

Background: The unfolded A1 subunit of cholera toxin (CT) enters the host cytosol by passing through a pore in the endoplasmic reticulum (ER) membrane.
Results: ATP-dependent refolding of CTA1 by Hsp90 is sufficient for toxin export to the cytosol.
Conclusion: Hsp90 couples CTA1 refolding with CTA1 extraction from the ER.
Significance: This work provides a molecular basis for toxin translocation into the host cytosol.
Keywords: Cholera Toxin, Fourier Transform IR (FTIR), Heat Shock Protein 90 (Hsp90), Host-pathogen Interaction, Protein Translocation, Surface Plasmon Resonance (SPR)
Abstract
Cholera toxin (CT) moves from the cell surface to the endoplasmic reticulum (ER) where the catalytic CTA1 subunit separates from the rest of the toxin. CTA1 then unfolds and passes through an ER translocon pore to reach its cytosolic target. Due to its intrinsic instability, cytosolic CTA1 must be refolded to achieve an active conformation. The cytosolic chaperone Hsp90 is involved with the ER to cytosol export of CTA1, but the mechanistic role of Hsp90 in CTA1 translocation remains unknown. Moreover, potential post-translocation roles for Hsp90 in modulating the activity of cytosolic CTA1 have not been explored. Here, we show by isotope-edited Fourier transform infrared spectroscopy that Hsp90 induces a gain-of-structure in disordered CTA1 at physiological temperature. Only the ATP-bound form of Hsp90 interacts with disordered CTA1, and refolding of CTA1 by Hsp90 is dependent upon ATP hydrolysis. In vitro reconstitution of the CTA1 translocation event likewise required ATP hydrolysis by Hsp90. Surface plasmon resonance experiments found that Hsp90 does not release CTA1, even after ATP hydrolysis and the return of CTA1 to a folded conformation. The interaction with Hsp90 allows disordered CTA1 to attain an active state, which is further enhanced by ADP-ribosylation factor 6, a host cofactor for CTA1. Our data indicate CTA1 translocation involves a process that couples the Hsp90-mediated refolding of CTA1 with CTA1 extraction from the ER. The molecular basis for toxin translocation elucidated in this study may also apply to several ADP-ribosylating toxins that move from the endosomes to the cytosol in an Hsp90-dependent process.
Introduction
Cholera toxin (CT)3 is an AB5 toxin containing two distinct subunits. The A subunit consists of two domains, A1 and A2, linked by a disulfide bond. The A1 domain is responsible for catalytic activity, and the A2 domain acts as a tether between the A1 domain and the B subunit. The homopentameric B subunit is a highly stable ring-like structure with a central pore that interacts with the A2 chain and contains binding sites for GM1 gangliosides on the plasma membrane of the host cell (1, 2). Binding to GM1 triggers toxin endocytosis from the cell surface and subsequent toxin delivery to the endoplasmic reticulum (ER) via retrograde vesicular transport (3). Reduction of the disulfide bond between the A1 and A2 domains occurs in the ER (4, 5). Reduced CTA1 is released from its non-covalent association with the holotoxin by protein-disulfide isomerase (PDI) (6–8). The isolated CTA1 domain then enters the cytosol and interacts with host ADP-ribosylation factors (ARFs) to constitutively activate the G protein stimulatory α-subunit (Gαs) through ADP-ribosylation (9–11). Activation of Gαs increases adenylate cyclase activity, leading to a massive increase in the amount of cAMP produced. The increased level of cAMP stimulates Cl− release and inhibits Na+ absorption in intestinal epithelial cells, causing an efflux of water that generates the profuse watery diarrhea associated with cholera (12).
The isolated CTA1 polypeptide is an unstable protein that assumes a disordered conformation at physiological temperature (13). The conformational instability of free CTA1 affects many steps of the intoxication process (14, 15). For example, CTA1 spontaneously unfolds after its PDI-mediated release from the holotoxin in the ER (6). Unfolded CTA1 is then exported to the cytosol through the ER-associated degradation (ERAD) pathway (16–20). The ER to cytosol translocation of unfolded proteins by ERAD is a normal process that prevents the accumulation of protein aggregates in the ER. Exported ERAD substrates move through protein-conducting channels in the ER membrane and are targeted for cytosolic degradation by the 26 S proteasome in a ubiquitin-dependent manner (21, 22). A number of AB toxins exploit this system for A chain passage into the cytosol (15, 23, 24). CTA1 and A chains from other ER-translocating toxins contain an arginine over lysine bias in their amino acid sequences. The lack of A chain lysine residues limits the number of ubiquitination sites, thereby inhibiting toxin degradation by the 26 S proteasome (24–28). CTA1 is still subject to ubiquitin-independent degradation by the core 20 S proteasome (13), a variant of the proteasome that can only degrade unfolded proteins (29). The relatively long half-life (t½ between 1 and 2 h) for cytosolic CTA1 suggests a stabilizing interaction with host factors may protect CTA1 from rapid, ubiquitin-independent proteasomal degradation (13).
CTA1 instability also plays a role in toxin extraction from the ER to the cytosol. An early model of toxin translocation proposed CTA1 spontaneously refolded as it emerged at the cytosolic face of the ER translocon pore (27). This would prevent back-sliding into the pore and would thus provide an ER to cytosol directionality to the translocation process. Yet the subsequent discovery of CTA1 conformational instability indicated the toxin could not independently refold at physiological temperature and therefore required a host factor(s) for extraction to the cytosol (13, 30). Although most ERAD export is mediated by the AAA ATPase p97 (31, 32), CTA1 translocation does not require this cytosolic protein (33, 34). The ER to cytosol translocation of unfolded CTA1 is instead facilitated by heat shock protein 90 (Hsp90), a cytosolic ATP-dependent chaperone that plays a role in multiple cellular events including protein folding, protein stabilization, and refolding of denatured proteins (35, 36). Loss of Hsp90 function trapped CTA1 in the ER (37). This was the first demonstration for Hsp90-dependent export of a soluble protein from the ER.
CTA1 passes through one or more ER translocon pores (38–42) in an unfolded state and must achieve an active conformation in the cytosol to modify its G protein target in the lipid rafts of the plasma membrane (43, 44). Refolding will not occur spontaneously because CTA1 is an unstable protein. In fact, the isolated CTA1 polypeptide has little to no enzymatic activity at 37 °C (45, 46). An interaction with ARF proteins will enhance the activity of folded CTA1 and is required for productive intoxication of cultured cells, but ARF alone cannot induce a gain-of-structure or gain-of-function in disordered CTA1 (47). Other host factors must therefore place cytosolic CTA1 in a folded conformation that can be further activated by ARF proteins. Lipid rafts were recently shown to exhibit a “lipochaperone” property that places disordered CTA1 in a folded, functional state at physiological temperature. Furthermore, lipid rafts are essential for the in vivo activity of cytosolic CTA1 (46). Other host factors that could refold and/or activate cytosolic CTA1 have yet to be identified. The high affinity interaction between CTA1 and Hsp90 (37), combined with the established chaperone activity of Hsp90, suggests Hsp90 could be another host factor linked to the cytosolic activity of CTA1.
To generate molecular detail regarding the co- and post-translocation roles of Hsp90 in CT intoxication, we performed a structure/function analysis on the interaction between Hsp90 and CTA1. Biophysical measurements provided a mechanistic basis for Hsp90-mediated extraction of CTA1 from the ER by demonstrating that Hsp90 can convert disordered CTA1 to a structured conformation. ATP hydrolysis was required for Hsp90 to refold CTA1 and to maintain a high-affinity interaction with the refolded toxin. ATP hydrolysis by Hsp90 was also required for the ER to cytosol export of CTA1. The Hsp90-mediated refolding of CTA1 thus appears to provide the driving force for toxin extraction from the ER: refolded CTA1 could not slide back into the translocon pore, thereby resulting in a unidirectional ER to cytosol export. Hsp90 remains associated with the refolded toxin and allows ARF6 to simulate the activity of CTA1 at 37 °C. These studies have elucidated a new, Hsp90-driven mechanism for toxin translocation that may apply to a broad range of ADP-ribosylating toxins.
EXPERIMENTAL PROCEDURES
Materials
CTA, ATP, GTP, geldanamycin, protein A-Sepharose, and antibodies to CTA and CT were purchased from Sigma. Antibodies against Hsp90 were purchased from Calbiochem (Darmstadt, Germany). The ARF6 antibody was from Santa Cruz Biotechnology Inc. (Dallas, TX). ATPγS was purchased from Enzo (Farmingdale, NY). AG50W-4X beads were purchased from Bio-Rad. Cell culture reagents including Lipofectamine were purchased from Invitrogen. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). Phosphate-buffered saline with 0.05% Tween 20 (PBST) tablets were purchased from Medicago (Research Triangle Park, NC). 35S-Labeled methionine was purchased from PerkinElmer Life Sciences (Waltham, MA). Hsp90 was purchased from Biovision (Milpitas, CA). Diethylamino(benzylidine-amino)guanidine (DEA-BAG) was synthesized as previously described (48). Uniformly 13C-labeled CTA1-His6 was produced as described in Ref. 6 and purified after an 18-h isopropyl 1-thio-β-d-galactopyranoside induction at 18 °C as described in Ref. 17. ARF6 was generated as previously described (49). Large unilamellar vesicles (LUVs) mimicking the composition of the plasma membrane or lipid rafts were generated as previously described (46).
Isotope-edited Fourier Transform Infrared (FTIR) Spectroscopy
Samples for FTIR measurement were prepared in D2O-based 10 mm sodium borate buffer (pD 7.0) containing 100 mm NaCl as previously described (6). 13C-Labeled CTA1 was used at 10 μm concentration, and a 2:1 molar ratio of Hsp90:CTA1 was used for all measurements involving both proteins. ATP and ATPγS, when present, were used at 1 mm concentration. Measurements were performed on a Jasco 4200 FTIR spectrometer (Easton, MD). As detailed in Ref. 50, amide I components at the following wavenumbers were assigned to specific secondary structures for 13C-labeled CTA1 in a D2O-based buffer: 1604 ± 4 cm−1, α-helix; 1577 ± 7 cm−1, β-sheet; 1593 ± 5 cm−1, irregular. Components around 1614 ± 4 cm−1 were assigned to turns and tabulated as “other” structures. Analysis of CTA1 secondary structure was performed as described in detail in the supplemental material of Ref. 6.
In Vitro ADP-ribosylation Assay
To monitor the catalytic activity of CTA1, the stated concentrations of CTA1-His6 were placed in a 200-μl assay buffer containing 200 mm potassium phosphate buffer (pH 7.5), 20 mm DTT, and 0.1 mg/ml of BSA. Hsp90 at a 2:1 molar ratio to CTA1, equimolar ARF6, and/or 800 μm lipid raft LUVs were also present as indicated, as were 1 mm ATP, ATPγS, or GTP. CTA1 samples were incubated for 30 min at 25 or 37 °C, followed by the addition of the indicated components for a further 1-h incubation at the indicated temperature. The ADP-ribosylation reaction was initiated by addition of 0.4 mg/ml of DEA-BAG and 25 μl of 10 mm NAD. Samples were then incubated at the indicated temperature for 2 h, during which CTA1 (if active) was able to ADP-ribosylate the DEA-BAG substrate. The reaction was stopped by the addition of 800 μl of 30% AG50W-4X bead slurry. The samples were vortexed for 30 s, followed by centrifugation at maximum speed for 10 min. ADP-ribosylation of DEA-BAG inhibits its ability to bind to AG50W-4X beads, so pelleting the resin leaves modified DEA-BAG in the supernatant. 400 μl of supernatant was removed, and the intrinsic fluorescence of DEA-BAG (excitation 361 nm, emission 440 nm) was read with a Bio-Tek (Winooski, VT) Synergy 2 plate reader.
Surface Plasmon Resonance (SPR)
Experiments were performed with a Reichert (Depew, NY) SR7000 SPR refractometer using the same basic protocol outlined in Ref. 52. After establishment of a baseline measurement (0 refractive index unit; RIU) corresponding to the mass of the sensor-bound CTA1, Hsp90, ARF6, or other ligands were perfused over the CTA1-coated sensor slide. The protein concentration was 1600 ng/ml in PBST. A 5-min perfusion of ligand was followed by a 5-min PBST wash. The flow rate for all steps was 41 μl/min. Reichert Labview software was used for data collection. The BioLogic (Campbell, Australia) Scrubber 2 software and WaveMetrics (Lake Oswego, OR) Igor Pro software were used to analyze the data and generate figures.
In Vitro Translocation Assay
CHO cells were seeded to 6-well plates in Ham's F-12 media supplemented with 10% FBS and incubated overnight at 37 °C with 5% CO2 to reach 60–70% confluence. Cells were washed twice with F-12 media, and 1 ml of 100 ng/ml of CT was added to each well. Cells were then incubated 2 h at 37 °C with 5% CO2. Cells were then washed twice with PBS, and 0.5 ml of PBS with 0.5 mm EDTA was added for a 5–10-min incubation at room temperature. 3 wells of cells per condition were then collected in a microcentrifuge tube and centrifuged at 2,400 × g for 2 min. PBS was removed, and cells and digitonin buffer (0.04% digitonin in HCN buffer with 10 μl/ml of protease inhibitor mixture) were allowed to incubate separately on ice for 10 min. Following this pre-incubation, 100 μl of digitonin buffer was added to each sample for 10 min. The samples were then centrifuged at 16,200 × g for 10 min. The supernatant, containing cytosolic components, and pellet, containing membrane bound components, were separated for further processing. Pelleted membranes were incubated with 800 mm NaCl or 2 m urea in PBS for 30 min at 4 °C to remove membrane-associated proteins. The pellet was then washed twice with PBS before purified Hsp90, ATP, Hsp90/ATP, or Hsp90/ATPγS in HCN buffer was added for a 1-h incubation at 37 °C. After a 5-min spin at 16,200 × g, the supernatant was collected and perfused over an SPR sensor slide coated with an anti-CTA antibody.
RESULTS
Hsp90 Refolds Disordered CTA1 in a Process Requiring ATP Hydrolysis
We performed isotope-edited FTIR spectroscopy to examine the effect of Hsp90 on the folding state of CTA1 (Fig. 1). Uniformly 13C-labeled CTA1 was used to differentiate between the spectra of CTA1 and unlabeled Hsp90. An ∼50 cm−1 shift in the spectra of the 13C-labeled protein allows it to be resolved from the spectra of the unlabeled protein, thus facilitating analysis of the individual proteins (6, 50, 53). The spectra of CTA1 alone (Fig. 1A) showed that, at 10 °C, CTA1 is in a folded conformation with ∼34% α-helical, 42% β-sheet, and 17% irregular (i.e. undefined) structure (Table 1). The amount of secondary and irregular structures in CTA1 at 10 °C was consistent with previous FTIR measurements of folded CTA1 (6) and the structural content of holotoxin-associated CTA1 as determined by x-ray crystallography (2, 54). As expected from the intrinsic instability of CTA1, increasing the temperature to 37 °C caused CTA1 to unfold: it lost at least half of its initial α-helical and β-sheet content, whereas the amount of irregular structure increased to 54% (Fig. 1B, Table 1). In the absence of ATP, Hsp90 cannot bind to CTA1 (37) and accordingly had no appreciable effect on the structure of CTA1 when added to the unfolded toxin at 37 °C (Fig. 1C, Table 1). However, the addition of Hsp90/ATP to unfolded CTA1 at 37 °C produced a substantial increase in both the α-helical and β-sheet content of CTA1 with a corresponding loss of irregular structure (Fig. 1D, Table 1). A 2:1 molar ratio of Hsp90:CTA1 was used for all studies because Hsp90 functions as a dimer. Exposure of unfolded CTA1 to Hsp90/ATP did not fully restore the toxin to its native conformation, but CTA1 did gain 11% α-helical content and 18% β-sheet content while losing 26% irregular structure. Hsp90/ATP thus induces a gain-of-structure in the disordered 37 °C conformation of CTA1. In contrast, Hsp90/ATPγS did not substantially alter the structure of disordered CTA1 (Fig. 1E, Table 1). ATPγS is a non-hydrolyzable form of ATP. Thus, the chaperone-driven refolding of disordered CTA1 required ATP hydrolysis by Hsp90. Control experiments using FTIR spectroscopy demonstrated that ATP and ATPγS had no direct effect on the structure of CTA1 (data not shown). Additional control experiments using SPR confirmed that Hsp90/ATPγS could, like Hsp90/ATP, bind to CTA1 at physiological temperature (data not shown).
FIGURE 1.

Hsp90/ATP induces a gain-of-structure in disordered CTA1. The FTIR spectrum of 13C-labeled CTA1 was recorded in the absence or presence of Hsp90. In the curve-fitting panels of the left columns, the dotted line represents the sum of all deconvoluted components (solid lines) from the measured spectrum (dashed line). The right columns present the respective second derivatives. CTA1 samples incubated with Hsp90 were first heated to 37 °C for 30 min prior to addition of a 2-fold molar excess of Hsp90. A, CTA1 structure at 10 °C. B, CTA1 structure at 37 °C. C, CTA1 structure in the presence of Hsp90 at 37 °C. D, CTA1 structure in the presence of Hsp90/ATP at 37 °C. E, CTA1 structure in the presence of Hsp90/ATPγS at 37 °C.
TABLE 1.
ATP-driven refolding of CTA1 by Hsp90
Deconvolution of amide I bands from the FTIR spectroscopy data of Fig. 1 was used to calculate the percentages of CTA1 structure under the stated conditions. The averages ± S.D. from three to four individual curve fittings are shown.
| Condition | % of CTA1 structure |
|||
|---|---|---|---|---|
| α-Helix | β-Sheet | Irregular | Other | |
| 10 °C | 34 ± 2 | 42 ± 2 | 17 ± 2 | 8 ± 0 |
| 37 °C | 17 ± 1 | 15 ± 2 | 54 ± 2 | 14 ± 1 |
| 37 °C + Hsp90 | 20 ± 1 | 19 ± 3 | 52 ± 3 | 9 ± 1 |
| 37 °C + Hsp90/ATP | 28 ± 1 | 33 ± 2 | 28 ± 3 | 11 ± 1 |
| 37 °C + Hsp90/ATPγS | 16 ± 1 | 12 ± 2 | 52 ± 2 | 19 ± 2 |
ATP Hydrolysis by Hsp90 Is Required for CTA1 Extraction from the ER
To establish a molecular basis for the role of Hsp90 in CTA1 translocation, we monitored the ER to cytosol export of CTA1 with a series of in vitro reconstitution experiments. CHO cells were exposed to CT for 2 h before selective permeabilization of the plasma membrane with digitonin. Distinct cytosolic and intact endomembrane fractions were then generated by centrifugation. The pelleted membranes were washed with 800 mm salt to remove any peripheral membrane-associated proteins. The minor pool of Hsp90, which is consistently detected in association with the membrane fraction of digitonin-permeabilized cells (17, 37, 55), was removed by this wash (Fig. 2A). Pelleted membranes were then washed twice with PBS before purified Hsp90, ATP, Hsp90/ATP, or Hsp90/ATPγS was added to the membrane fraction for 1 h at 37 °C. After a 5-min spin at 16,200 × g, the new supernatant from the membrane fraction was collected and perfused over a SPR sensor coated with an anti-CTA1 antibody. CTA1 translocation across the ER membrane would place it in the supernatant and would accordingly generate a positive SPR signal. Membrane fractions incubated with Hsp90 alone, ATP alone, or Hsp90/ATPγS did not produce a positive signal for CTA1 export (Fig. 2B). However, the addition of Hsp90/ATP promoted CTA1 translocation from the ER membrane and accordingly generated a positive SPR signal (Fig. 2B). Identical results were obtained when the pelleted membranes were washed with 2 m urea instead of 800 mm salt (Fig. 2, C and D). We also found the addition of ATP alone could support CTA1 translocation from ER membranes that had not been washed with salt or urea (Fig. 2D), which was consistent with the results from a previous in vitro CTA1 translocation assay (42). The inability of Hsp90/ATPγS to support CTA1 translocation demonstrated that ATP hydrolysis by Hsp90 was required for toxin export from the ER. Because ATP hydrolysis by Hsp90 is also required to refold CTA1, our collective observations support a model of toxin translocation in which Hsp90 couples CTA1 refolding with CTA1 extraction from the ER.
FIGURE 2.

ATP hydrolysis by Hsp90 is required for CTA1 extraction from the ER. CHO cells incubated with 100 ng/ml of CT for 2 h at 37 °C were treated with digitonin to selectively permeabilize the plasma membrane. Following centrifugation, the intact membrane pellet was collected for further study. A, Western blot analysis was used to detect Hsp90 and PDI in association with unwashed or salt-washed membrane pellets. Hsp90 and PDI protein standards (Std.) were used as controls. B, purified Hsp90, ATP, Hsp90/ATP, or Hsp90/ATPγS in HCN buffer was added to the salt-washed membrane pellet for 1 h at 37 °C. To detect the exported pool of CTA1, supernatant samples obtained after centrifugation were perfused over an SPR sensor coated with an anti-CTA1 antibody. C, Western blot analysis was used to detect Hsp90 and PDI in association with unwashed or urea-washed membrane pellets. Hsp90 and PDI protein standards (Std.) were used as controls. D, purified Hsp90, Hsp90/ATP, or Hsp90/ATPγS in HCN buffer was added to the urea-washed membrane pellet for 1 h at 37 °C. ATP alone was also added to a membrane pellet that had not been washed with urea or salt. To detect the exported pool of CTA1, supernatant samples obtained after centrifugation were perfused over an SPR sensor coated with an anti-CTA1 antibody.
Hsp90 Preferentially Binds to Disordered CTA1 but Is Not Released after CTA1 Refolding
A series of SPR experiments was performed to characterize the interaction between Hsp90/ATP and CTA1 (Fig. 3). Hsp90/ATP was perfused over a CTA1-coated sensor slide at 15 °C, a temperature that maintains CTA1 in a folded conformation (13). A minimal interaction between Hsp90 and CTA1 was recorded at this temperature (Fig. 3A, dotted line). The temperature was then increased to 37 °C to promote the unfolding of CTA1. As expected, Hsp90/ATP bound readily to CTA1 at 37 °C (Fig. 3A, solid line). Hsp90 thus appeared to specifically recognize the unfolded conformation of CTA1. In support of this interpretation, we found that Hsp90/ATP could bind to heat-denatured CTA1 at 15 °C (Fig. 3B). This demonstrated Hsp90/ATP is functional at low temperature, so the weak interaction between Hsp90/ATP and folded CTA1 at 15 °C could be attributed to the conformation of CTA1 rather than to the temperature of the experiment.
FIGURE 3.
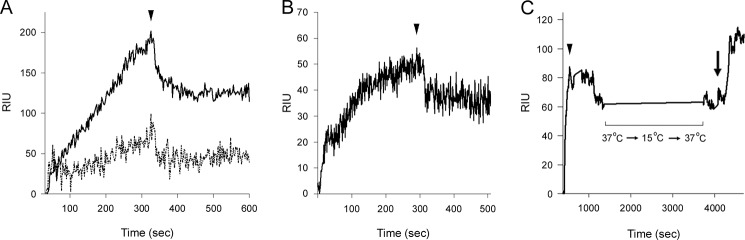
Hsp90/ATP binds unfolded CTA1 and is not released after CTA1 refolding. A, Hsp90/ATP was perfused over a CTA1-coated SPR sensor at 15 °C (dashed line) or 37 °C (solid line). The arrowhead denotes removal of Hsp90/ATP from the perfusion buffer. B, Hsp90/ATP was perfused at 15 °C over a SPR sensor coated with heat-denatured CTA1. The arrowhead denotes removal of Hsp90/ATP from the perfusion buffer. C, Hsp90/ATP was perfused over a CTA1-coated SPR sensor at 37 °C and was removed from the perfusion buffer after 300 s (arrowhead). This was followed by a stepwise temperature decrease to 15 °C, which was held for 10 min, before returning the temperature to 37 °C. Upon return to 37 °C, the continued association of Hsp90 with CTA1 was verified by the addition of an anti-Hsp90 antibody to the perfusion buffer (arrow). Note that the time scale for panel C is distinct from the time intervals in panels A and B.
Hsp90/ATP interacts specifically with disordered CTA1 (Figs. 3, A and B) and will refold the toxin in an ATP-dependent process (Fig. 1, Table 1), yet toxin refolding does not appear to result in the dissociation of Hsp90. To further examine this possibility, we perfused Hsp90/ATP over a CTA1-bound sensor slide at 37 °C (Fig. 3C). After Hsp90/ATP was removed from the perfusion buffer, the Hsp90-CTA1 complex was cooled to 15 °C in a stepwise manner. This process has previously been shown to promote the refolding of disordered CTA1 (13). The temperature was then increased back to 37 °C to allow direct comparison of the RIU signal before and after cooling (temperature has a direct effect on the RIU signal (56)). We recorded identical RIU signals before and after cooling, which indicated Hsp90 did not appreciably dissociate from the temperature-stabilized CTA1 subunit over the time course of the experiment. The continued presence of Hsp90 on the CTA1-coated sensor slide was verified with an anti-Hsp90 antibody that generated a positive signal when perfused over the sensor slide. Thus, neither the chaperone- nor temperature-induced refolding of CTA1 appeared to displace Hsp90 from its binding partner.
Additional SPR experiments were performed to see if specific host factors could promote the dissociation of Hsp90 from CTA1. For these experiments, Hsp90/ATP was perfused over a CTA1-coated sensor slide at 37 °C. Subsequent perfusions of purified Hsp90 cofactors, the 20S proteasome, 26 S proteasome, LUVs mimicking the composition of the plasma membrane or lipid rafts, or the cytosolic fraction from ∼3.6 × 106 cells failed to separate Hsp90 from CTA1 (Table 2). Although these in vitro studies may not fully replicate the in vivo condition, they suggest Hsp90 does not dissociate from CTA1 after extracting the toxin from the ER.
TABLE 2.
Host factors do not displace Hsp90 from CTA1
Hsp90/ATP was perfused over a CTA1-coated SPR sensor at 37 °C. After plateau of the resulting RIU signal, Hsp90 was removed from the perfusion buffer and replaced with one of the listed host factors for a 300-s perfusion. The continued association of Hsp90 with CTA1 was then confirmed with the addition of an anti-Hsp90 antibody to the perfusion buffer, which provided a positive signal in every case.
| Hsp90 cofactors | CTA1 cofactors | Other host factors |
|---|---|---|
| Hsp40 | ARF6 | Cytosolic extract |
| Hsc70 | Lipid raft LUVs | Plasma membrane LUVs |
| Hop | 20 S proteasome | |
| Aha | 26 S proteasome | |
| p23 |
Hsp90 Does Not Protect Cytosolic CTA1 from Degradation
The cytosolic pool of CTA1 is degraded by a ubiquitin-independent proteasomal mechanism with a half-life between 1 and 2 h (13, 49). Degradation likely involves processing by the core 20 S proteasome, which can only interact with disordered/unfolded substrates (13, 29). Thus, the Hsp90-mediated refolding of CTA1 could potentially protect the toxin from proteasomal degradation. Furthermore, Hsp90 can directly associate with the proteasome and thereby inhibit protein degradation (57, 58). To determine whether the continued association between Hsp90 and CTA1 had an effect on the in vivo turnover of CTA1, we used a plasmid-based system to express CTA1 directly in the host cytosol (59). The Hsp90 inhibitor geldanamycin was then used to inhibit the function of Hsp90 in CTA1-expressing cells. Exogenous application of the CT holotoxin could not be used for this experiment because geldanamycin blocks CTA1 translocation from the ER to the cytosol (37), thus preventing subsequent interactions between CTA1 and the proteasome. Plasmid-expressed CTA1 was pulse-labeled with [35S]methionine and chased for up to 4 h. The radiolabeled pool of CTA1 immunoprecipitated at 0, 0.5, 1, 2, or 4 h of chase was visualized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis with the Bio-Rad Personal Molecular Imager and quantified with the Bio-Rad Quantity One software. In both untreated and geldanamycin-treated cells, CTA1 exhibited a half-life between 1 and 2 h and was degraded with equivalent kinetics (data not shown). These observations indicate the association of CTA1 with Hsp90 does not affect the half-life of the toxin.
Hsp90/ATP Induces a Gain-of-function in Disordered CTA1
The Hsp90-mediated refolding of CTA1 could potentially place the toxin in a functional conformation at 37 °C. To determine the effect of Hsp90 on the activity of CTA1, we performed an in vitro toxicity assay using DEA-BAG as a substrate for the ADP-ribosyltransferase activity of CTA1 (Fig. 4). The fluorescent output from this assay is directly proportional to the amount of substrate modified by CTA1 (60). As shown in Fig. 4A, the folded conformation of CTA1 at 25 °C (circles) produced a concentration-dependent increase in the amount of modified DEA-BAG. CTA1 activity at 25 °C was slightly elevated in the presence of Hsp90/ATP (squares). When our ADP-ribosylation assay was repeated at 37 °C, the disordered conformation of CTA1 did not produce a concentration-dependent increase in the amount of modified DEA-BAG (Fig. 4B, circles). The baseline signal observed under this condition represents a background reading, as we have found CTA1 and heat-denatured CTA1 produce similar DEA-BAG responses at 37 °C (47). Addition of Hsp90 alone (triangles) or Hsp90/ATPγS (inverted triangles) to the disordered conformation of CTA1 at 37 °C did not restore enzymatic activity to the toxin (Fig. 4B). However, the addition of Hsp90/ATP to disordered CTA1 (squares) produced a dramatic, concentration-dependent increase in toxin activity. The gain-of-structure in disordered CTA1 resulting from its interaction with Hsp90/ATP thus generated a corresponding gain-of-function for the toxin ADP-ribosyltransferase activity. This gain-of-function was dependent upon ATP hydrolysis by Hsp90, which was consistent with the structural requirements for CTA1 refolding by Hsp90.
FIGURE 4.
Hsp90/ATP increases the in vitro ADP-ribosylation activity of CTA1. 2-Fold dilutions of CTA1 were incubated with DEA-BAG, a substrate for ADP-ribosylation, in the absence or presence of 2-fold molar excess Hsp90. Substrate modification by CTA1 results in a fluorescent signal that is proportional to the level of toxin activity. A, CTA1 was incubated in the absence or presence of Hsp90/ATP at 25 °C, a temperature that maintains the toxin in a folded conformation. B, CTA1 was placed in a disordered conformation by incubation at 37 °C for 30 min. Hsp90, Hsp90/ATP, or Hsp90/ATPγS was then added for a 1-h incubation at 37 °C before initiating the ADP-ribosylation reaction with the addition of NAD and DEA-BAG. Error bars indicate S.E. of 12–16 replications from 4 independent experiments.
ARF6/GTP Can Bind and Activate the Hsp90-associated CTA1 Polypeptide
The GTP-bound form of ARF is an allosteric activator of CTA1 (10, 11), and in vivo CT intoxication requires an interaction between ARF and CTA1 (47). To determine whether ARF6/GTP could associate with the CTA1-Hsp90 complex, sequential additions of Hsp90/ATP and ARF6/GTP were perfused over a CTA1-bound SPR sensor slide (Fig. 5A). As indicated in the sensorgram, the presence of Hsp90/ATP did not interfere with the binding of ARF6/GTP. The stable association of Hsp90 and ARF6 with CTA1 was verified by the capture of anti-Hsp90 and anti-ARF6 antibodies on the sensor slide at the end of the experiment (data not shown). We then performed an in vitro toxicity assay to determine whether ARF6/GTP could enhance the enzymatic activity of Hsp90-associated CTA1 (Fig. 5B). We found that ARF6 stimulation of CTA1 activity was not inhibited by the presence of Hsp90. Rather, the addition of both ARF6/GTP and Hsp90/ATP enhanced the activity of CTA1 (Fig. 5B, triangles) above that obtained with Hsp90/ATP alone (Fig. 5B, squares). Consistent with our previous observations (47), ARF6 alone could not induce a gain-of-activity in the disordered conformation of CTA1 (Fig. 5B, circles). These collective observations indicate optimal toxin activity involves contributions from both Hsp90 and ARF6: Hsp90 refolds CTA1 to a stable conformation, which is then followed by ARF6 activation.
FIGURE 5.
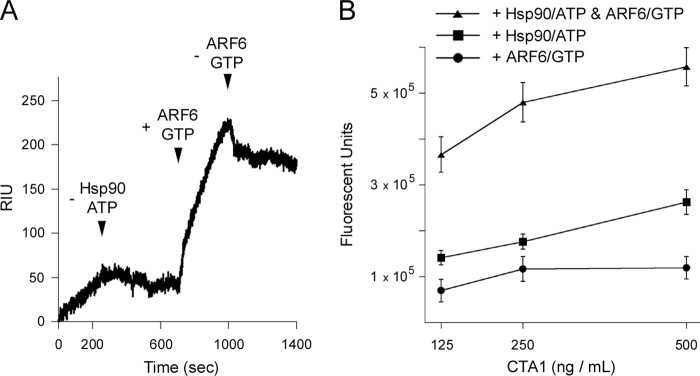
Hsp90/ATP does not affect the ability of ARF6/GTP to bind and activate CTA1. A, Hsp90/ATP was perfused over a CTA1-coated SPR sensor at 37 °C and was removed from perfusion buffer as indicated by the first arrowhead. ARF6/GTP was subsequently perfused over the CTA1-Hsp90 complex (indicated by the second arrowhead) and was removed from the perfusion buffer as denoted by the third arrowhead. B, 2-fold dilutions of CTA1 were incubated at 37 °C for 30 min before the addition of ARF6/GTP, Hsp90/ATP, or both Hsp90/ATP and ARF6/GTP. After a 1-h incubation at 37 °C, NAD and DEA-BAG were added to initiate the ADP-ribosylation reaction. Error bars indicate S.E. of 8–12 replications from 3 independent experiments.
CTA1 Activity Is Enhanced by Host Factors in a Sequence-dependent Manner
The G protein target of CTA1 is located in plasma membrane lipid rafts that have been shown to induce a gain-of-structure and function in disordered CTA1 (46). Previous experiments verified that lipid rafts did not dislodge Hsp90 from CTA1 (Table 2). To determine whether Hsp90-associated CTA1 could interact with lipid rafts, CTA1 and Hsp90/ATP were incubated at 37 °C for 10 min to allow complex formation and subsequently perfused over a SPR sensor slide coated with lipid raft LUVs (Fig. 6A). The resulting increase in RIU indicated the complex of CTA1 and Hsp90 could bind to lipid rafts. The bound components were then probed with anti-CTA and anti-Hsp90 antibodies, verifying the presence of both proteins on the lipid raft sensor (data not shown).
FIGURE 6.
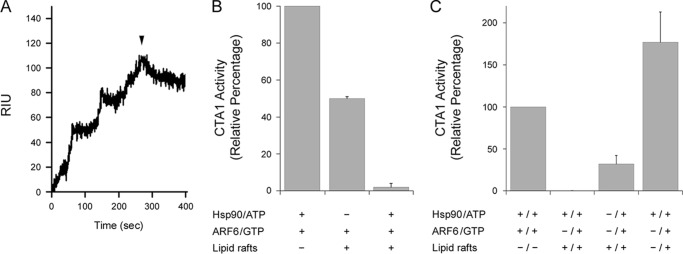
CTA1 activity is enhanced by host factors in a sequential manner. A, Hsp90/ATP and CTA1 were incubated at 37 °C for 10 min at a molar ratio of 2:1 to promote complex formation. The complex was then perfused over a SPR sensor coated with LUVs mimicking the composition of a lipid raft. The arrowhead indicates when the Hsp90-CTA1 complex was removed from the perfusion buffer. B, CTA1 samples (1 μg/ml) were incubated for 30 min at 37 °C before the addition of the indicated host factors for 1 h at 37 °C. NAD and DEA-BAG were then added for 2 h at 37 °C before CTA1 activity against the DEA-BAG substrate was recorded. Data represent the averages ± ranges of 2 independent experiments with 4 replications each. C, CTA1 samples (1 μg/ml) were incubated for 30 min at 37 °C before the addition of a host factor or factors as denoted by the + sign to the left of the slash. After 1 h at 37 °C, additional host factors were added for another hour at 37 °C as denoted by new + signs to the right of the slash. NAD and DEA-BAG were then added for 2 h at 37 °C before CTA1 activity against the DEA-BAG substrate was recorded. Data represent the averages ± ranges of 2 independent experiments with 4 replications each.
To assess the relative contributions of host factors to CTA1 activity, various combinations of Hsp90/ATP, ARF6/GTP, and lipid raft LUVs were added to CTA1 in our ADP-ribosylation assay (Fig. 6, B and C). Experiments were performed at 37 °C to mimic physiological temperature. In Fig. 6B, host factors were added to CTA1 after the toxin had been warmed to 37 °C for 30 min. Combinations of both ARF6/Hsp90 and ARF6/lipid rafts returned the disordered toxin to a functional state. However, toxin activity in the presence of ARF6/Hsp90 was around 2-fold greater than the level of activity recorded for the CTA1 sample exposed to a combination of ARF6 and lipid rafts. We hypothesized that a combination of ARF6, Hsp90, and lipid rafts would restore the greatest level of activity to disordered CTA1, but, surprisingly, no activity was observed from the CTA1 sample incubated simultaneously with all three host factors.
In vivo, CTA1 will initially interact with Hsp90 at the ER membrane before encountering the lipid raft environment where its Gαs target is located. Furthermore, our data suggest Hsp90 remains bound to the cytosolic pool of CTA1 after facilitating the translocation event. We therefore hypothesized that sequential interactions between CTA1 and Hsp90, followed by contact with lipid rafts, would allow the refolded toxin to maintain a functional conformation that could be further activated by ARFs. To test this model, we warmed CTA1 to 37 °C and then added Hsp90/ATP, lipid rafts, or both. After 1 h at 37 °C, ARF6/GTP and (if applicable) the other missing host factor were added for another hour at 37 °C. The ADP-ribosylation reaction was then initiated with the addition of substrate and NAD and allowed to proceed for 2 h at 37 °C. As a reference point for previous experiments, we also added Hsp90/ATP and ARF6/GTP simultaneously to disordered CTA1 at 37 °C. Toxin activity under this condition was set at 100%, and results from other conditions were expressed as percentages of this value (Fig. 6C). No toxin activity was recorded when disordered CTA1 was exposed to a combination of Hsp90 and lipid rafts before the addition of ARF6, but some level of activity was obtained when lipid rafts were added before Hsp90 and ARF6. A much greater level of activity was recorded when Hsp90 was added before lipid rafts and ARF6. This sequence of additions also promoted a greater level of activity than was recorded for the simultaneous exposure to Hsp90 and ARF6, which indicated lipid rafts could enhance activity of an Hsp90-ARF6-CTA1 complex. Collectively, our data suggest the order of host factor interaction is important for CTA1 to achieve maximum activity.
DISCUSSION
CT binds the cell surface, is internalized, and undergoes retrograde transport to the ER where CTA1 is separated from CTA2/CTB5. Due to the thermal instability of CTA1, it spontaneously unfolds following holotoxin disassembly. The ERAD system then recognizes unfolded CTA1 and exports the toxin to the cytosol. We previously reported that the cytosolic chaperone Hsp90 is active in the ER to cytosol export of CTA1. Our current structure/function analysis provides a molecular basis for the Hsp90-driven extraction of CTA1 from the ER and suggests a post-translocation role for Hsp90 in promoting CTA1 activity.
As shown by isotope-edited FTIR spectroscopy, Hsp90 refolds CTA1 in a process that requires ATP hydrolysis by Hsp90 (Fig. 1, Table 1). Translocation of CTA1 from the ER is also dependent on ATP hydrolysis by Hsp90 (Fig. 2). These data provide experimental support for a new model of toxin translocation involving a mechanism in which Hsp90 couples CTA1 refolding with CTA1 extraction from the ER. Refolded CTA1 would not slide back into the translocon pore, thus resulting in unidirectional movement of CTA1 from the ER to the cytosol.
Our initial studies on Hsp90-CTA1 interactions were inspired by reports documenting the role of Hsp90 in the endosome to cytosol translocation of certain AB-type, ADP-ribosylating toxins (61–63). Cells lacking functional Hsp90 due to drug treatment or RNAi sequester these toxins in the lumen of the endosomes and are accordingly resistant to intoxication (61–65). However, the molecular mechanism for toxin translocation across the endosomal membrane has not been determined. Our mechanistic studies on Hsp90-CTA1 interactions provide a possible molecular basis for the Hsp90-dependent movement of toxin A chains across the endosomal membrane: as with CTA1, Hsp90 could facilitate translocation from the endosomes by coupling toxin refolding with toxin extraction from the endosomes. Although this model requires experimental validation, it suggests our studies on the modulation of CTA1 structure/function by Hsp90 could be broadly applicable to the family of AB-type, ADP-ribosylating toxins. Our observations may also provide a foundation to understand the p97-independent translocation of select endogenous ERAD substrates (66, 67).
Hsp90/ATP preferentially interacts with unfolded CTA1 (Fig. 3A) and thus acts as a typical chaperone in this fashion. An unusual feature of the Hsp90-CTA1 interaction occurs when Hsp90 couples its refolding of CTA1 to CTA1 extraction from the cytosol. Another unusual feature of the Hsp90-CTA1 interaction involves the continued association of Hsp90 with refolded CTA1. This association was not disrupted by the presence of specific binding partners for CTA1 or Hsp90. Even exposure to a cytosolic extract did not dislodge Hsp90 from refolded CTA1 (Table 2). It is possible we have not identified proper conditions for the separation of Hsp90 from CTA1, but our observations suggest Hsp90 remains bound to CTA1 after toxin translocation to the cytosol. Attempts to detect a co-immunoprecipitated cytosolic complex of Hsp90 and CTA1 by Western blot or SPR were unsuccessful, possibly because of the extremely low quantity of CTA1 that reaches the cytosol of intoxicated cells and the background signal from our anti-Hsp90 antibody. However, with the available data, we posit Hsp90 remains associated with CTA1 after toxin extraction from the ER. Due to the intrinsic instability of CTA1, dissociation from Hsp90 in the host cytosol would place the toxin in an unfolded, inactive, and protease-sensitive conformation. The exploitation of cellular processes by CT may therefore involve a mechanism that promotes productive intoxication by preserving a post-translocation/post-refolding interaction between CTA1 and Hsp90.
The continued association between Hsp90 and refolded CTA1 has an impact on the activity of CTA1. In vitro ADP-ribosylation assays documented a gain-of-function at physiological temperature that resulted from the Hsp90-mediated refolding of disordered CTA1. This gain-of-function was dependent on ATP hydrolysis (Fig. 4B), consistent with the requirement of ATP hydrolysis for the Hsp90-mediated refolding of CTA1 (Fig. 1). ARF6 could bind to the CTA1-Hsp90 complex (Fig. 5A) and increased the activity of CTA1 above the gain-of-function obtained through the addition of Hsp90/ATP alone (Fig. 5B). Lipid rafts also induce a gain-of-function in disordered CTA1 at physiological temperature (46), yet no ADP-ribosylation activity was detected after the simultaneous addition of Hsp90/ATP, ARF6/GTP, and lipid rafts to disordered CTA1 (Fig. 6B). In contrast, substantial CTA1 activity was obtained when ARF6 and lipid rafts were added to a pre-formed CTA1-Hsp90 complex (Fig. 6C). These collective observations indicate the order of host factor interaction is important for CTA1 activity and are consistent with the in vivo sequence of CT intoxication: CTA1 first encounters the membrane-associated pool of Hsp90 during the translocation event, likely remains associated with Hsp90 in the cytosol, and then recruits ARF proteins to enhance the ADP-ribosylation of Gαs in the lipid raft microenvironment of the plasma membrane.
Western blot analysis from a previous study failed to detect Hsp90 in cytosolic fractions that facilitated in vitro CTA1 translocation, leading some to question the link between Hsp90 and CTA1 translocation (68). However, this interpretation is based upon a negative result from an assay that did not use a salt wash to strip Hsp90 from the membrane fraction. We have used a range of experimental approaches to document the essential role of Hsp90 in CTA1 translocation and cholera intoxication. Protein-protein interactions monitored by SPR have demonstrated Hsp90 specifically recognizes the unfolded conformation of CTA1 in an ATP-dependent process. Biophysical and biochemical assays have shown that ATP hydrolysis by Hsp90 is required for Hsp90 to induce a gain-of-structure and gain-of-function in disordered CTA1. In vitro reconstitution of the CTA1 translocation event likewise required ATP hydrolysis by Hsp90: the addition of Hsp90/ATP but neither Hsp90 nor Hsp90/ATPγS to salt- or urea-washed membranes was sufficient for CTA1 export. ATPγS has been previously reported to block in vitro CTA1 translocation, although the affected host protein was not identified (68). Multiple cytosolic host factors may be active in the CTA1 translocation event, but our in vitro reconstitution assay has shown, for the first time, that Hsp90/ATP is sufficient for toxin export to the cytosol. In previous work we demonstrated cells lacking functional Hsp90 due to RNAi or drug treatment trap CTA1 in the ER lumen and are therefore resistant to cholera intoxication (37). Thus, through the combination of in vivo and in vitro assays, we have established that Hsp90 is necessary and sufficient for CTA1 translocation. We have also provided experimental support for a model of Hsp90-driven translocation that couples toxin refolding with toxin export from the ER. Further elucidation of this model should generate additional insights into a new mechanism of translocation with potential applications to other ADP-ribosylating toxins and p97-independent ERAD substrates.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AI099493 from the NIAID (to K. T.).
- CT
- cholera toxin
- ARF
- ADP-ribosylation factor
- DEA-BAG
- diethylamino(benzylidine-amino)guanidine
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- Hsp90
- heat shock protein 90
- LUV
- large unilamellar vesicle
- RIU
- refractive index unit
- SPR
- surface plasmon resonance
- PDI
- protein-disulfide isomerase
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. De Haan L., Hirst T. R. (2004) Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (review). Mol. Membr. Biol. 21, 77–92 [DOI] [PubMed] [Google Scholar]
- 2. Zhang R. G., Scott D. L., Westbrook M. L., Nance S., Spangler B. D., Shipley G. G., Westbrook E. M. (1995) The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 251, 563–573 [DOI] [PubMed] [Google Scholar]
- 3. Wernick N. L., Chinnapen D. J., Cho J. A., Lencer W. I. (2010) Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins 2, 310–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Majoul I., Ferrari D., Söling H. D. (1997) Reduction of protein disulfide bonds in an oxidizing environment. The disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett. 401, 104–108 [DOI] [PubMed] [Google Scholar]
- 5. Orlandi P. A. (1997) Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J. Biol. Chem. 272, 4591–4599 [PubMed] [Google Scholar]
- 6. Taylor M., Banerjee T., Ray S., Tatulian S. A., Teter K. (2011) Protein-disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J. Biol. Chem. 286, 22090–22100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor M., Burress H., Banerjee T., Ray S., Curtis D., Tatulian S. A., Teter K. (2014) Substrate-induced unfolding of protein disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathogens 10, e1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsai B., Rodighiero C., Lencer W. I., Rapoport T. A. (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104, 937–948 [DOI] [PubMed] [Google Scholar]
- 9. O'Neal C. J., Jobling M. G., Holmes R. K., Hol W. G. (2005) Structural basis for the activation of cholera toxin by human ARF6-GTP. Science 309, 1093–1096 [DOI] [PubMed] [Google Scholar]
- 10. Kahn R. A., Gilman A. G. (1984) Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J. Biol. Chem. 259, 6228–6234 [PubMed] [Google Scholar]
- 11. Welsh C. F., Moss J., Vaughan M. (1994) ADP-ribosylation factors: a family of approximately 20-kDa guanine nucleotide-binding proteins that activate cholera toxin. Mol Cell Biochem. 138, 157–166 [DOI] [PubMed] [Google Scholar]
- 12. Harris J. B., LaRocque R. C., Qadri F., Ryan E. T., Calderwood S. B. (2012) Cholera. Lancet 379, 2466–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pande A. H., Scaglione P., Taylor M., Nemec K. N., Tuthill S., Moe D., Holmes R. K., Tatulian S. A., Teter K. (2007) Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 374, 1114–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Teter K. (2013) Cholera toxin interactions with host cell stress proteins. in Moonlighting Cell Stress Proteins in Microbial Infections (Henderson B., ed), pp. 323–338, Springer, New York [Google Scholar]
- 15. Teter K. (2013) Toxin instability and its role in toxin translocation from the endoplasmic reticulum to the cytosol. Biomolecules 3, 997–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Banerjee T., Pande A., Jobling M. G., Taylor M., Massey S., Holmes R. K., Tatulian S. A., Teter K. (2010) Contribution of subdomain structure to the thermal stability of the cholera toxin A1 subunit. Biochemistry 49, 8839–8846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Massey S., Banerjee T., Pande A. H., Taylor M., Tatulian S. A., Teter K. (2009) Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J. Mol. Biol. 393, 1083–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taylor M., Banerjee T., Navarro-Garcia F., Huerta J., Massey S., Burlingame M., Pande A. H., Tatulian S. A., Teter K. (2011) A therapeutic chemical chaperone inhibits cholera intoxication and unfolding/translocation of the cholera toxin A1 subunit. PLoS ONE 6, e18825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teter K., Holmes R. K. (2002) Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect. Immun. 70, 6172–6179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teter K., Jobling M. G., Holmes R. K. (2003) A class of mutant CHO cells resistant to cholera toxin rapidly degrades the catalytic polypeptide of cholera toxin and exhibits increased endoplasmic reticulum-associated degradation. Traffic 4, 232–242 [DOI] [PubMed] [Google Scholar]
- 21. Bagola K., Mehnert M., Jarosch E., Sommer T. (2011) Protein dislocation from the ER. Biochim. Biophys. Acta 1808, 925–936 [DOI] [PubMed] [Google Scholar]
- 22. Nakatsukasa K., Brodsky J. L. (2008) The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 9, 861–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spooner R. A., Lord J. M. (2012) How ricin and Shiga toxin reach the cytosol of target cells: retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 357, 19–40 [DOI] [PubMed] [Google Scholar]
- 24. Hazes B., Read R. J. (1997) Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36, 11051–11054 [DOI] [PubMed] [Google Scholar]
- 25. Deeks E. D., Cook J. P., Day P. J., Smith D. C., Roberts L. M., Lord J. M. (2002) The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 41, 3405–3413 [DOI] [PubMed] [Google Scholar]
- 26. Marshall R. S., Jolliffe N. A., Ceriotti A., Snowden C. J., Lord J. M., Frigerio L., Roberts L. M. (2008) The role of CDC48 in the retro-translocation of non-ubiquitinated toxin substrates in plant cells. J. Biol. Chem. 283, 15869–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodighiero C., Tsai B., Rapoport T. A., Lencer W. I. (2002) Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 3, 1222–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Worthington Z. E., Carbonetti N. H. (2007) Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect. Immun. 75, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orlowski M., Wilk S. (2003) Ubiquitin-independent proteolytic functions of the proteasome. Arch. Biochem. Biophys. 415, 1–5 [DOI] [PubMed] [Google Scholar]
- 30. Ampapathi R. S., Creath A. L., Lou D. I., Craft J. W., Jr., Blanke S. R., Legge G. B. (2008) Order-disorder-order transitions mediate the activation of cholera toxin. J. Mol. Biol. 377, 748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bar-Nun S. (2005) The role of p97/Cdc48p in endoplasmic reticulum-associated degradation: from the immune system to yeast. Curr. Top. Microbiol. Immunol. 300, 95–125 [DOI] [PubMed] [Google Scholar]
- 32. Elkabetz Y., Shapira I., Rabinovich E., Bar-Nun S. (2004) Distinct steps in dislocation of luminal endoplasmic reticulum-associated degradation substrates: roles of endoplamic reticulum-bound p97/Cdc48p and proteasome. J. Biol. Chem. 279, 3980–3989 [DOI] [PubMed] [Google Scholar]
- 33. Kothe M., Ye Y., Wagner J. S., De Luca H. E., Kern E., Rapoport T. A., Lencer W. I. (2005) Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J. Biol. Chem. 280, 28127–28132 [DOI] [PubMed] [Google Scholar]
- 34. McConnell E., Lass A., Wójcik C. (2007) Ufd1-Npl4 is a negative regulator of cholera toxin retrotranslocation. Biochem. Biophys. Res. Commun. 355, 1087–1090 [DOI] [PubMed] [Google Scholar]
- 35. Sreedhar A. S., Kalmár E., Csermely P., Shen Y. F. (2004) Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 562, 11–15 [DOI] [PubMed] [Google Scholar]
- 36. Csermely P., Schnaider T., Soti C., Prohászka Z., Nardai G. (1998) The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 79, 129–168 [DOI] [PubMed] [Google Scholar]
- 37. Taylor M., Navarro-Garcia F., Huerta J., Burress H., Massey S., Ireton K., Teter K. (2010) Hsp90 is required for transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 285, 31261–31267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bernardi K. M., Forster M. L., Lencer W. I., Tsai B. (2008) Derlin-1 facilitates the retro-translocation of cholera toxin. Mol. Biol. Cell 19, 877–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bernardi K. M., Williams J. M., Kikkert M., van Voorden S., Wiertz E. J., Ye Y., Tsai B. (2010) The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol. Biol. Cell 21, 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dixit G., Mikoryak C., Hayslett T., Bhat A., Draper R. K. (2008) Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp. Biol. Med. (Maywood) 233, 163–175 [DOI] [PubMed] [Google Scholar]
- 41. Saslowsky D. E., Cho J. A., Chinnapen H., Massol R. H., Chinnapen D. J., Wagner J. S., De Luca H. E., Kam W., Paw B. H., Lencer W. I. (2010) Intoxication of zebrafish and mammalian cells by cholera toxin depends on the flotillin/reggie proteins but not Derlin-1 or -2. J. Clin. Invest. 120, 4399–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmitz A., Herrgen H., Winkeler A., Herzog V. (2000) Cholera toxin is exported from microsomes by the Sec61p complex. J. Cell Biol. 148, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allen J. A., Halverson-Tamboli R. A., Rasenick M. M. (2007) Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140 [DOI] [PubMed] [Google Scholar]
- 44. Ostrom R. S., Insel P. A. (2004) The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br. J. Pharmacol. 143, 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murayama T., Tsai S. C., Adamik R., Moss J., Vaughan M. (1993) Effects of temperature on ADP-ribosylation factor stimulation of cholera toxin activity. Biochemistry 32, 561–566 [DOI] [PubMed] [Google Scholar]
- 46. Ray S., Taylor M., Banerjee T., Tatulian S. A., Teter K. (2012) Lipid rafts alter the stability and activity of the cholera toxin A1 subunit. J. Biol. Chem. 287, 30395–30405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Banerjee T., Taylor M., Jobling M. G., Burress H., Yang Z., Serrano A., Holmes R. K., Tatulian S. A., Teter K. (2014) ADP-ribosylation factor 6 acts as an allosteric activator for the folded but not disordered cholera toxin A1 polypeptide. Mol. Microbiol. 10.1111/mmi.12807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reddy S., Taylor M., Zhao M., Cherubin P., Geden S., Ray S., Francis D., Teter K. (2013) Grape extracts inhibit multiple events in the cell biology of cholera intoxication. PLoS One 8, e73390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Teter K., Jobling M. G., Sentz D., Holmes R. K. (2006) The cholera toxin A13 subdomain is essential for interaction with ADP-ribosylation factor 6 and full toxic activity but is not required for translocation from the endoplasmic reticulum to the cytosol. Infect. Immun. 74, 2259–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tatulian S. A. (2013) Structural characterization of membrane proteins and peptides by FTIR and ATR-FTIR spectroscopy. in Lipid-Protein Interactions: Methods and Protocols (Kleinschmidt J. H., ed), pp. 177–218, Humana Press, New York: [DOI] [PubMed] [Google Scholar]
- 51. Deleted in proof.
- 52. Massey S., Burress H., Taylor M., Nemec K. N., Ray S., Haslam D. B., Teter K. (2011) Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect. Immun. 79, 4739–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tatulian S. A. (2010) Structural analysis of proteins by isotope-edited FTIR spectroscopy. Spectroscopy Int. J. 24, 37–43 [Google Scholar]
- 54. O'Neal C. J., Amaya E. I., Jobling M. G., Holmes R. K., Hol W. G. (2004) Crystal structures of an intrinsically active cholera toxin mutant yield insight into the toxin activation mechanism. Biochemistry 43, 3772–3782 [DOI] [PubMed] [Google Scholar]
- 55. Bernardi K. M., Williams J. M., Inoue T., Schultz A., Tsai B. (2013) A deubiquitinase negatively regulates retro-translocation of nonubiquitinated substrates. Mol. Biol. Cell 24, 3545–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ozdemir S., Turhan-Sayan G. (2003) Temperature effects on surface plasmon resonance: design considerations for an optical temperature sensor. J. Lightwave Technol. 21, 805–814 [Google Scholar]
- 57. Wagner B. J., Margolis J. W. (1995) Age-dependent association of isolated bovine lens multicatalytic proteinase complex (proteasome) with heat-shock protein 90, an endogenous inhibitor. Arch Biochem. Biophys 323, 455–462 [DOI] [PubMed] [Google Scholar]
- 58. Tsubuki S., Saito Y., Kawashima S. (1994) Purification and characterization of an endogenous inhibitor specific to the Z-Leu-Leu-Leu-MCA degrading activity in proteasome and its identification as heat-shock protein 90. FEBS Lett. 344, 229–233 [DOI] [PubMed] [Google Scholar]
- 59. Teter K., Jobling M. G., Holmes R. K. (2004) Vesicular transport is not required for the cytoplasmic pool of cholera toxin to interact with the stimulatory α subunit of the heterotrimeric G protein. Infect. Immun. 72, 6826–6835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Soman G., Narayanan J., Martin B. L., Graves D. J. (1986) Use of substituted (benzylidineamino)guanidines in the study of guanidino group specific ADP-ribosyltransferase. Biochemistry 25, 4113–4119 [DOI] [PubMed] [Google Scholar]
- 61. Haug G., Aktories K., Barth H. (2004) The host cell chaperone Hsp90 is necessary for cytotoxic action of the binary ι-like toxins. Infect. Immun. 72, 3066–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Haug G., Leemhuis J., Tiemann D., Meyer D. K., Aktories K., Barth H. (2003) The host cell chaperone Hsp90 is essential for translocation of the binary Clostridium botulinum C2 toxin into the cytosol. J. Biol. Chem. 278, 32266–32274 [DOI] [PubMed] [Google Scholar]
- 63. Ratts R., Zeng H., Berg E. A., Blue C., McComb M. E., Costello C. E., vanderSpek J. C., Murphy J. R. (2003) The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J. Cell Biol. 160, 1139–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kaiser E., Kroll C., Ernst K., Schwan C., Popoff M., Fischer G., Buchner J., Aktories K., Barth H. (2011) Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect. Immun. 79, 3913–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lang A. E., Ernst K., Lee H., Papatheodorou P., Schwan C., Barth H., Aktories K. (2014) The chaperone Hsp90 and PPIases of the cyclophilin and FKBP families facilitate membrane translocation of Photorhabdus luminescens ADP-ribosyltransferases. Cell Microbiol. 16, 490–503 [DOI] [PubMed] [Google Scholar]
- 66. Carlson E. J., Pitonzo D., Skach W. R. (2006) p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 25, 4557–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wójcik C., Rowicka M., Kudlicki A., Nowis D., McConnell E., Kujawa M., DeMartino G. N. (2006) Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 17, 4606–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moore P., He K., Tsai B. (2013) Establishment of an in vitro transport assay that reveals mechanistic differences in cytosolic events controlling cholera toxin and T-cell receptor α retro-translocation. PLoS One 8, e75801. [DOI] [PMC free article] [PubMed] [Google Scholar]