaperoning of syntaxin-1 to the priming of SNARE-mediated membrane fusion. We hypothesize that the flexible α-helical structure containing Pro-335 (29) has to be hinged to maintain the folding of this domain-3a and thus subsequent tight binding with closed syntaxin-1A, and to perform syntaxin-1A chaperoning activity (Fig. 9). Then, during the later stage of exocytosis, the flexible α-helical structure of Munc18-1 extends to release monomeric syntaxin-1 and binds to the assembled SNARE complex to prime exocytosis. We speculate that the P335A mutation destabilizes the binding to syntaxin-1, whereas it facilitates the binding to synaptobrevin-2 within the SNARE complex or another region in the four-helical bundle of the SNARE complex by favoring the extended conformation of Munc18-1. As a result, it facilitates the priming activity. However, a biochemical study that examined the binary interaction between Munc18-1 and synaptobrevin-2 does not detect an increase in the binding by the P335A mutation (17). Therefore, further research needs to be conducted to elucidate the biochemical mechanisms that underlie how the P335A mutation facilitates priming and/or exocytosis. Testing the ability of the P335A mutant to restore synaptic exocytosis of Munc18-1 null neurons would also be necessary before concluding that it is truly a gain-of-function mutant in priming exocytosis.
FIGURE 9.
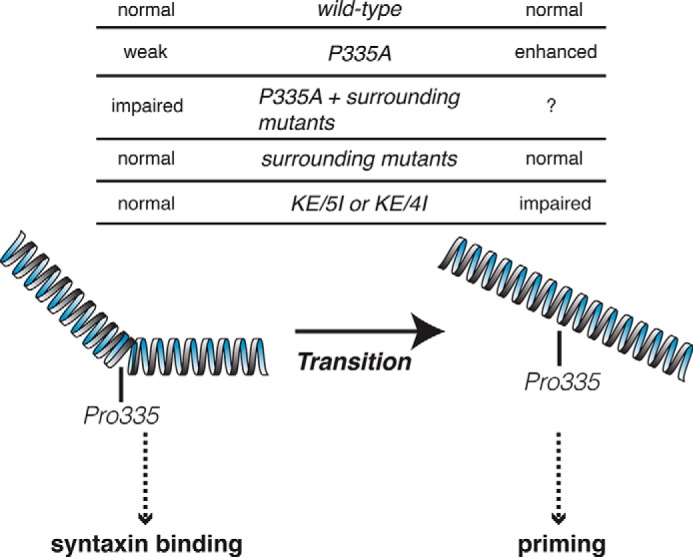
A proposed model in which Pro-335 in domain-3a of Munc18-1 regulates the balance of dual functions. The flexible α-helical structure containing Pro-335 has to be hinged to maintain the folding of this protein and tight binding with closed syntaxin-1A and to perform syntaxin-1A chaperoning activity (left). Then, during the later stage of exocytosis or priming, it extends to release the monomeric syntaxin-1 and to bind to the assembled SNARE complex to prime exocytosis (right). P335A mutation destabilizes the binding to syntaxin-1, whereas it facilitates priming. P335A with mutations in surrounding residues abolish the binding with syntaxin-1, impairing the chaperoning function. In contrast, the insertion mutants (KE/5I or KE/4I) retain intact Pro-335 and Gln-336, which allow the tight binding to closed syntaxin-1A. However, due to the insertion, the conformational change, namely extension of α-helix, could be impaired, resulting in impaired priming.
Surprisingly, the priming-defective mutants that we previously found and confirmed in this study (Fig. 8) contain the mutations of K332E/K333E (11), which are common with the chaperoning mutants. Unlike the additional point mutations P335A/Q336A/Y337L that the chaperone-defective mutant harbors, the priming mutants have an insertion of 4 (EMPQ) or 5 (EEMPQ) residues in the α-helical region. We would like to provide the following hypothesis to explain why either insertion causes a priming defect instead of a chaperoning defect (Fig. 9). Our insertion mutants retain intact Pro-335 and Gln-336, which allow the tight binding to closed syntaxin-1A; however, due to the insertion of 4 residues (EMPQ) together with K332E/K333E mutations, the conformational change, namely the extension of α-helix, could be impaired. This impairment hinders the interaction with synaptobrevin-2 or the SNARE complex (11). Independent of whether this hypothesis is correct or incorrect, the fact that mutations in overlapping regions in domain-3a (Lys-332 and Lys-333 residues) have selective effects on the priming and chaperoning functions of Munc18-1 suggests that these two functions may be more strongly coupled than previously recognized. Future structural study that incorporates the mutants we have identified here will provide new insights into how Munc18-1 domain-3a contributes to both syntaxin-1 chaperoning and priming exocytosis and how these dual functions are coupled or balanced.
Acknowledgment
We thank Dr. H. Gaisano for the reagents used in this study.
This research was supported by the Natural Sciences and Engineering Research Council of Canada (Grant 298461-09), the Heart and Stroke Foundation (Grants NA6217 and T6700), and the Canadian Institute of Health Research (Grants MOP-93665 and 130573).
The following mutant designations are used throughout: KE/5I, K332E/K333E mutations with 5-residue (EEMPQ) insertion; KE/4I, K332E/K333E mutations with 4-residue (EMPQ) insertion; KE/3I, K332E/K333E mutations with 3-residue (MPQ) insertion; KKQY, K332E/K333E/Q336A/Y337L multiple mutant; KKPQY, K332E/K333E/P335A/Q336A/Y337L multiple mutant.
- NA
- noradrenaline
- EmGFP
- emerald green fluorescent protein
- PSS
- physiological saline solution
- DKD
- double knockdown.
REFERENCES
- 1. Verhage M., Maia A. S., Plomp J. J., Brussaard A. B., Heeroma J. H., Vermeer H., Toonen R. F., Hammer R. E., van den Berg T. K., Missler M., Geuze H. J., Südhof T. C. (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287, 864–869 [DOI] [PubMed] [Google Scholar]
- 2. Voets T., Toonen R. F., Brian E. C., de Wit H., Moser T., Rettig J., Südhof T. C., Neher E., Verhage M. (2001) Munc18-1 promotes large dense-core vesicle docking. Neuron 31, 581–591 [DOI] [PubMed] [Google Scholar]
- 3. Harrison S. D., Broadie K., van de Goor J., Rubin G. M. (1994) Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron 13, 555–566 [DOI] [PubMed] [Google Scholar]
- 4. Hosono R., Hekimi S., Kamiya Y., Sassa T., Murakami S., Nishiwaki K., Miwa J., Taketo A., Kodaira K. I. (1992) The unc-18 gene encodes a novel protein affecting the kinetics of acetylcholine metabolism in the nematode Caenorhabditis elegans. J. Neurochem. 58, 1517–1525 [DOI] [PubMed] [Google Scholar]
- 5. Meijer M., Burkhardt P., de Wit H., Toonen R. F., Fasshauer D., Verhage M. (2012) Munc18-1 mutations that strongly impair SNARE-complex binding support normal synaptic transmission. EMBO J. 31, 2156–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deák F., Xu Y., Chang W. P., Dulubova I., Khvotchev M., Liu X., Südhof T. C., Rizo J. (2009) Munc18-1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J. Cell Biol. 184, 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gulyás-Kovács A., de Wit H., Milosevic I., Kochubey O., Toonen R., Klingauf J., Verhage M., Sørensen J. (2007) Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J. Neurosci. 27, 8676–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han L., Jiang T., Han G. A., Malintan N. T., Xie L., Wang L., Tse F. W., Gaisano H. Y., Collins B. M., Meunier F. A., Sugita S. (2009) Rescue of Munc18-1 and -2 double knockdown reveals the essential functions of interaction between Munc18 and closed syntaxin in PC12 cells. Mol. Biol. Cell 20, 4962–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han G. A., Malintan N. T., Saw N. M., Li L., Han L., Meunier F. A., Collins B. M., Sugita S. (2011) Munc18-1 domain-1 controls vesicle docking and secretion by interacting with syntaxin-1 and chaperoning it to the plasma membrane. Mol. Biol. Cell 22, 4134–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malintan N. T., Nguyen T. H., Han L., Latham C. F., Osborne S. L., Wen P. J., Lim S. J., Sugita S., Collins B. M., Meunier F. A. (2009) Abrogating Munc18-1-SNARE complex interaction has limited impact on exocytosis in PC12 cells. J. Biol. Chem. 284, 21637–21646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han G. A., Bin N. R., Kang S. Y., Han L., Sugita S. (2013) Domain 3a of Munc18-1 plays a crucial role at the priming stage of exocytosis. J. Cell Sci. 126, 2361–2371 [DOI] [PubMed] [Google Scholar]
- 12. Martin S., Tomatis V. M., Papadopulos A., Christie M. P., Malintan N. T., Gormal R. S., Sugita S., Martin J. L., Collins B. M., Meunier F. A. (2013) The Munc18-1 domain 3a loop is essential for neuroexocytosis but not for syntaxin-1A transport to the plasma membrane. J. Cell Sci. 126, 2353–2360 [DOI] [PubMed] [Google Scholar]
- 13. Shen J., Tareste D. C., Paumet F., Rothman J. E., Melia T. J. (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128, 183–195 [DOI] [PubMed] [Google Scholar]
- 14. Tareste D., Shen J., Melia T. J., Rothman J. E. (2008) SNAREpin/Munc18 promotes adhesion and fusion of large vesicles to giant membranes. Proc. Natl. Acad. Sci. U.S.A. 105, 2380–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rathore S. S., Bend E. G., Yu H., Hammarlund M., Jorgensen E. M., Shen J. (2010) Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc. Natl. Acad. Sci. U.S.A. 107, 22399–22406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen J., Rathore S. S., Khandan L., Rothman J. E. (2010) SNARE bundle and syntaxin N-peptide constitute a minimal complement for Munc18-1 activation of membrane fusion. J. Cell Biol. 190, 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parisotto D., Pfau M., Scheutzow A., Wild K., Mayer M. P., Malsam J., Sinning I., Söllner T. H. (2014) An extended helical conformation in domain 3a of Munc18-1 provides a template for SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex assembly. J. Biol. Chem. 289, 9639–9650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Han G. A., Malintan N. T., Collins B. M., Meunier F. A., Sugita S. (2010) Munc18-1 as a key regulator of neurosecretion. J. Neurochem. 115, 10.1111/j.1471-4159.2010.06900.x [DOI] [PubMed] [Google Scholar]
- 19. Rowe J., Corradi N., Malosio M. L., Taverna E., Halban P., Meldolesi J., Rosa P. (1999) Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J. Cell Sci. 112, 1865–1877 [DOI] [PubMed] [Google Scholar]
- 20. Rowe J., Calegari F., Taverna E., Longhi R., Rosa P. (2001) Syntaxin 1A is delivered to the apical and basolateral domains of epithelial cells: the role of munc-18 proteins. J. Cell Sci. 114, 3323–3332 [DOI] [PubMed] [Google Scholar]
- 21. Medine C. N., Rickman C., Chamberlain L. H., Duncan R. R. (2007) Munc18-1 prevents the formation of ectopic SNARE complexes in living cells. J. Cell Sci. 120, 4407–4415 [DOI] [PubMed] [Google Scholar]
- 22. Arunachalam L., Han L., Tassew N. G., He Y., Wang L., Xie L., Fujita Y., Kwan E., Davletov B., Monnier P. P., Gaisano H. Y., Sugita S. (2008) Munc18-1 is critical for plasma membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Mol. Biol. Cell 19, 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McEwen J. M., Kaplan J. M. (2008) UNC-18 promotes both the anterograde trafficking and synaptic function of syntaxin. Mol. Biol. Cell 19, 3836–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rodkey T. L., Liu S., Barry M., McNew J. A. (2008) Munc18a scaffolds SNARE assembly to promote membrane fusion. Mol. Biol. Cell 19, 5422–5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Südhof T. C., Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dulubova I., Khvotchev M., Liu S., Huryeva I., Südhof T. C., Rizo J. (2007) Munc18-1 binds directly to the neuronal SNARE complex. Proc. Natl. Acad. Sci. U.S.A. 104, 2697–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rickman C., Medine C. N., Bergmann A., Duncan R. R. (2007) Functionally and spatially distinct modes of Munc18-syntaxin 1 interaction. J. Biol. Chem. 282, 12097–12103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu S. H., Latham C. F., Gee C. L., James D. E., Martin J. L. (2007) Structure of the Munc18c/Syntaxin4 N-peptide complex defines universal features of the N-peptide binding mode of Sec1/Munc18 proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 8773–8778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu S. H., Christie M. P., Saez N. J., Latham C. F., Jarrott R., Lua L. H., Collins B. M., Martin J. L. (2011) Possible roles for Munc18-1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc. Natl. Acad. Sci. U.S.A. 108, 1040–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Y., Su L., Rizo J. (2010) Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry 49, 1568–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Misura K. M., Scheller R. H., Weis W. I. (2000) Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 404, 355–362 [DOI] [PubMed] [Google Scholar]
- 32. Barnstable C. J., Hofstein R., Akagawa K. (1985) A marker of early amacrine cell development in rat retina. Brain Res. 352, 286–290 [DOI] [PubMed] [Google Scholar]
- 33. Vojtek A. B., Hollenberg S. M., Cooper J. A. (1993) Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74, 205–214 [DOI] [PubMed] [Google Scholar]
- 34. Schiestl R. H., Gietz R. D. (1989) High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet. 16, 339–346 [DOI] [PubMed] [Google Scholar]
- 35. Dulubova I., Sugita S., Hill S., Hosaka M., Fernandez I., Südhof T. C., Rizo J. (1999) A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 18, 4372–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Okamoto M., Südhof T. C. (1997) Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J. Biol. Chem. 272, 31459–31464 [DOI] [PubMed] [Google Scholar]