

Background: Arrestin mediates G protein-independent signaling and internalization of the D2 receptor.
Results: A D2 receptor mutant with modestly diminished ability to recruit arrestin and β2-adaptin did not internalize in response to agonists.
Conclusion: Arrestin-mediated recruitment of receptor to AP2 is not sufficient for internalization.
Significance: Receptor mutants lacking specific functions are tools for analysis of signaling mechanisms.
Keywords: Adenylate Cyclase (Adenylyl Cyclase), Arrestin, Bioluminescence Resonance Energy Transfer (BRET), Dopamine Receptor, G Protein, beta2-Adaptin
Abstract
Arrestins mediate desensitization and internalization of G protein-coupled receptors and also direct receptor signaling toward heterotrimeric G protein-independent signaling pathways. We previously identified a four-residue segment (residues 212–215) of the dopamine D2 receptor that is necessary for arrestin binding in an in vitro heterologous expression system but that also impairs receptor expression. We now describe the characterization of additional mutations at that arrestin binding site in the third intracellular loop. Mutating two (residues 214 and 215) or three (residues 213–215) of the four residues to alanine partially decreased agonist-induced recruitment of arrestin3 without altering activation of a G protein. Arrestin-dependent receptor internalization, which requires arrestin binding to β2-adaptin (the β2 subunit of the clathrin-associated adaptor protein AP2) and clathrin, was disproportionately affected by the three-residue mutation, with no agonist-induced internalization observed even in the presence of overexpressed arrestin or G protein-coupled receptor kinase 2. The disjunction between arrestin recruitment and internalization could not be explained by alterations in the time course of the receptor-arrestin interaction, the recruitment of G protein-coupled receptor kinase 2, or the receptor-induced interaction between arrestin and β2-adaptin, suggesting that the mutation impairs a property of the internalization complex that has not yet been identified.
Introduction
Dopamine receptors belong to the large superfamily of G protein-coupled receptors (GPCRs)4 and consist of five receptor subtypes (D1–5) that are categorized into two subgroups (D1-like and D2-like) defined by several criteria, including primary structure, signaling properties, and pharmacological profile. Dopaminergic neurotransmission via the D2 receptor contributes to the rewarding properties of drugs of abuse (1) and is the target of therapeutics used to treat disorders such as Parkinson's disease (2) and schizophrenia (3). The dopamine D2 receptor activates heterotrimeric Gi/o/z proteins that decrease the production of cyclic AMP (4–7). Activated D2 receptors are then phosphorylated by G protein receptor kinase 2/3 (GRK2/3) on serine and threonine residues in IL3 of the receptor, and subsequent receptor internalization is mediated through recruitment of arrestin3 (8–11).
As well as being crucial for termination of G protein-dependent signaling and receptor internalization, arrestins act as scaffolds for signaling proteins (12). Agonist-induced recruitment of arrestin by a GPCR may activate a unique set of signaling pathways that have distinctive behavioral consequences. For example, the D2 receptor mediates arrestin-dependent dephosphorylation of the protein kinase Akt by protein phosphatase 2A and a net increase in glycogen synthase kinase 3β activity, which contributes to amphetamine-induced locomotor activation (13, 14). This signaling pathway may be involved in the pathophysiology of schizophrenia (15), suggesting that it would be valuable to develop tools, such as biased ligands (16, 17) or a D2 receptor that preferentially activates either G protein- or arrestin-dependent pathways, to investigate pathway-specific behaviors.
In the standard model of arrestin-mediated desensitization, GPCR phosphorylation by GRK increases the affinity of arrestin for the receptor, leading to subsequent receptor desensitization and internalization (18, 19). However, although IL3 of the D2 receptor is the site of GRK2 phosphorylation and directly interacts with arrestin3, receptor phosphorylation by GRK2 may not be required for either arrestin3 recruitment or receptor internalization (20).
We have shown previously that arrestin3 binding to the D2 receptor is greatly reduced when amino acids 212–215 (IYIV), located within IL3 of the D2 receptor, were mutated to alanine residues. This mutation abolishes agonist-induced receptor internalization, with less effect on G protein-mediated signaling (9). This mutation also greatly impairs trafficking of the receptor to the cell membrane, thereby reducing its value as a tool to study the relationships between specific signaling pathways and dopamine-dependent behaviors. The purpose of this study was to develop a D2 receptor mutant that is significantly impaired in arrestin3 recruitment yet maintains G protein signaling and receptor expression.
Herein we describe the creation and characterization of a mutant D2 dopamine receptor that has modestly diminished agonist-induced recruitment of GRK2 and arrestin3 yet maintains G protein-dependent signaling and normal expression. Surprisingly, despite retaining substantial ability to recruit arrestin3 and to promote arrestin3-dependent interaction with β2-adaptin (the β2 subunit of the clathrin-associated adaptor protein AP2), this mutant D2 receptor failed to undergo agonist-induced receptor internalization.
EXPERIMENTAL PROCEDURES
Materials
Quinpirole, dopamine hydrochloride, (+)-butaclamol, haloperidol, (S)-(−)-sulpiride, and G418 were purchased from Sigma-Aldrich. Phosphatase inhibitor mixture set II and protease inhibitor mixture set III were purchased from Calbiochem, and coelenterazine h was from Dalton Chemical Laboratories (Toronto, Canada). The radioligands [3H](−)-sulpiride and [3H]YM-09151-2 were purchased from PerkinElmer Life Sciences. Rabbit anti-actin, mouse anti-arrestin3, rabbit anti-GRK2, and mouse anti-c-Myc antibodies were purchased from Santa Cruz Biotechnology, Inc., and mouse anti-FLAG M2 was purchased from Sigma-Aldrich. Alexa Fluor® 488 goat anti-mouse IgG, Alexa Fluor® 647 goat anti-mouse IgG, and Alexa Fluor® 568 goat anti-rabbit IgG were purchased from Invitrogen.
Plasmids
For the G protein activation bioluminescence resonance energy transfer (BRET) assay, pcDNA3.1 plasmids were used carrying c-Myc-tagged wild type or mutant rat D2L (D2-WT, A2, A3, or A4), Renilla luciferase 8 (Rluc8) inserted at the position 91 of Gαi1 (Gαi1-91-Rluc8) (21), and mVenus fragments V1 and V2 fused to Gβ1 and Gγ2, respectively (V1-β1 and V2-γ2) (22). For the BRET-based inhibition of cAMP assay, pcDNA3.1 plasmids carrying D2-WT or A4 and CAMYEL (ATCC) (23) were used. For the recruitment BRET assays, FLAG-tagged wild type human D2L receptors fused to Rluc8 (D2-Rluc8) (24), mVenus fused to human arrestin3 (mVenus-Arr3), mVenus fused to bovine GRK2 (GRK2-Venus), human β2-adaptin-EYFP (β2-AP-EYFP) (25), and pcDNA3.1 plasmids were used. Human arrestin3 fused to Rluc8 at its C terminus with a SRPPVAT amino acid linker (Arr3-Rluc8) was created in pcDNA3.1. The arrestin translocation assay used Rluc8-arrestin3 with an SH3-binding peptide (Sp1) at its C terminus (Rluc8-arrestin3-Sp1) and a doubly palmitoylated fragment of GAP43 linked to citrine and an SH3 domain through a serine- and glycine-rich linker (mem-linker-citrine-SH3). For internalization and immunoblotting assays, plasmids carrying rat D2L receptor, wild type and mutants of arrestin3, and GRK2 were used.
Mutagenesis
Multiple point mutations of both the D2L receptor, D2-Rluc8, and arrestin were constructed using the QuikChange® Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA) per the manufacturer's instructions to introduce alanine substitution of residues 214 and 215 (A2), 213–215 (A3), or 212–215 (A4) within IL3. Residues of arrestin3 were deleted (373LIEFD377) or mutated (R395E) alone or in combination using this method. All mutations were confirmed by DNA sequencing.
Cell Culture and Transfections
HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 5% bovine calf serum (Thermo Scientific, Logan, UT), 5% fetal clone serum (Thermo Scientific), and 1% penicillin-streptomycin solution (Thermo Scientific) at 37 °C with 10% CO2. HEK293T cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (Corning Inc.) at 37 °C with 10% CO2. For the internalization and Western blot studies, stable expression of either wild type or mutant D2L receptors in HEK293 cells was obtained by transfecting 6 μg of D2 receptor cDNA using Lipofectamine 2000 (Invitrogen) per the manufacturer's recommended protocol. Pools of stably transfected cells were then maintained under selective pressure using 600 μg/ml G418 for 3 weeks, after which the amount of G418 in the growth medium was decreased to 300 μg/ml. Transient expression of arrestin3, GRK2, and/or pcDNA3.1 into HEK293 cells stably expressing wild type or mutant D2L receptors was obtained by transfecting 3 μg of cDNA using Lipofectamine 2000 (Invitrogen). After overnight incubation at 37 °C, transfection complexes were rinsed off, and the cells were split into new plates and incubated for an additional 48 h before conducting experiments. Protein expression was confirmed by immunoblotting.
For all BRET studies, constructs were transiently transfected into either HEK293 or HEK293T cells using polyethyleneimine (PEI; Polysciences, Inc., Warrington, PA), as described previously (22). The quantity of transfected DNA was adjusted so that the levels of membrane expression of wild type and mutant D2 receptors were similar as determined by radioligand binding assay or FACS, as described below.
For the arrestin3 mutant internalization studies, 2 μg of GRK2 were contransfected with 2 μg of pcDNA3.1, Arr3-WT, Arr3-ΔLIEFD, Arr3-R395E, or Arr3-ΔLIEFD/R395E into HEK293 cells stably expressing wild-type D2L receptors using PEI at a ratio of 2 μl of PEI (1 μg/μl) per 1 μg of cDNA. After overnight incubation at 37 °C, cells were washed with PBS, split into new plates, and incubated for an additional 48 h before conducting experiments.
BRET
HEK293 or HEK293T cells were transiently co-transfected with plasmids as described above. For mVenus-Arr3 and β2-AP-EYFP recruitment to the receptor, cells were harvested and split into two plates, one for radioligand binding assays and one for BRET assays. Forty-eight hours after transfection, cells were harvested, resuspended in assay buffer (PBS with Ca2+, MgCl2, and 0.2 μm ascorbic acid), and plated at ∼150,000 cells/well in 96-well OptiPlates (PerkinElmer Life Sciences). Vehicle or quinpirole was added with 2 μm coelenterazine h and incubated at 25 °C. Emission of the donor (460 nm) and the acceptor (535 nm) was then measured once at 10 min or every 5 min for 30 min using a VictorTM X Light luminescence reader (PerkinElmer Life Sciences), and the BRET ratio was calculated as described previously (26). Cells used in the G protein activation, GRK2 recruitment to receptor, and arrestin3 recruitment to β2-AP-EYFP BRET assays were prepared and assayed as described previously (22) and were measured 2, 2, and 20 min, respectively, after the addition of quinpirole.
For the arrestin translocation assay, pcDNA3.1 encoding wild type or mutant D2 receptors was cotransfected with plasmids containing GRK2, Rluc8-arrestin3-Sp1, and a membrane marker mem-linker-citrine-SH3. The Sp1 and SH3 helper peptides, adapted from the helper interaction FRET system (27), were used to enhance the interaction between arrestin recruited to the plasma membrane by receptor and the membrane marker, thus increasing the dynamic range of the assay. Dopamine- and quinpirole-induced arrestin recruitment to the membrane was measured after 10 min of stimulation.
D2-mediated inhibition of cAMP was measured using a BRET-based cAMP sensor, CAMYEL. Cells transiently transfected with D2-WT or A4 receptor and CAMYEL were preincubated for 10 min with 10 μm forskolin (Sigma) to stimulate cAMP production, followed by the addition of quinpirole. Quinpirole-mediated inhibition of forskolin-induced cAMP was measured after 10 min.
For all BRET studies, the vehicle BRET ratio was subtracted from the quinpirole BRET ratio and presented as a percentage of the Emax fit of quinpirole at the wild type receptor. Data were analyzed by nonlinear regression using Prism (GraphPad, San Diego, CA). To depict the variability in the activity of D2-WT and mutant receptors across replicate experiments, we calculated the mean of the Emax values for D2-WT, converted the Emax value for each replicate to a percentage of that value, and then determined the mean ± S.E. for all replicates.
D2L Receptor Radioligand Binding
To compare membrane expression of D2 receptors, cells were lysed in ice-cold hypotonic buffer (1 mm HEPES, 2 mm EDTA, pH 7.4) for 20 min at 4 °C, scraped from the plate, and centrifuged at 17,000 × g at 4 °C for 20 min. The resulting pellet was resuspended in TBS (50 mm Tris, 120 mm NaCl, pH 7.4) and homogenized for 10 s using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). Protein concentration of the membranes was determined using a BCA protein assay kit (Thermo Scientific) and 5 μg of protein was incubated in TBS containing 0.02% BSA (Fisher) at a volume of 1 ml with [3H]YM-09151-2 for 1 h at 25 °C. Nonspecific binding was assessed using (+)-butaclamol (2 μm). Membranes were then harvested, and radioactivity was measured as described previously (9). Data were analyzed by nonlinear regression using GraphPad Prism to determine Kd and Bmax values.
FACS
Transfected cells expressing N-terminal FLAG- or c-Myc-tagged D2 receptor were dissociated, and surface receptors were labeled using mouse anti-FLAG or anti-c-Myc antibodies and goat anti-mouse-Alexa647 antibodies diluted 1:400 in PBS (with 0.1% BSA and 0.1% NaN3) and quantitated using a C6 Flow Cytometer (Accuri). This FACS assay was used to equalize receptor expression in the assays for G protein activation (Figs. 1 and 3, A and B), inhibition of cAMP (Fig. 1D), GRK2-Venus recruitment (Fig. 6), and the interaction between arrestin and β2-adaptin (Fig. 9).
FIGURE 1.

D2-A4-mediated inhibition of cyclic AMP accumulation and G protein activation. A, HEK293T cells transfected with D2-WT or D2-A4 and CAMYEL, a BRET-based cAMP sensor. The inhibition of forskolin (10 μm)-stimulated cAMP by quinpirole at the indicated concentrations was measured after 10 min. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± S.E.). B, maximal inhibition of forskolin-stimulated cAMP by D2-A4 in response to quinpirole or dopamine stimulation determined from concentration-response curves and expressed as a percentage of the activation by D2-WT. Each bar represents the mean ± S.E. (error bars) of three independent experiments. *, p < 0.05 compared with D2-WT. C, schematic of the heterotrimeric G protein BRET biosensor. G protein activation is detected as a decrease in BRET between the Gαi1-Rluc8 donor and complemented mVenus-Gβ1γ2 acceptor. D, HEK293T cells transfected with the components of the G protein biosensor depicted in C and D2-WT or D2-A4 receptors were incubated with quinpirole at the indicated concentrations for 2 min before measuring the G protein BRET response. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± S.E.). E, maximal G protein activation by D2-A4 in response to quinpirole or dopamine stimulation determined from concentration response curves and expressed as a percentage of the activation by D2-WT. Each bar represents the mean ± S.E. of three independent experiments. *, p < 0.05; **, p < 0.01 compared with D2-WT within the same treatment group, Student's t test. For the cylic AMP experiments, D2-A4 expression was 104 ± 18% that of the wild-type receptor, whereas for the G protein activation experiments, D2-A4 expression was 80 ± 3% that of the wild type receptor.
FIGURE 3.

D2-WT-, D2-A3-, and D2-A2-mediated G protein activation and arrestin3 recruitment. A, D2-WT-, D2-A3-, and D2-A2-mediated G protein activation was assessed in HEK293T cells co-transfected with Gαi1-Rluc8, V1-Gβ1, V2-Gγ2, and receptor and then incubated with quinpirole at the indicated concentrations for 2 min before measuring the G protein BRET response. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± S.E. (error bars)). B, maximal G protein activation by D2-A3 and D2-A2 in response to quinpirole or dopamine stimulation, determined from the concentration response curves and expressed as a percentage of the response to D2-WT. G protein activation by the mutants was not significantly different from that by WT as determined by Tukey's multiple comparison test. D2-A3 and D2-A2 were expressed at 100 ± 5 and 78 ± 7% of the wild type receptor, respectively. C, D2-WT-, D2-A3-, and D2-A2-mediated arrestin3 recruitment in HEK293 cells transfected with the indicated receptor fused to Rluc8 and mVenus-Arr3 and incubated with agonist at the indicated concentrations for 10 min. Values are expressed as the mean ± S.E. of seven (D2-WT) or four (D2-A3 and D2-A2) independent experiments. D, maximal mVenus-Arr3 recruitment by D2-A3 and D2-A2 in response to quinpirole or dopamine stimulation, determined from the individual concentration-response curves and expressed as a percentage of the maximal response to D2-WT. *, p < 0.05 compared with D2-WT within the same treatment group, Tukey's multiple comparison test. Receptor density and basal BRET for D2-WT-Rluc8 were 1290 ± 151 fmol/mg protein and 0.07 ± 0.007; for D2-A3-Rluc8, they were 1223 ± 160 fmol/mg protein and 0.14 ± 0.006; and for D2-A2-Rluc8, they were 1300 ± 60 fmol/mg protein and 0.10 ± 0.008. E, D2-WT-, D2-A3-, and D2-A2-mediated arrestin3 translocation to the membrane in HEK293 cells transfected with the indicated receptor, GRK2, Rluc8-arrestin3-Sp1, and mem-linker-citrine-SH3 and incubated with agonist at the indicated concentrations for 10 min. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± S.E.). F, maximal Rluc8-arrestin3-SP1 membrane translocation in response to quinpirole or dopamine stimulation of untagged D2-WT, D2-A3, or D2-A2 receptors. Basal BRET was 0.64 ± 0.020 for D2-WT, 0.64 ± 0.017 for D2-A3, and 0.63 ± 0.023 for D2-A2.
FIGURE 6.
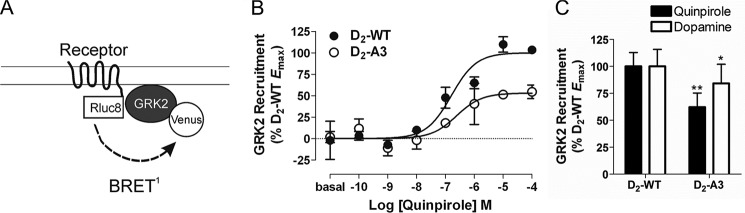
Agonist-induced recruitment of GRK2. A, schematic of the BRET biosensor used to measure receptor-mediated recruitment of GRK2. B, HEK293T cells transfected with GRK2-Venus and D2-WT-Rluc8 or D2-A3-Rluc8 were incubated with the indicated concentrations of quinpirole for 2 min before measuring BRET between the receptor and GRK2. Dose-response curves are representative of five independent experiments performed with triplicate samples (mean ± S.E. (error bars)). C, maximal GRK2-Venus recruitment by D2-A3 in response to quinpirole or dopamine stimulation was determined from the concentration-response curves and is expressed as a percentage of the maximal response to D2-WT. Each bar represents the mean ± S.E. of four or five independent experiments. *, p < 0.05; **, p < 0.01 compared with D2-WT within the same treatment group, paired Student's t test. Expression of D2-A3-Rluc8 was at 105 ± 16% of the wild type receptor. Basal BRET values for D2-WT and A3 were 0.513 ± 0.006 and 0.513 ± 0.005, respectively.
FIGURE 9.
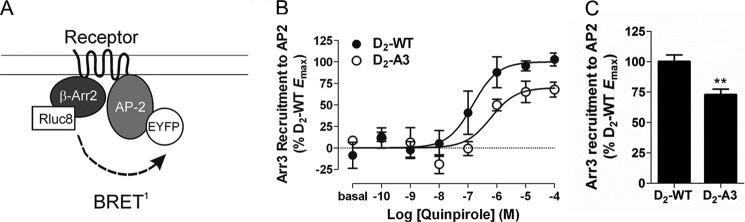
Receptor-mediated interaction between arrestin3 and β2-adaptin. A, schematic of the BRET biosensor used to measure the interaction between arrestin3 and the β2 subunit of AP2. B, cells transiently expressing D2-WT or D2-A3 were co-transfected with Arr3-Rluc8 and β2-AP2-EYFP, and the interaction between Arr3-Rluc8 and β2-AP2-EYFP was measured after quinpirole stimulation at the indicated concentrations for 20 min. Dose-response curves are representative of three independent experiments performed with triplicate samples (mean ± S.E. (error bars)). C, maximal D2-WT- or D2-A3-mediated BRET between Arr3-Rluc8 and β2-AP2-EYFP expressed as a percentage of the maximal response with D2-WT. Each bar represents the mean ± S.E. of three independent experiments. The D2-A3-mediated increase in the arrestin3/β2-adaptin interaction was significantly different from that by D2-WT as determined by Student's t test (**, p < 0.01). Expression of D2-A3 was at 88 ± 5% of the wild type receptor. Basal BRET values for D2-WT and A3 were 0.492 ± 0.013 and 0.495 ± 0.014, respectively.
Internalization Assay
D2 receptor internalization was measured by using a whole-cell [3H]sulpiride binding assay described previously (28). HEK293 cells stably expressing wild-type or mutant D2 receptors were transiently transfected with pcDNA3.1, wild type or mutant arrestin3, and/or GRK2 as described above. After overnight incubation with the transfection complexes, cells were harvested and split into three plates, one for confirmation of arrestin3 and/or GRK2 protein expression (described below) and two for the internalization assay. Forty-eight hours after transfection, cells were incubated with either a vehicle control, quinpirole (10 μm), or dopamine (10 μm) for 30 min at 37 °C, washed three times with ice-cold PBS, and scraped into 2 ml of cold assay buffer (PBS containing 2 mm EDTA and 0.01% BSA). The total number of cells in each condition was determined using a NucleoCounter (Chemometec, Lillerød, Denmark), and 300,000 cells were incubated in assay buffer at a volume of 1 ml with the membrane-impermeant antagonist [3H](−)-sulpiride (final concentration, 5 nm) for 3 h at 4 °C with or without unlabeled haloperidol (10 μm). Membranes were harvested, and radioactivity was measured as described previously (9).
Immunoblot
To confirm expression of arrestin3 and/or GRK2, cells were washed twice with cold PBS and lysed in ice-cold radioimmune precipitation assay lysis buffer (Millipore, Billerica, MA) containing phosphatase inhibitor mixture set II (1:100) and protease inhibitor mixture set III (1:200) for 20 min with gentle agitation at 4 °C. Samples were then centrifuged for 15 min (14,000 × g, 4 °C), and the protein concentration of the supernatant was determined using a BCA protein assay. Proteins (30–40 μg total) were separated by SDS-PAGE on CriterionTM 10% Tris-HCl precast gels (Bio-Rad) and transferred onto PVDF membranes (Millipore). Membranes were washed once in TBS, blocked in blocking buffer (TBS containing 5% BSA) for 1 h at room temperature, and incubated in mouse anti-β-arrestin2 (1:300) and rabbit anti-GRK2 (1:1000) antibodies overnight at 4 °C. After primary antibody incubation, membranes were washed three times for 5 min in TBS containing 0.1% Tween 20 (TBST; Sigma-Aldrich), incubated with Alexa Fluor® 488 goat anti-mouse (1:500) and Alexa Fluor® 568 goat anti-rabbit (1:500) secondary antibodies diluted in blocking buffer for 2 h at room temperature, and then washed three times for 5 min in TBST and once for 5 min in TBS. Fluorescence was detected using a Typhoon 9410 variable mode imager (GE Healthcare). Antibody was then stripped off the membrane using RestoreTM Western blot stripping buffer (Thermo Scientific) per the manufacturer's protocol and vigorously washed four times for 5 min in TBS. The membrane was then blocked in blocking buffer for 1 h at room temperature and incubated with rabbit anti-actin (1:500) diluted in blocking buffer overnight at 4 °C. Membranes were then washed, incubated in Alexa Fluor® 568 goat anti-rabbit, and washed again, and fluorescence was detected as described above.
RESULTS
D2-A4 Is Not Suitable for in Vivo Analyses of D2-mediated Pathways
Mutation of amino acid residues 212–215 in IL3 of the D2 receptor to alanines (D2-A4) creates a receptor that does not recruit arrestin3 and retains some G protein signaling but is poorly expressed at the cell membrane (9). To confirm these results, radioligand binding was used to determine D2-A4 membrane expression, and BRET-based assays were used to monitor receptor-mediated G protein activation and arrestin3 recruitment. When a constant amount of plasmid DNA encoding wild type D2 receptor (D2-WT) or D2-A4 was transiently transfected into HEK293 cells, D2-A4 was expressed at ∼10% of the density of D2-WT (data not shown). For the experiments in Fig. 1, D2-A4 DNA was adjusted to obtain expression equivalent to D2-WT.
The ability of D2-WT and D2-A4 to inhibit forskolin-mediated activation of adenylyl cyclase was measured to determine the capacity of each receptor to signal through G proteins. Quinpirole stimulation of D2-WT produced a dose-dependent increase in the inhibition of cyclic AMP accumulation (Fig. 1A), whereas activation of D2-A4 with quinpirole resulted in less inhibition of adenylyl cyclase activity (Fig. 1, A and B). This suggests that D2-A4 is a poor activator of G proteins. To confirm this hypothesis, D2-A4-mediated G protein activation was measured using a BRET assay in which agonist-bound receptor induces a separation of the Gα energy donor (Gαi1-91-Rluc8) from the complemented Gβγ acceptor (mVenus-Gβ1γ2), thus decreasing the BRET signal (Fig. 1C) (22). Activation of D2-WT produced a dose-dependent increase in G protein activation (Fig. 1D), whereas activation of D2-A4 with either quinpirole or dopamine resulted in significantly less G protein activation than seen at the wild-type receptor (Fig. 1, D and E). In combination, the cyclic AMP and BRET data suggest that D2-A4 is a poor G protein activator relative to D2-WT, which appears to differ from our previous results using cells stably expressing D2-A4 and D2-WT; under those conditions, agonist stimulation of D2-A4 resulted in a decrease in cAMP that was similar to that observed for the wild type receptor, but a decrease in the potency of dopamine in one experiment in that earlier work suggests that a receptor reserve might have obscured a partial loss of coupling efficiency (9).
We used a BRET assay (24) to monitor the recruitment of mVenus-arrestin3 to Rluc8-tagged-D2-WT and D2-A4 receptors (Fig. 2A). Activation of the wild-type receptor by quinpirole produced a dose-dependent increase in the BRET ratio (Fig. 2B), which is indicative of the interaction between the receptor and arrestin3 (26). As we reported previously, quinpirole activation of D2-A4 produced essentially no increase in the BRET ratio (Fig. 2, B and C) even when membrane expression of D2-A4 was similar to the wild type receptor.
FIGURE 2.
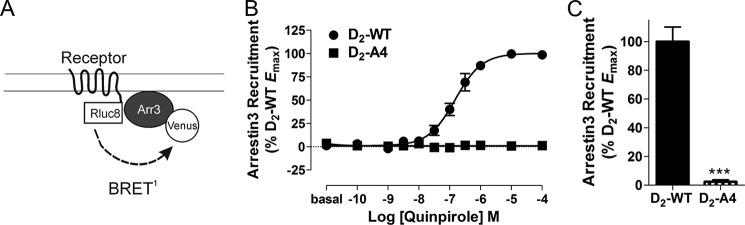
D2-A4-mediated arrestin3 recruitment. A, schematic of the BRET biosensor used to measure recruitment of arrestin to receptor. B, D2-WT-Rluc8- and D2-A4-Rluc8-mediated mVenus-Arr3 recruitment in response to incubation with quinpirole at the indicated concentrations for 10 min before measuring receptor/arrestin3 BRET. Values are expressed as the mean ± S.E. (error bars) of four independent experiments. C, maximal mVenus-Arr3 recruitment by D2-A4-Rluc8 in response to quinpirole stimulation, expressed as a percentage of the activation by D2-WT. Because a curve could not be fit to the data for D2-A4, the BRET response in the presence of 100 μm quinpirole was used as the maximal response. ***, p < 0.001 compared with D2-WT, Student's t test. Receptor density and the basal BRET for D2-WT-Rluc8 were 569 ± 111 fmol/mg protein and 0.03 ± 0.005, and for D2-A4 Rluc8, they were 722 ± 209 fmol/mg protein and 0.05 ± 0.016, respectively.
Taken together, these results suggest that despite its inability to recruit arrestin, the D2-A4 receptor would be a poor candidate for studying pathway-specific D2 receptor-mediated behavior, given the impairment of its expression and G protein signaling. Based on these results, we sought to create a receptor that has an impaired interaction with arrestin3 but unimpaired ability to activate G proteins and to be expressed at the cell membrane.
D2-A3 and D2-A2 Receptor G Protein Activation and Arrestin3 Recruitment
Residues 213–215 or residues 214 and 215 within IL3 of the D2 receptor were mutated to alanines (D2-A3 and D2-A2, respectively), and receptor activation of G protein and interaction with arrestin3 were evaluated using the techniques described above. Agonist stimulation of D2-A3 and D2-A2 produced a dose-dependent increase (Fig. 3A) in G protein activation that was similar to that of D2-WT (Fig. 3B). In contrast, the extent of quinpirole-induced recruitment of arrestin3 to the receptor was significantly less for the D2-A3 receptor than for D2-WT, with a trend for reduced recruitment of arrestin by the D2-A2 receptor mutant (59 and 76% of the wild type receptor, respectively; Fig. 3, C and D). Dopamine stimulation also produced modestly reduced arrestin3 recruitment to the receptor by the D2-A3 and D2-A2 receptor mutants that was not statistically different from recruitment by D2-WT (Fig. 3D). To monitor receptor-induced translocation of arrestin to the plasma membrane in a manner that does not require Rluc8 to be attached to the C terminus of the receptor, matching the internalization assay used below, we developed an assay in which untagged receptors were cotransfected with BRET sensors Rluc8-arrestin3-Sp1 and a plasma membrane marker, mem-linker-citrine-SH3 (see “Experimental Procedures”). Relative arrestin translocation to the membrane induced by the wild type D2 receptor, D2-A3, and D2-A2 closely matched the recruitment to the receptor observed using Rluc8-tagged receptors, except that none of the reductions in agonist-induced translocation of arrestin to the membrane was significant (Fig. 3, E and F). Sulpiride (10 μm) completely blocked arrestin translocation in response to activation of all three receptors, demonstrating the specificity of the response in our novel BRET arrestin translocation assay (data not shown). Importantly, despite modest reductions in arrestin membrane translocation and recruitment to the receptor, both D2-A3 and D2-A2 receptors still robustly recruited arrestin3 in response to agonist stimulation.
Receptor Internalization
One of the functional consequences of arrestin3 interaction with the D2 receptor is receptor internalization. Agonist-induced receptor internalization was measured using a whole-cell binding assay in HEK293 cells that stably express D2-WT, D2-A4, D2-A3, or D2-A2. We hypothesized that the amount of agonist-induced receptor endocytosis would be positively correlated with agonist-induced recruitment of arrestin3. Indeed, after treatment with quinpirole for 30 min, cell surface expression of the D2-WT receptor was significantly decreased (17 and 30% in the absence and presence of overexpressed arrestin3, respectively; Fig. 4A), whereas D2-A4 did not internalize at all in response to agonist stimulation, even when arrestin3 was overexpressed (Fig. 4, A and B). Quinpirole stimulation of D2-A2 receptors produced receptor internalization that was 44% of internalization of the wild-type receptor (Fig. 4A). Overexpression of arrestin3 with the D2-A2 receptor increased agonist-induced receptor internalization, consistent with the ability of this receptor to recruit arrestin3, although even in the presence of overexpressed arrestin, quinpirole-induced internalization of D2-A2 was less than that of D2-WT (Fig. 4A). Surprisingly, although D2-A3 also partially recruited arrestin, the D2-A3 mutant did not undergo either dopamine or quinpirole-induced internalization, even in cells that overexpressed arrestin3 (Fig. 4, A and B).
FIGURE 4.
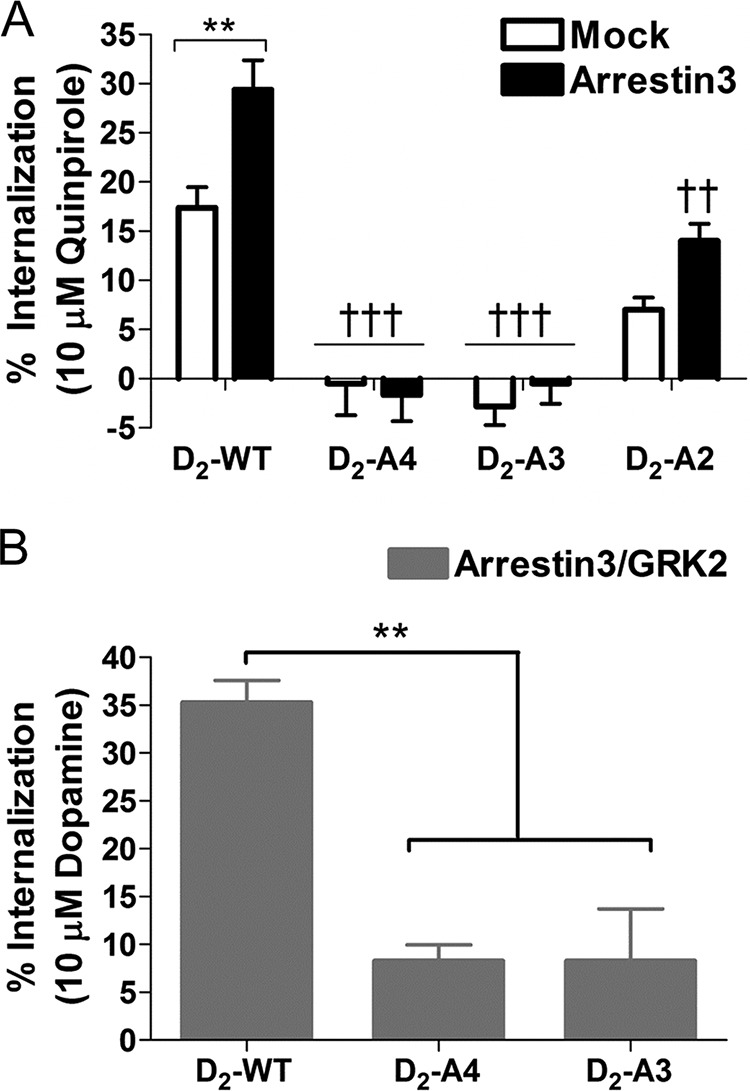
Agonist-induced receptor internalization. A, HEK293 cells stably expressing D2-WT, D2-A4, D2-A3, or D2-A2 transiently transfected with or without arrestin3 were treated with quinpirole (10 μm, 30 min) prior to measuring the loss of surface receptors using a whole-cell binding assay. Each bar represents the mean ± S.E. (error bars) (n = 5–8) of the quinpirole-induced decrease in binding. **, p < 0.01, mock transfection versus arrestin3; ††, p < 0.01; †††, p < 0.001 compared with D2-WT (same transfection), Tukey's multiple comparison test. B, HEK293 cells stably expressing D2-WT, D2-A4, or D2-A3 transiently transfected with arrestin3 and GRK2 were treated with dopamine (10 μm, 30 min) prior to measuring the loss of surface receptors using a whole-cell binding assay. Each bar represents the mean ± S.E. (n = 4) of the dopamine-induced decrease in binding. **, p < 0.01, compared with D2-WT, Tukey's multiple comparison test.
Kinetics of Arrestin3 Recruitment to D2 Receptor
The discrepancy between the amount of arrestin3 recruited to the D2-A3 receptor and amount of receptor internalized could be due to a difference in the time of measurement between the assays (10 min of agonist stimulation in the arrestin recruitment BRET assay and 30 min in the internalization assay) (i.e. the interaction of arrestin3 and D2-A3 might be relatively transient and absent after 30 min of agonist stimulation). To address this possibility, the time course of the BRET response to quinpirole or dopamine was determined for the D2-WT and D2-A3 receptors. The onset and gradual decay of the response appeared similar for the two receptors, with the D2-A3 BRET response being significantly less than the response to the wild type receptor after 10–30 min of quinpirole stimulation (Fig. 5A). Notably, the ratio of the BRET response for D2-A3 to the response for D2-WT was stable from 10 min (0.69) to 30 min (0.66) (Fig. 5A). Similar results were observed for the time course of the response of the two receptors to dopamine (data not shown). Thus, the failure of D2-A3 to internalize does not reflect a more transient association with arrestin3.
FIGURE 5.
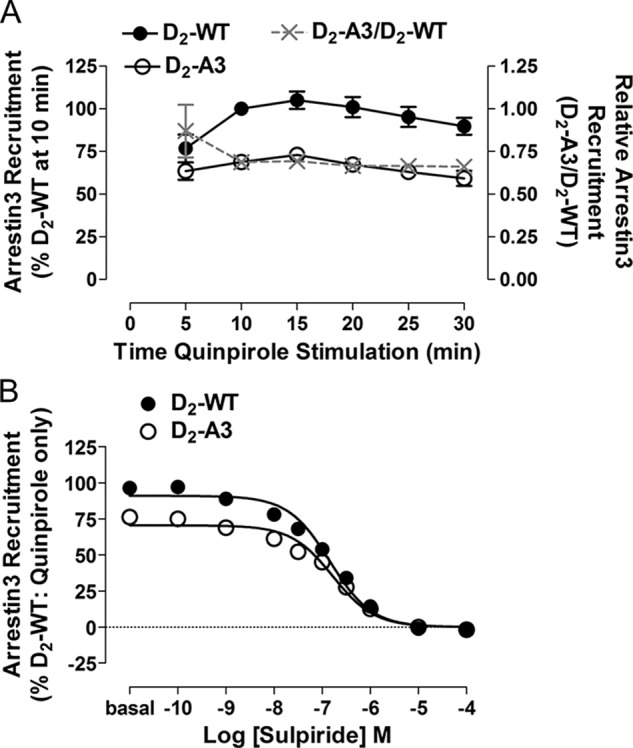
Characterization of quinpirole-induced recruitment of arrestin3. A, time course of mVenus-Arr3 recruitment to D2-WT-Rluc8 and D2-A3-Rluc8 receptors during treatment with quinpirole (1 μm). Each point represents the mean ± S.E. (error bars) of four independent experiments. Also depicted in the figure is the ratio of the responses mediated by D2-A3 and D2-WT (A3/WT) at each time point. Expression of D2-WT-Rluc8 was 964 ± 569 fmol/mg protein, and expression of D2-A3-Rluc8 was 730 ± 143 fmol/mg protein. B, inhibition of the quinpirole-induced recruitment of arrestin3 by the antagonist sulpiride. HEK293 cells expressing D2-WT-Rluc8 or D2-A3-Rluc8 with mVenus-Arr3 were pretreated with (S)-(−)-sulpiride at the indicated concentrations for 15 min at room temperature before the addition of quinpirole (10 μm, 10 min). Results are expressed as a percentage of quinpirole-induced BRET for D2-WT in the absence of sulpiride. Each value represents the mean ± S.E. of four independent experiments. Receptor density and the basal BRET for D2-WT-Rluc8 were 733 ± 41 fmol/mg protein and 0.06 ± 0.005, and the values for D2-A3-Rluc8 were 698 ± 87 fmol/mg protein and 0.12 ± 0.007.
D2-A3 Interaction with Arrestin3 Occurs at the Cell Membrane
Because the BRET response being measured is from populations of whole cells rather than at the subcellular level (29), the interaction between D2-A3 and Arr3 may be occurring at a location other than the cell membrane (i.e. within intracellular compartments, from which, by definition, internalization cannot occur). To confirm that agonist-induced BRET takes place at the cell membrane, we pretreated cells expressing D2-WT or D2-A3 with the membrane-impermeable D2 receptor antagonist, (S)-(−)-sulpiride (30), before treatment with quinpirole (1 μm). When the cells were pretreated with increasing concentrations of sulpiride, there was a dose-dependent decrease in quinpirole-induced arrestin3 interactions with both receptors (Fig. 5B). As would be expected, D2-A3 recruited significantly less arrestin3 than D2-WT in response to quinpirole (72 ± 3%, p = 0.02, Student's t test). The potency of sulpiride, however, was similar (Table 1), and the BRET signal was completely abolished in both the D2-WT and D2-A3 receptor-expressing cells at concentrations of sulpiride above 1 μm. These results suggest that quinpirole-induced recruitment of arrestin3 is due to activation of receptors on the cell membrane and not receptors expressed in intracellular compartments that are not subject to internalization.
TABLE 1.
pEC50 values from G protein activation and recruitment assays
Quinpirole dose-response curves were generated, and the pEC50 values were determined using GraphPad Prism. Each value represents the mean ± S.E. of 3–7 independent experiments. The figure in which the corresponding dose-response curves are depicted is also indicated.
| Assay | Figure | Receptor |
|||
|---|---|---|---|---|---|
| WT | A4 | A3 | A2 | ||
| cAMP inhibition (quinpirole) | Fig. 1A | 8.12 ± 0.44 | 8.17 ± 0.43 | ||
| G protein activation (quinpirole) | Figs. 1D and 3A | 6.27 ± 0.10 | 6.24 ± 0.36 | 6.64 ± 0.14 | 6.26 ± 0.05 |
| G protein activation (dopamine) | NSa | 7.00 ± 0.09 | 6.46 ± 0.31 | 7.23 ± 0.08 | 6.91 ± 0.17 |
| Arrestin3 recruitment (quinpirole) | Figs. 2B and 3C | 6.78 ± 0.06 | NDb | 6.76 ± 0.07 | 6.89 ± 0.08 |
| Arrestin3 recruitment (dopamine) | NS | 7.13 ± 0.04 | 7.32 ± 0.06 | 7.14 ± 0.05 | |
| Arrestin3 translocation (quinpirole) | Fig. 3E | 6.46 ± 0.09 | 6.63 ± 0.10 | 6.50 ± 0.10 | |
| Arrestin3 translocation (dopamine) | NS | 6.61 ± 0.08 | 6.60 ± 0.17 | 6.66 ± 0.13 | |
| Arrestin3 recruitment (quinpirole + sulpiride) | Fig. 5B | 6.84 ± 0.07 | 6.78 ± 0.03 | ||
| GRK2 recruitment (quinpirole) | Fig. 6B | 6.70 ± 0.05 | 7.00 ± 0.64 | ||
| GRK2 recruitment (dopamine) | NS | 6.74 ± 0.03 | 6.92 ± 0.07 | ||
| Arrestin3 recruitment (−GRK2, quinpirole) | Fig. 7A | 6.99 ± 0.02 | 6.84 ± 0.04 | ||
| Arrestin3 recruitment (+GRK2, quinpirole) | Fig. 7A | 7.55 ± 0.05c | 7.52 ± 0.04c | ||
| β2-Adaptin recruitment to arrestin3 (quinpirole) | Fig. 9B | 6.61 ± 0.13 | 6.50 ± 0.21 | ||
| β2-Adaptin recruitment to receptor (quinpirole) | Fig. 10B | 7.12 ± 0.19 | 7.02 ± 0.24 | ||
| β2-Adaptin recruitment to receptor (dopamine) | Fig. 10D | 7.49 ± 0.08 | 7.51 ± 0.05 | ||
a NS, not shown.
b ND, not detectable.
c p < 0 .001, compared with the same receptor without GRK2 (−GRK2), Tukey's multiple-comparison test.
GRK2 Recruitment to the D2-WT and D2-A3 Receptors
Typically, phosphorylation of GPCRs by GRKs produces an increased affinity of arrestin for the receptor, resulting in endocytosis (31). We hypothesized that the A3 mutation within the D2 receptor may impair the ability of the receptor to interact with GRK2, subsequently disrupting the ability of the receptor to recruit arrestin3 and undergo receptor internalization. To test this hypothesis, we first measured the amount of BRET between GRK2-Venus and D2-WT or D2-A3 receptors fused to Rluc8 (Fig. 6A). Quinpirole produced a dose-dependent increase in recruitment of GRK2 to D2-WT and, to a lesser extent, to D2-A3 (62% of WT) (Fig. 6, B and C). Similarly, dopamine-induced recruitment of GRK2 to D2-A3 was significantly reduced when compared with recruitment to D2-WT (Fig. 6C).
The functional consequences of GRK2 interactions with D2-WT and D2-A3 were then assessed by overexpressing GRK2 and measuring recruitment of arrestin3 and agonist-induced receptor internalization. GRK2 was overexpressed in cells expressing D2-WT-Rluc8 or D2-A3-Rluc8 and mVenus-Arr3 (Fig. 2A). Similar to previous experiments, quinpirole stimulation induced less recruitment of Arr3 to D2-A3 than to D2-WT (Fig. 7A). Interestingly, when GRK2 was overexpressed with D2-WT and D2-A3, at both receptors there was an almost 5-fold leftward shift in the quinpirole dose-response curve (Fig. 7A and Table 1) and an increase in the maximal quinpirole-induced interaction with arrestin3 (53 and 37%, respectively; Fig. 7, A and B). Furthermore, when GRK2 was overexpressed with D2-A3, the Emax for quinpirole-induced recruitment of arrestin3 to the receptor was indistinguishable from that for D2-WT expressed alone (i.e. without GRK2; Fig. 7A). These results suggest that although the A3 mutation partially disrupts interactions with GRK2, GRK2-enhanced recruitment of arrestin3 remains intact.
FIGURE 7.

Influence of GRK2 on receptor recruitment of arrestin3 and internalization. A, HEK293 cells transfected with D2-WT-Rluc8 or D2-A3-Rluc8 and mVenus-Arr3, with or without co-transfected GRK2, were treated with quinpirole at the indicated concentrations for 10 min prior to measuring receptor/arrestin BRET. Each value represents the mean ± S.E. (error bars) of four independent experiments. B, maximal mVenus-Arr3 recruitment in cells containing D2-WT-Rluc8 or D2-A3-Rluc8 with GRK2 overexpression, determined from the individual concentration-response curves and expressed as a percentage of the response to each receptor in the absence of overexpressed GRK2. ***, p < 0.001 compared with without GRK2, Tukey's multiple comparison test. Receptor density and basal BRET were 964 ± 56 fmol/mg protein and 0.07 ± 0.003 for D2-WT-Rluc8, 890 ± 56 fmol/mg protein and 0.07 ± 0.002 for D2-WT-Rluc8 with GRK2, 730 ± 143 fmol/mg protein and 0.11 ± 0.006 for D2-A3-Rluc8, and 639 ± 75 fmol/mg protein and 0.11 ± 0.007 for D2-A3-Rluc8 with GRK2. C, representative immunoblot (WB) of GRK2, arrestin3 (Arr3), and actin immunoreactivity in cells stably expressing D2-WT or D2-A3 with or without overexpression of GRK2 or arrestin3. D, HEK293 cells stably expressing D2-WT or D2-A3 were transiently transfected with pcDNA3.1, pcDNA3.1 and GRK2, or arrestin3 and GRK2. Quinpirole (10 μm, 30 min)-induced receptor internalization was measured using a whole-cell binding assay. Each bar represents the mean ± S.E. (error bars) (n = 6–8). *, p < 0.05; ***, p < 0.001 comparing among transfection conditions; †††, p < 0.001 compared with D2-WT for the same transfection conditions, Tukey's multiple comparison test.
Overexpression of GRK2 increased the amount of agonist-induced internalization of D2-WT (Fig. 7D), and internalization was further enhanced by overexpression of both GRK2 and arrestin3. Similar to previous results, quinpirole failed to induce internalization of D2-A3 even when GRK2 and arrestin3 were overexpressed. Although overexpression of GRK2 enhanced D2-A3-mediated recruitment of arrestin3 to a level comparable with that of D2-WT alone (Fig. 7, A and B, and Table 1), overexpression of GRK2 alone or in combination with arrestin3 failed to rescue internalization of D2-A3 (Fig. 7D).
The Role of Arrestin Interactions with Clathrin and AP2 in D2 Receptor Internalization
Arrestin-mediated endocytosis of GPCRs, such as β2-adrenergic receptor, has been shown to depend on the interaction of arrestin with clathrin and the endocytic adaptor AP2, which targets the receptors to clathrin-coated pits (32). A possible explanation for our results is that the A3 mutation in D2 disrupts agonist-induced receptor endocytosis by preventing the association of the receptor or arrestin3 with these endocytic proteins. The primary clathrin binding site in arrestin is an LϕXϕ(D/E) motif (33), where X is any residue and ϕ is a bulky hydrophobic residue, whereas AP2 binds to a positively charged arginine residue; both binding sites are located in the C-terminal region of arrestin (34). Dominant negative variants of arrestin carrying either deletions or mutations of these binding sites are deficient in mediating internalization and have been used to probe the mechanisms of arrestin-dependent internalization of GPCRs (33–35).
To investigate the role of clathrin and AP2 in arrestin-dependent internalization of the D2 receptor, HEK293 cells stably expressing D2-WT were transfected with GRK2 and either Arr3-WT, Arr3-ΔLIEFD, Arr3-R395E, Arr3-ΔLIEFD/R395E, or the control plasmid pcDNA3.1. Incubating cells expressing D2-WT and Arr3-WT with quinpirole (10 μm, 30 min) produced robust internalization of the receptor that was significantly greater than cells expressing the control plasmid (Fig. 8). In contrast, none of the three arrestin mutants increased D2 receptor internalization over the amount of internalization observed in the absence of overexpressed arrestin, indicating that, indeed, interactions with both AP2 and clathrin are required for arrestin3-mediated internalization of the D2 receptor.
FIGURE 8.
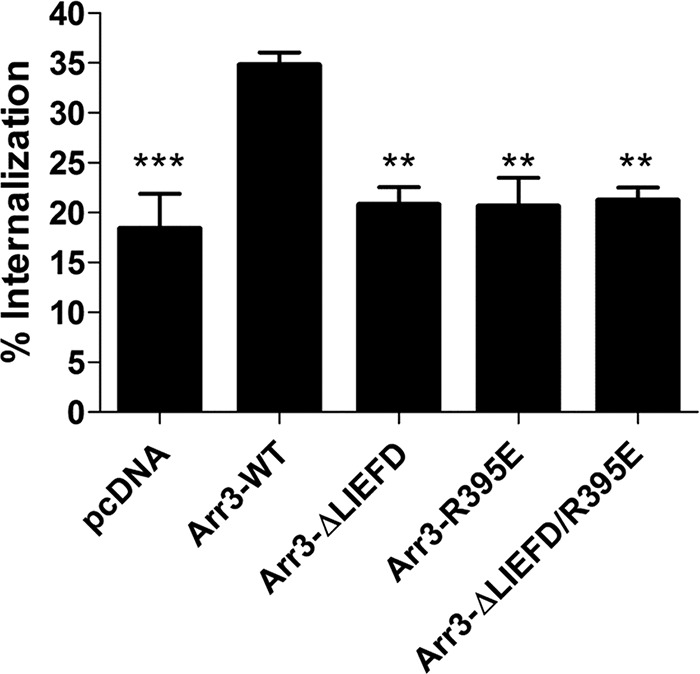
Clathrin- and AP-2-mediated internalization of D2-WT. HEK293 cells stably expressing D2-WT were transfected with GRK2 and either pcDNA3.1, wild type arrestin3 (Arr3-WT), or arrestin3 mutants: Arr3-ΔLIEFD, Arr3-R395E, or Arr3-ΔLIEFD/R395E. Cells were treated with quinpirole (10 μm) for 30 min, and internalization was assessed using the whole-cell binding technique. Each bar represents the mean ± S.E. (error bars) of five independent experiments. **, p < 0.01; ***, p < 0.001 compared with Arr3-WT, Tukey's multiple comparison test.
β2-Adaptin Interaction with the Receptor-Arrestin Complex
Adaptor protein complexes are essential for cargo selection and subsequent clathrin-mediated endocytosis (36). For example, agonist stimulation of some GPCRs has been shown to induce a direct interaction between arrestin3 and β2-adaptin to initiate clathrin-mediated receptor endocytosis (25, 37), although this has not been studied in the D2 receptor. We hypothesized that the failure of D2-A3 to internalize in response to quinpirole may be due to its inability to induce the interaction between arrestin3 and β2-adaptin or to directly interact with adaptor protein complexes, despite having the ability to partially recruit GRK2 and arrestin3.
To test this hypothesis, the interaction between Arr3-Rluc8 and β2-adaptin-EYFP was measured as described previously (25) in cells containing untagged D2-WT or D2-A3 receptors (Fig. 9A). Similar to the interaction between receptor and arrestin, quinpirole stimulation of D2-WT and D2-A3 produced a dose-dependent increase in the interaction between arrestin3 and β2-adaptin, and the maximal response was significantly less for the mutant receptor (Fig. 9, B and C). The reduced interaction between β2-adaptin and arrestin3 mediated by D2-A3 receptor is most likely due to the reduced ability of the receptor to interact with arrestin3.
We sought to determine whether the agonist-induced interaction between arrestin3 and β2-adaptin produced an interaction between D2-WT and β2-adaptin, critical for internalization, that might not be observed between D2-A3 and β2-adaptin. HEK293 cells were transiently transfected with D2-WT-Rluc8 or D2-A3-Rluc8 and β2-adaptin-EYFP, and the interaction between the two proteins in response to quinpirole stimulation was monitored using BRET (Fig. 10A). Quinpirole stimulation of either D2-WT or D2-A3 produced a negligible increase in the interaction between the receptor and β2-adaptin (Fig. 10B). When the receptor-β2-adaptin BRET pair was co-transfected with arrestin3 and GRK2, however, robust recruitment of β2-adaptin was seen in response to agonist stimulation (Fig. 10, B and D). The BRET observed was significantly lower for D2-A3 than for the wild type D2 receptor (Fig. 10, C and E). The reduced response for D2-A3 is similar in magnitude to the mutant's reduced ability to recruit arrestin3 (Fig. 3, C and D) and promote the interaction between arrestin3 and β2-adaptin (Fig. 9, B and C). These data indicate that D2-A3 has a reduced ability to recruit arrestin3, resulting in a diminished but still quite substantial ability to bind β2-adaptin.
FIGURE 10.

Interaction between receptors and β2-adaptin. A, schematic of the BRET biosensor used to detect receptor recruitment of β2-AP2-EYFP. B, HEK293 cells transiently transfected with either D2-WT-Rluc8 or D2-A3-Rluc8 and β2-AP2-EYFP were co-transfected with a control vector or arrestin3 and GRK2 (+Arr3/GRK2). Cells were then treated with quinpirole at the indicated concentrations for 20 min, and β2-AP2-EYFP recruitment was measured using BRET. Each value represents the mean ± S.E. (error bars) of four independent experiments. C, maximal β2-AP2-EYFP recruitment in cells expressing D2-WT or D2-A3 with (+Arr3/GRK2) or without (−Arr3/GRK2) arrestin3 and GRK2 determined from the individual concentration-response curves and expressed as a percentage of the maximal response to D2-WT + Arr3/GRK2. Because a curve could not be fit to data from most of the experiments without added Arr3/GRK2, the BRET response in the presence of 10 μm quinpirole was used as the maximal response. **, p < 0.01; ***, p < 0.001, Tukey's multiple comparison test. Receptor density and basal BRET were 686 ± 120 fmol/mg protein and 0.001 ± 0.0003 for D2-WT-Rluc8, 834 ± 133 fmol/mg protein and 0.002 ± 0.001 for D2-WT-Rluc8 + Arr3/GRK2, 1209 ± 335 fmol/mg protein and 0.001 ± 0.001 for D2-A3-Rluc8, and 1327 ± 19 fmol/mg protein and 0.004 ± 0.001 for D2-A3-Rluc8 + Arr3/GRK2. D, HEK293 cells transiently transfected with either D2-WT-Rluc8 or D2-A3-Rluc8 and β2-AP2-EYFP were co-transfected with arrestin3 and GRK2. Cells were then treated with dopamine at the indicated concentrations for 20 min, and β2-AP2-EYFP recruitment was measured using BRET. Each value represents the mean ± S.E. of four independent experiments. E, maximal β2-AP-EYFP recruitment in cells expressing D2-WT or D2-A3 with arrestin3 and GRK2 determined from the individual concentration-response curves and expressed as a percentage of the maximal response of D2-WT. **, p < 0.01, Student's t test. Receptor density and basal BRET were 1069 ± 177 fmol/mg protein and 0.003 ± 0.001 for D2-WT-Rluc8 and 1231 ± 158 fmol/mg protein and 0.004 ± 0.001 for D2-A3-Rluc8.
DISCUSSION
We previously identified a four-residue segment of the D2 receptor, residues 212–215 in IL3, that is required for recruitment of arrestin and receptor internalization but not for G protein-dependent signaling (9), a finding that we confirmed in the present study using agonist-induced BRET between the D2 receptor and arrestin3 to measure arrestin recruitment. The D2-A4 mutant also demonstrated a considerable reduction in its ability to activate heterotrimeric G proteins, as determined by measuring the inhibition of cyclic AMP accumulation and BRET between Gαi1 and mVenus-Gβ1γ2. Thus, we made more limited mutations in an attempt to selectively impair recruitment of arrestin. Instead, we identified a mutant that failed to undergo agonist-induced internalization despite retaining a substantial ability to recruit arrestin3.
Mutants D2-A3 and D2-A2 were created with residues 213–215 or residues 214 and 215, respectively, substituted with alanine. In contrast to D2-A4, both of these mutants had only modestly decreased ability to recruit arrestin or induce translocation to the membrane, which was more evident with the synthetic agonist quinpirole than with dopamine and significantly decreased only for quinpirole at D2-A3-Rluc8. Both of the mutants were able to fully activate Gαi.
As we reported previously (9), the agonist quinpirole was unable to induce internalization of the D2-A4 receptor, compared with robust internalization of the wild type receptor that was enhanced by overexpression of arrestin3. Treatment with quinpirole induced partial internalization of D2-A2 that was commensurate with the partial ability of that mutant receptor to recruit arrestin. In contrast, D2-A3 displayed little or no quinpirole- or dopamine-induced internalization, despite retaining substantial ability to recruit arrestin.
We evaluated several hypotheses for the disjunction between the ability of D2-A3 to recruit arrestin and its inability to be internalized. One hypothesis was that, although D2-A3 recruited a substantial amount of arrestin3 when measured after 10 min of agonist treatment, the interaction between D2-A3 and arrestin3 was less stable than that between the wild type D2 receptor and arrestin3. We determined, however, that the time course of the recruitment of arrestin3 by the wild type and mutant receptors was indistinguishable for both dopamine and quinpirole. A second hypothesis was that the interaction between arrestin3 was taking place somewhere other than at the cell membrane and therefore would not lead to the receptor being internalized; however, the ability of the hydrophilic antagonist sulpiride to inhibit the quinpirole-induced response indicated that the response came from receptors at the plasma membrane. A third hypothesis was that the mutation impaired the ability of GRK2 to bind to the receptor, and, indeed, we determined that the BRET response between the D2 receptor and GRK2 was decreased by the A3 mutation. In vitro data using fragments of the receptor strongly support the hypothesis that the A3/A4 sequence is required for binding of arrestin3 to IL3 of the D2 receptor (9); it is, however, unknown whether the same is true for GRK2. Despite the reduction in the agonist-induced BRET response, overexpression of GRK2 enhanced quinpirole-induced arrestin3 recruitment to D2-A3 by approximately the same percentage as recruitment of arrestin3 to the wild type D2 receptor, and quinpirole concentration-response curves for both receptors were shifted to the left in the presence of added GRK2, suggesting that altered binding of GRK2 to D2-A3 does not underlie its inability to be internalized.
Knockdown of either GRK2 or arrestin3 significantly reduces the amount of agonist-induced D2 receptor internalization (10, 38), and overexpression of either protein enhances the amount of internalization seen in response to agonist (8, 20, 39, 40). Furthermore, this response is potentiated when GRK2 and arrestin3 are overexpressed together (8, 20, 39). Thus, it is possible that the reduced internalization of D2-A3 simply reflects its diminished GRK2 and arrestin3 recruitment, with a partial recruitment deficit somehow translating into a more severe internalization deficit. However, because overexpression of GRK2 increased the amount of arrestin3 recruitment by D2-A3 to the level recruited by the wild type receptor, as quantified by the BRET assay, overexpression of GRK2 would be predicted to increase agonist-induced internalization of D2-A3 to the same level observed for the wild type receptor in the absence of overexpressed GRK2. This was found not to be the case because little or no internalization of D2-A3 was detected even when arrestin3 recruitment was normalized by GRK2 overexpression.
Arrestin-mediated endocytosis via clathrin-coated pits requires binding of arrestin to both clathrin and AP2, and deletion of the clathrin binding motif (Arr3-ΔLIEFD) or mutation of the arginine that is necessary for binding to AP2 (Arr3-R395E) prevents binding to clathrin and AP2, respectively, and greatly impairs agonist-mediated internalization of the β2-adrenergic receptor (35). Using these mutants, we determined that arrestin-dependent D2 receptor internalization was dependent on the interaction of arrestin3 with both AP2 and clathrin and that preventing either interaction produced complete inhibition of arrestin3-mediated internalization.
AP2 is a heterotetramer that consists of two large subunits (α2 and β2), a medium subunit (μ2), and a small subunit (σ2); the β2 (β2-adaptin) and μ2 subunits determine cargo selection by direct interaction with the cargo or accessory proteins (41). Agonist stimulation of GPCRs induces a direct interaction between receptor-bound arrestin3 and β2-adaptin to initiate clathrin-mediated receptor endocytosis (25, 37). We tested the hypotheses that the A3 mutation within the D2 receptor prevents the receptor from promoting the interaction between β2-adaptin and arrestin3 or alters an arrestin-mediated interaction between the receptor and β2-adaptin. Either effect could underlie this receptor's inability to internalize in response to quinpirole. Thus, we measured BRET between either arrestin3 or the D2 receptor and β2-adaptin-EYFP. The quinpirole-induced interaction between arrestin3 and β2-adaptin was reduced but still quite substantial in cells expressing D2-A3 compared with D2-WT. Further, although a very limited quinpirole-induced change in BRET was observed when receptor and β2-adaptin were co-expressed, the addition of arrestin3 and GRK2 enabled a robust quinpirole- or dopamine-induced interaction between these proteins. This is the first time, to our knowledge, that an energy transfer methodology has been used to measure the interaction between a GPCR and β2-adaptin. The BRET response between D2-A3 and β2-adaptin was also robust but significantly less than the response to the wild type receptor. That this closely recapitulated the mutation-induced decrease in arrestin3 recruitment indicates that it reflects the latter process rather than a specific inability of D2-A3 to induce an interaction between arrestin3 and β2-adaptin. The extremely low BRET response between the D2 receptor and β2-adaptin in the absence of overexpressed arrestin and GRK2 suggests that functional interaction of overexpressed receptor with endogenous arrestins and GRKs in HEK293 cells is very low; nevertheless, agonist-induced internalization of D2-WT without overexpressed arrestin/GRK was greater than internalization of D2-A3 with overexpressed arrestin/GRK when robust interaction with β2-adaptin was observed. This comparison between the wild type and mutant receptors highlights the qualitatively distinct inability of D2-A3 to undergo agonist-induced internalization.
In conclusion, D2-A3 is a partially signaling-biased receptor that fully activates G protein but has a moderately decreased ability to recruit arrestin3 and GRK2. D2-A3 was also deficient in arrestin-dependent interaction with β2-adaptin, most likely reflecting diminished recruitment of arrestin3 to the receptor. Despite a significant capability for recruitment of arrestin and β2-adaptin, D2-A3 displayed virtually no agonist-induced internalization even in the presence of overexpressed arrestin3 and GRK2. Arrestin probably serves as a scaffold to bring the receptor and AP2 sufficiently close to allow BRET between the C terminus of the receptor and β2-adaptin. Nonetheless, in the case of D2-A3, the precise conformation and interactions between these components may not be sufficient for internalization. Furthermore, despite the proximity between D2-A3 and β2-adaptin, D2-A3 may not be able to directly interact with other subunits of AP2 (42, 43) or with another protein or lipid required for wild type receptor internalization. The inability of D2-A3 to internalize may also reflect characteristics of the dynamic interaction among GPCR, arrestin, and AP2 that are not captured in these BRET assays. Regardless of the molecular mechanism, the identification of this mutant provides us with a construct suitable for exploring the physiological roles of D2 receptor internalization in more complex systems and in vivo.
Acknowledgments
We thank Drs. Michel Bouvier for β2-adaptin-EYFP, Celine Gales for Gαi1-91-Rluc8, Marc Caron for rat arrestin3, and Jeffrey Benovic and David Sibley for GRK2.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 MH045372 (to K. A. N.); T32 DA007262 (to C. C. C.); and K05 DA022413, R01 MH054137, and R01 DA015170 (to J. A. J.). This work was also supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development.
- GPCR
- G protein-coupled receptor
- AP2
- clathrin-associated adaptor protein complex 2
- Arr3-Rluc8
- human arrestin3 with Rluc8 fused to its C terminus
- β2-adaptin
- the β-subunit of AP2
- BRET
- bioluminescence resonance energy transfer
- CAMYEL
- cAMP sensor using YFP-Epac-RLuc
- D2-A2
- D2 receptor with residues 214 and 215 converted to alanine
- D2-A3
- D2 receptor with residues 213–215 converted to alanine
- D2-A4
- D2 receptor with residues 212–215 converted to alanine
- Rluc8
- Renilla luciferase 8
- D2-Rluc8
- human D2 receptor with Rluc8 fused to its C terminus
- D2-WT
- wild type D2 receptor
- EYFP
- enhanced yellow fluorescent protein
- GRK
- G protein-coupled receptor kinase
- IL3
- intracellular loop 3 of a GPCR
- mem-linker-citrine-SH3
- a doubly palmitoylated fragment of GAP43 linked to citrine and an SH3 domain through a serine- and glycine-rich linker
- Sp1
- SH3 domain-binding peptide used in helper interaction BRET
- V1-Gβ1
- fragment of mVenus fused to the heterotrimeric G protein β1 subunit
- V2-Gγ2
- fragment of mVenus fused to the heterotrimeric G protein γ2 subunit
- SH3
- Src homology 3.
REFERENCES
- 1. Baik J. H. (2013) Dopamine signaling in reward-related behaviors. Front. Neural Circuits 7, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jenner P. (2003) Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson's disease. Curr. Opin. Neurol. 16, Suppl. 1, S3–S7 [DOI] [PubMed] [Google Scholar]
- 3. Strange P. G. (2008) Antipsychotic drug action: antagonism, inverse agonism or partial agonism. Trends Pharmacol. Sci. 29, 314–321 [DOI] [PubMed] [Google Scholar]
- 4. Ohara K., Haga K., Berstein G., Haga T., Ichiyama A. (1988) The interaction between D-2 dopamine receptors and GTP-binding proteins. Mol. Pharmacol. 33, 290–296 [PubMed] [Google Scholar]
- 5. Obadiah J., Avidor-Reiss T., Fishburn C. S., Carmon S., Bayewitch M., Vogel Z., Fuchs S., Levavi-Sivan B. (1999) Adenylyl cyclase interaction with the D2 dopamine receptor family; differential coupling to Gi, Gz, and Gs. Cell Mol. Neurobiol. 19, 653–664 [DOI] [PubMed] [Google Scholar]
- 6. Neve K. A., Seamans J. K., Trantham-Davidson H. (2004) Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 24, 165–205 [DOI] [PubMed] [Google Scholar]
- 7. Leck K. J., Blaha C. D., Matthaei K. I., Forster G. L., Holgate J., Hendry I. A. (2006) Gz proteins are functionally coupled to dopamine D2-like receptors in vivo. Neuropharmacology 51, 597–605 [DOI] [PubMed] [Google Scholar]
- 8. Kim K. M., Valenzano K. J., Robinson S. R., Yao W. D., Barak L. S., Caron M. G. (2001) Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and β-arrestins. J. Biol. Chem. 276, 37409–37414 [DOI] [PubMed] [Google Scholar]
- 9. Lan H., Liu Y., Bell M. I., Gurevich V. V., Neve K. A. (2009) A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Mol. Pharmacol. 75, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Namkung Y., Dipace C., Urizar E., Javitch J. A., Sibley D. R. (2009) G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J. Biol. Chem. 284, 34103–34115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skinbjerg M., Ariano M. A., Thorsell A., Heilig M., Halldin C., Innis R. B., Sibley D. R. (2009) Arrestin3 mediates D2 dopamine receptor internalization. Synapse 63, 621–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajagopal S., Rajagopal K., Lefkowitz R. J. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beaulieu J. M., Sotnikova T. D., Yao W.-D., Kockeritz L., Woodgett J. R., Gainetdinov R. R., Caron M. G. (2004) Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beaulieu J. M., Sotnikova T. D., Marion S., Lefkowitz R. J., Gainetdinov R. R., Caron M. G. (2005) An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 [DOI] [PubMed] [Google Scholar]
- 15. Beaulieu J. M., Gainetdinov R. R., Caron M. G. (2009) Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347 [DOI] [PubMed] [Google Scholar]
- 16. Urban J. D., Clarke W. P., von Zastrow M., Nichols D. E., Kobilka B., Weinstein H., Javitch J. A., Roth B. L., Christopoulos A., Sexton P. M., Miller K. J., Spedding M., Mailman R. B. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1–13 [DOI] [PubMed] [Google Scholar]
- 17. Allen J. A., Yost J. M., Setola V., Chen X., Sassano M. F., Chen M., Peterson S., Yadav P. N., Huang X. P., Feng B., Jensen N. H., Che X., Bai X., Frye S. V., Wetsel W. C., Caron M. G., Javitch J. A., Roth B. L., Jin J. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 108, 18488–18493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurevich V. V., Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25, 105–111 [DOI] [PubMed] [Google Scholar]
- 19. Violin J. D., Ren X. R., Lefkowitz R. J. (2006) G-protein-coupled receptor kinase specificity for β-arrestin recruitment to the β2-adrenergic receptor revealed by fluorescence resonance energy transfer. J. Biol. Chem. 281, 20577–20588 [DOI] [PubMed] [Google Scholar]
- 20. Namkung Y., Dipace C., Javitch J. A., Sibley D. R. (2009) G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J. Biol. Chem. 284, 15038–15051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saulière A., Bellot M., Paris H., Denis C., Finana F., Hansen J. T., Altié M. F., Seguelas M. H., Pathak A., Hansen J. L., Sénard J. M., Galés C. (2012) Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat. Chem. Biol. 8, 622–630 [DOI] [PubMed] [Google Scholar]
- 22. Newman A. H., Beuming T., Banala A. K., Donthamsetti P., Pongetti K., LaBounty A., Levy B., Cao J., Michino M., Luedtke R. R., Javitch J. A., Shi L. (2012) Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 55, 6689–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang L. I., Collins J., Davis R., Lin K. M., DeCamp D., Roach T., Hsueh R., Rebres R. A., Ross E. M., Taussig R., Fraser I., Sternweis P. C. (2007) Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 282, 10576–10584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klewe I. V., Nielsen S. M., Tarpø L., Urizar E., Dipace C., Javitch J. A., Gether U., Egebjerg J., Christensen K. V. (2008) Recruitment of β-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology 54, 1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamdan F. F., Rochdi M. D., Breton B., Fessart D., Michaud D. E., Charest P. G., Laporte S. A., Bouvier M. (2007) Unraveling G protein-coupled receptor endocytosis pathways using real-time monitoring of agonist-promoted interaction between β-arrestins and AP-2. J. Biol. Chem. 282, 29089–29100 [DOI] [PubMed] [Google Scholar]
- 26. Pfleger K. D., Seeber R. M., Eidne K. A. (2006) Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat. Protoc. 1, 337–345 [DOI] [PubMed] [Google Scholar]
- 27. Grünberg R., Burnier J. V., Ferrar T., Beltran-Sastre V., Stricher F., van der Sloot A. M., Garcia-Olivas R., Mallabiabarrena A., Sanjuan X., Zimmermann T., Serrano L. (2013) Engineering of weak helper interactions for high-efficiency FRET probes. Nat. Methods 10, 1021–1027 [DOI] [PubMed] [Google Scholar]
- 28. Itokawa M., Toru M., Ito K., Tsuga H., Kameyama K., Haga T., Arinami T., Hamaguchi H. (1996) Sequestration of the short and long isoforms of dopamine D2 receptors expressed in Chinese hamster ovary cells. Mol. Pharmacol. 49, 560–566 [PubMed] [Google Scholar]
- 29. Coulon V., Audet M., Homburger V., Bockaert J., Fagni L., Bouvier M., Perroy J. (2008) Subcellular imaging of dynamic protein interactions by bioluminescence resonance energy transfer. Biophys. J. 94, 1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo N., Guo W., Kralikova M., Jiang M., Schieren I., Narendran R., Slifstein M., Abi-Dargham A., Laruelle M., Javitch J. A., Rayport S. (2010) Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology 35, 806–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reiter E., Lefkowitz R. J. (2006) GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 32. Kang D. S., Tian X., Benovic J. L. (2014) Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krupnick J. G., Goodman O. B., Jr., Keen J. H., Benovic J. L. (1997) Arrestin/clathrin interaction: localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J. Biol. Chem. 272, 15011–15016 [DOI] [PubMed] [Google Scholar]
- 34. Laporte S. A., Oakley R. H., Holt J. A., Barak L. S., Caron M. G. (2000) The interaction of β-arrestin with the AP-2 adaptor is required for the clustering of β2-adrenergic receptor into clathrin-coated pits. J. Biol. Chem. 275, 23120–23126 [DOI] [PubMed] [Google Scholar]
- 35. Kim Y. M., Benovic J. L. (2002) Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J. Biol. Chem. 277, 30760–30768 [DOI] [PubMed] [Google Scholar]
- 36. McMahon H. T., Boucrot E. (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 12, 517–533 [DOI] [PubMed] [Google Scholar]
- 37. Laporte S. A., Miller W. E., Kim K. M., Caron M. G. (2002) β-Arrestin/AP-2 interaction in G protein-coupled receptor internalization: identification of a β-arrestin binding site in β2-adaptin. J. Biol. Chem. 277, 9247–9254 [DOI] [PubMed] [Google Scholar]
- 38. Macey T. A., Gurevich V. V., Neve K. A. (2004) Preferential interaction between the dopamine D2 receptor and arrestin2 in neostriatal neurons. Mol. Pharmacol. 66, 1635–1642 [DOI] [PubMed] [Google Scholar]
- 39. Cho D. I., Zheng M., Min C., Kwon K. J., Shin C. Y., Choi H. K., Kim K. M. (2013) ARF6 and GASP-1 are post-endocytic sorting proteins selectively involved in the intracellular trafficking of dopamine D2 receptors mediated by GRK and PKC in transfected cells. Br. J. Pharmacol. 168, 1355–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iwata K., Ito K., Fukuzaki A., Inaki K., Haga T. (1999) Dynamin and Rab5 regulate GRK2-dependent internalization of dopamine D2 receptors. Eur. J. Biochem. 263, 596–602 [DOI] [PubMed] [Google Scholar]
- 41. Robinson M. S., Bonifacino J. S. (2001) Adaptor-related proteins. Curr. Opin. Cell Biol. 13, 444–453 [DOI] [PubMed] [Google Scholar]
- 42. Diviani D., Lattion A. L., Abuin L., Staub O., Cotecchia S. (2003) The adaptor complex 2 directly interacts with the α1b-adrenergic receptor and plays a role in receptor endocytosis. J. Biol. Chem. 278, 19331–19340 [DOI] [PubMed] [Google Scholar]
- 43. Paing M. M., Temple B. R., Trejo J. (2004) A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J. Biol. Chem. 279, 21938–21947 [DOI] [PubMed] [Google Scholar]