

Background: The transmembrane metalloproteinase ADAM8 promotes tumor growth and metastasis in Triple-Negative breast cancer.
Results: Three sites of N-glycosylation controlled ADAM8 processing, localization, stability, and activity.
Conclusion: N-Glycans play important roles in ADAM8 structure and function.
Significance: New insights into mechanisms regulating ADAM8 processing and activity may be exploited in future therapeutic strategies for Triple-Negative breast cancer.
Keywords: ADAM, Breast Cancer, N-linked Glycosylation, Post-translational Modification (PTM), Protein Turnover
Abstract
The transmembrane ADAM8 (A Disintegrin And Metalloproteinase 8) protein is abundantly expressed in human breast tumors and derived metastases compared with normal breast tissue, and plays critical roles in aggressive Triple-Negative breast cancers (TNBCs). During ADAM8 maturation, the inactive proform dimerizes or multimerizes and autocatalytically removes the prodomain leading to the formation of the active, processed form. ADAM8 is a glycoprotein; however, little was known about the structure or functional role of these sugar moieties. Here, we report that in estrogen receptor (ER)α-negative, but not -positive, breast cancer cells ADAM8 contains N-glycosylation, which is required for its correct processing and activation. Consistently ADAM8 dimers were detected on the surface of ERα-negative breast cancer cells but not on ERα-positive ones. Site-directed mutagenesis confirmed four N-glycosylazhytion sites (Asn-67, Asn-91, Asn-436, and Asn-612) in human ADAM8. The Asn-67 and Asn-91 prodomain sites contained high mannose, whereas complex type N-glycosylation was observed on Asn-436 and Asn-612 in the active and remnant forms. The Asn-91 and Asn-612 sites were essential for its correct processing and cell surface localization, in particular its exit from the Golgi and endoplasmic reticulum, respectively. The N436Q mutation led to decreased ADAM8 stability due to enhanced lysosomal degradation. In contrast, mutation of the Asn-67 site had only modest effects on enzyme stability and processing. Thus, N-glycosylation is essential for processing, localization, stability, and activity of ADAM8.
Introduction
ADAM8, a member of “A Disintegrin And Metalloproteinase” (ADAM)2 family of type I transmembrane glycoproteins, encodes a protein of 824 amino acids. Active ADAM8 is characterized by four extracellular domains: metalloproteinase (MP), disintegrin (DI), cysteine-rich (CRD), and an EGF-like domain (ELD) (1). These are followed by the transmembrane region and a cytoplasmic tail. ADAM8 is synthesized as an inactive 120 kDa proform, with an inhibitory N-terminal prodomain. Upon dimerization or multimerization, ADAM8 autocatalytically removes the prodomain leading to the formation of the 90 kDa active, processed form, which can be further processed to the 60 kDa remnant form by removal of the MP domain (2). In addition to autocatalysis, the MP domain can also modulate cellular signals through its sheddase activity by cleaving cell surface proteins including cytokines, growth factors, and their receptors, as well as components of the extracellular matrix (3–7). The 46-kDa type II membrane form of CD23 (mCD23), which is a well-known ADAM8 substrate, contains a C-terminal tail in the extracellular space (8, 9). ADAM8 cleavage of mCD23 releases a 29-kDa C-terminal soluble fragment (sCD23). In parallel, ADAM8 is also directly involved in protein interaction through its DI domain, which has been shown to be essential for binding to integrins and specifically the subsequent activation of the β1-integrin pathway (7).
The ability of ADAM8-null mice to develop normally suggests ADAM8 is a non-essential protein under physiological conditions (3, 10), although, it has been implicated in allergic diseases and asthmatic conditions (11), and in osteoclast differentiation (12). Importantly, overexpression of ADAM8 has been correlated with invasiveness and metastasis of several solid tumors, including lung adenocarcinoma (13, 14), pancreatic cancer (15), high-grade glioma (16), and squamous head and neck cell carcinoma (17). Recently, our laboratory demonstrated that ADAM8 is overexpressed in breast tumors and their metastases, particularly in Triple-Negative breast cancers (TNBCs) (7). Furthermore, ADAM8 was implicated in various steps of breast tumorigenesis and validated as a target for antibody-based targeted treatment of mammary tumors derived from TNBCs.
N-Glycosylation is a post-translational modification important for several physiological processes, including protein processing in the endoplasmic reticulum, trafficking through the Golgi, and protein folding and stability (18–23). Altered N-glycosylation of proteins has been linked to cancer progression (24, 25). Notably, N-glycosylation participates in the adherence of circulating tumor cells to the microvascular bed essential for extravasation and facilitates metastasis from the blood or lymph to other organs (26). Consistently, treatment of the highly aggressive sarcoma cell line S4MH with the N-glycosylation inhibitor tunicamycin resulted in loss of tumor cell adherence to endothelial cells and a significant reduction in the metastatic capacity (27). Murine ADAM8 was shown to be N-glycosylated (28). However, the sites of these post-translational modifications were not mapped or their functional roles elucidated. Here, we report that human ADAM8 is glycosylated in estrogen receptor (ER)α-negative human breast cancer cells and four sites of N-glycosylation sites demonstrated: Asn-67 and Asn-91 in the prodomain, and Asn-436 and Asn-612 in the DI and CRD, respectively. Using site-directed mutagenesis, immunofluorescence, and biochemical analysis, N-glycosylation at these sites was shown to be essential for the regulation of the cellular localization, stability, processing, and activity of ADAM8.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
N-Glycosidase F (PNGaseF) (P0704S) and endoglycosidase H (EndoH) (P0702S) were purchased from New England Biolabs (Ipswich, MA). Tunicamycin and cycloheximide were from Sigma-Aldrich and dissolved in DMSO (ABO3091) (American Bioanalytical, Natick, MA) and distilled water, respectively. Chloroquine (02193919) was purchased from MP Biomedicals (Solon, OH). Antibodies directed against the human ADAM8 cytoplasmic tail (rabbit; B4068) and ectodomain (goat; AF1031) were from LSBio (Seattle, WA) and R&D Systems (Minneapolis, MA), respectively. Antibodies against β-actin (AC-15) and β-tubulin (TUB 2.1) were from Sigma-Aldrich. Antibodies against LAMP 2 and M6PR were purchased from the Developmental Studies Hybridoma Bank at the University of Iowa. Pre-adsorbed goat anti-mouse IgG secondary antibody conjugated to horseradish peroxidase (HRP) (sc-2055), anti-c-Myc (9E10 sc-40), and anti-HA (sc-805) antibodies were purchased from Santa Cruz Biotechnology. Goat anti-rabbit IgG HRP (170–6515) and immobilized streptavidin-agarose (20349) were obtained from Bio-Rad and Thermo Scientific (Rockford, IL), respectively. Reduced serum medium Opti-MEM (31985–070), Alexa fluor 488 chicken anti-mouse IgG (A-21200), and Alexa fluor 594 chicken anti-rabbit IgG (A-21442) were from Invitrogen (Carlsbad, CA). Chemical cross-linking reagent bis (sulfosuccinimidyl) suberate (BS3), was purchased from Pierce.
Cell Lines and Culture Conditions
Human ERα-negative non-tumoral mammary epithelial MCF-10A and MDA-MB-231 breast cancer cells and ERα-positive breast cancer cell lines MCF-7, ZR-75, and BT474 were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in the medium recommended by ATCC. ERα-negative human mammary epithelial cells (HMEC), and ERα-positive MDA-MB-361 breast cancer cells were kindly provided by Charlotte Kuperwasser (Tufts University School of Medicine, Boston MA) and were maintained in mammalian epithelial growth medium (Lonza, Walksville, MD) and DMEM, respectively, supplemented with 10% fetal bovine serum (FBS) (Invitrogen). The human ERα-negative Inflammatory Breast Cancer line SUM-149 was generously provided by Stephen Ethier (University of Michigan Medical School, Ann Arbor, MI) and maintained as described (29). Human embryonic kidney (HEK)-293 cells were kindly provided by Nader Rahimi (Boston University School of Medicine, Boston, MA) and maintained in DMEM supplemented with 10% FBS. For growth under hypoxic conditions, cells were incubated at 1% oxygen, 95% nitrogen, and 5% carbon dioxide for 24 h at 37 °C. Stable clones of MDA-MB-231 with ADAM8-specific shRNA (shA8) or control shRNA (shCtrl) were kindly provided by Joerg Bartsch (Philipps-University Marburg, Department of Neurosurgery, Marburg, Germany) and maintained as described (7).
Cloned DNAs
CD23b plasmid was purchased from Addgene (Cambridge, MA) and had been deposited by Zena Werb (University of California at San Francisco). Full-length human ADAM8 (hADAM8) cDNA (MGC:134985; GenBankTM: BC115404.1) was kindly provided by Joerg Bartsch. Full-length hADAM8 was inserted into the EcoRI/HindIII site of pCDNA 3.1/Myc-His (−)Ver B vector using forward (5′- GAC GAA TTC CCG GCC ATG CGC GGC CTC GGG-3′) and reverse (5′-AAA GCT TCC GTA GGG TGC TGT GGG AGC TCC-3′) primers. To introduce mutations into the four putative sites of N-glycosylation, site-directed mutagenesis of the hADAM8 Myc/His pcDNA was performed to replace the indicated asparagines with glutamine using the overlapping extension method (30). The primers used in this study are as follows: ADAM8 N67Q, sense 5′-GCC ACA GGG CAC CAA TTC ACC CTC CAC-3′, and antisense 5′-GTG GAG GGT GAA TTG GTG CCC TGT GGC-3′; ADAM8 N91Q, sense 5′-TAT ACG GCT GCC CAA GGC TCC GAG GTG-3′, and antisense 5′-CAC CTC GGA GCC TTG GGC AGC CGT ATA-3′; ADAM8 N436Q, sense 5′-AAC CGC TGC TGC CAA TCT ACC ACC TGC-3′, and antisense 5′-GCA GGT GGT AGA TTG GCA GCA GCG GTT-3′; ADAM8 N612Q, sense 5′-TAC AGA TCC AGC CAA TGC TCT GCC CAG-3′, and antisense 5′-CTG GGC AGA GCA TTG GCT GGA TCT GTA-3′. To prepare an enzymatically inactive mutant of hADAM8, a point mutation of glutamate to glutamine E335Q was inserted using the following primers: 5′-ACC ATG GCC CAT CAG ATG GGC CAC AAC-3′ and 5′-GTT GTG GCC CAT CTG ATG GGC CAT GGT-3′. All isolated constructs were confirmed by DNA sequencing before use.
Immunoblotting
Cells were washed in 1X PBS, scraped and incubated in Lysis buffer (10 mm Tris-HCl, pH 7.5, 5 mm EDTA, 50 mm NaCl, 50 mm NaF, 1% Triton X-100) supplemented with protease and phosphatase inhibitors (PI78442, Thermo Scientific), as well as 5 mm EDTA and 10 mm phenanthroline to inhibit the autocatalytic activity of ADAM8. Lysates were sonicated and centrifuged at 16,000 × g for 15 min. Protein concentrations were calculated using a DC Protein Assay Reagent (Bio-Rad). Samples (50 μg) of these whole-cell extracts (WCEs) were separated in 8% polyacrylamide-SDS gels and analyzed by Western blotting as previously described (31). Molecular mass markers were included on each gel (1610394, Bio-Rad).
Glycosidase Digestion
To remove complex/hybrid N-glycans, protein extracts were digested with PNGaseF. Briefly, cell lysates (50 μg/μl) were incubated in 5 μl of 10× denaturation buffer (5% SDS, 0.4 m dithiothreitol (DTT) (New England Biolabs)) for 10 min at 90 °C and 5 μl of PNGase F at 500 units/μl in reaction buffer (0.5 m Sodium phosphate, pH 7.5) containing 10% Nonidet P-40 (which counteracts SDS inhibition of PNGase F) was added and the mixture incubated at 37 °C for 1 h. EndoH was used to remove high mannose N-glycans. Following the denaturation of proteins in the cell lysates as above, extracts were incubated with 50 units/μl per reaction of EndoH in 0.5 m sodium citrate, pH 5.5 at 37 °C for 1 h. Digested samples were subjected to immunoblotting.
Chemical Cross-linking
MCF-7 and MDA-MB-231 cells were grown to confluence in P60 dishes, washed in 1x PBS three times and cross-linked with membrane-non-permeable, thiol-non-cleavable cross-linker reagent BS3 at a final concentration of 2 mm for 30 min at room temperature. Then the cross linking reaction was stopped by addition of 50 mm glycine, pH 7.5 and the incubation continued for 15 min at room temperature. Following addition of Lysis buffer, the cell lysates were subjected to immunoblotting as described above. Experiments were repeated three times.
Transient Transfection
HEK-293 cells were plated at 5 × 105 cells/ml and after 24 h were transfected using the polyethylenimine (PEI) reagent (Polysciences, Warrington, PA). Briefly, 1 μg of DNA in 2 μl PEI and 500 μl Opti-MEM (31985-070, Invitrogen) was incubated for 15 min at room temperature, and then added to the culture plates. After 48 h, cells were lysed as described in “Immunoblotting.” For MDA-MB-231 and MCF-7 cells, cultures were plated at 10 × 105 cells/ml, and 4 μg of DNA were used for transfection, as above.
Cell-surface Biotinylation
Following transfection, HEK-293 cells were washed with cold PBS and incubated for 30 min at 4 °C with EZ-Link Sulfo-NHS-Biotin (21217, Thermo Scientific). Cultures were washed with 0.1 m glycine in PBS, and the cells were incubated in Lysis buffer (see “Immunoblotting”). Samples (500 μg) were incubated with 50 μl of streptavidin-agarose beads (20349, Thermo Scientific) for 2 h at 4 °C. The beads were then washed with the Lysis buffer, and the proteins released by boiling in 30 μl of 2× sample buffer (200 mm Tris HCl, pH 6.8, 40 mm DTT, 8% SDS, 0.4% bromphenol blue, 40% glycerol), and subjected to immunoblotting. The assay was performed three times.
Immunofluorescence
HEK-293 cells or SUM-149 (3 × 105 cells/ml) were plated on cover slides and transfected with empty vector (EV) DNA or plasmids expressing wild-type (WT) or the indicated N-glycosylation mutant of hADAM8, as above. After 24 h, the slides were washed with PBS and fixed with 2% electron microscopy-grade paraformaldehyde (EMS) at room temperature for 15 min. The slides were rinsed in PBS and stored in 70% ethanol at 4 °C for 16 h. To re-hydrate, slides were incubated at room temperature in PBS for 10 min, and the cells permeabilized with 0.5% Triton X-100 for 15 min. Following washing, slides were subjected to blocking with 2% (w/v) bovine serum albumin (Sigma-Aldrich) in PBS for 30 min and then incubated with the indicated specific primary antibodies for 1 h at room temperature. The slides were then washed and incubated with the secondary antibody (Alexa fluor 488 chicken anti-mouse IgG (A-21200, Invitrogen) and/or Alexa fluor 594 chicken anti-rabbit IgG (A-21442, Invitrogen)) for 1 h at room temperature. Following washing, the slides were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200, Vector Laboratories, Burligame, CA). Images were taken using a Nikon 80i Upright Research Fluorescence Scope (40x objective) with Andoe Clara-E Camera and processed with NIS-Elements Basic software.
Fluorescence-activated Cell Sorting (FACS) of ADAM8 Surface Expression
HEK-293 cells were plated at 5 × 105 cells/ml and transfected after 24 h, as described above. After 48 h, cells were detached using 1 mm EDTA in PBS and resuspended in 5 × 105 cells/100 μl of PBS. The cells were then incubated with 4 μg of goat anti-ADAM8 ectodomain antibody (AF1031, R&D Systems) for 30 min at room temperature. Cells were then washed twice in PBS and incubated with 1 μg of secondary antibody (Alexa fluor 488 chicken anti-goat IgG) for 30 min at room temperature. After washing the cells twice with PBS, 0.25 μg in 1 μl of propidium iodide (P16063, Invitrogen) was added to each tube. The cells were immediately analyzed by flow cytometry (FACSCalibur, Becton Dickinson). The data were analyzed using Summitv4.3 software. FACS analysis experiments were performed three independent times.
ADAM8 Stability and Activity Assays
To assess ADAM8 stability, cultures were treated with 25 μg/ml cycloheximide (dissolved in DMSO) or an equivalent amount of carrier DMSO (control) for the indicated times, in the absence or presence of the lysosomal inhibitor chloroquine at 50 μm. Extracts were prepared and subjected to Western blotting. Intensity of the ADAM8 proform and β-tubulin bands were quantified using ImageJ densitometry software. The amounts of ADAM8 proform normalized to the β-tubulin loading control are presented as percentage of the control sample. To test for ADAM8 activity, HEK-293 cells were plated at 5 × 105 cells/ml in P60 dishes, and after 24 h, co-transfected with 1 μg DNA of a plasmid encoding C-terminal HA-tagged CD23b and 1 μg of ADAM8 expression vector or EV pCDNA 3.1 Version B DNA. 6 h after transfection, the medium was replaced with culture medium without FBS. The serum-free medium was harvested after 16 h, and the cell debris removed by centrifugation. The supernatants were concentrated using Amicon-ultra 4 centrifugal filter units (UFC 801024, Millipore, Billerica, MA). The remaining cells attached to the dishes were washed in PBS, scraped, lysed, and subjected to immunoblotting as described above. Stable clones of MDA-MB-231 (shCtrl or shA8) were transfected with either 1 μg of EV or C-terminal HA-tagged CD23b and ADAM8 activity assay performed as described above. These assays were performed three independent times.
RESULTS
ADAM8 Undergoes N-Glycosylation in Human ERα-negative Breast Cancer Cell Lines
To assess the expression of ADAM8 and its various forms in breast cancer cells, WCEs from ERα-negative (MDA-MB-231 and SUM-149), and ERα-positive (MCF-7 and ZR-75) breast cancer cells were analyzed by Western blotting (Fig. 1A). The proform as well as the active and remnant forms of ADAM8 were all seen in ERα-negative breast cancer cells, although in somewhat different ratios, indicating processing was occurring in these lines. In contrast, ERα-positive cell lines had essentially only one ADAM8 band that ran slightly slower than the unprocessed proform seen in the ERα-negative lines (Fig. 1A). To determine whether the inability to process ADAM8 was common to ERα-positive breast cancer cells, two additional ERα-positive lines BT474 and MDA-MB-361 were examined. Again, only a more slowly migrating ADAM8 band was seen in ERα-positive BT474 and MDA-MB-361 cells (Fig. 1B). We next asked what forms of ADAM8 are expressed in untransformed breast epithelial cells, in particular ERα-negative HMEC and MCF-10A cells. The expression patterns of ADAM8 proform and active forms in the ERα-negative untransformed HMEC and MCF-10A cells was similar to the ERα-negative breast cancer lines, although their levels were lower especially the processed forms of ADAM8, as expected (Fig. 1C) (7). Thus, ADAM8 processing appears differentially regulated in ERα-negative versus -positive lines, and we next investigated whether these results were due to alternative post-translational modifications.
FIGURE 1.

ADAM8 undergoes N-glycosylation in ERα-negative breast cancer cells. A–C, samples of WCEs (50 μg) from the indicated human ERα-negative (ERα−) or ERα-positive (ERα+) breast cancer cells or untransformed breast epithelial lines (HMEC and MCF-10A) were examined by Western blotting for ADAM8 expression (LSBio antibody), and for β-tubulin as a loading control. D–F, samples of WCEs (50 μg) from the indicated ERα-negative or ERα-positive human breast cancer cell lines were compared before (−) or following (+) PNGaseF digestion using Western blotting for ADAM8 (D–E) or E-cadherin (F), and for β-tubulin. The white dashed line indicates the migratory position of the proform in ERα-negative lines. Positions of molecular mass markers are indicated. *, ns: non-specific band. MDA-231, MDA-MB-231; MDA-361, MDA-MB-361; Remt, Remnant; E-Cadh: E-cadherin.
Since ectopically expressed mouse ADAM8 in COS-7 cells was N-glycosylated (28), we sought to determine the glycosylation status of endogenous ADAM8 in the ERα-negative versus -positive human breast cancer cells. WCEs were subjected to treatment with PNGase F, which cleaves most N-glycans, including high mannose, hybrid, and complex structures at asparagine residues of N-linked glycoproteins. In both MDA-MB-231 and SUM-149 ERα-negative cells, the proform and active forms of endogenous ADAM8 displayed increased mobility with PNGaseF treatment, demonstrating that these forms were N-glycosylated (Fig. 1D). The remnant form was also susceptible to PNGaseF digestion, however, the resulting band was quite diffuse (Fig. 1D, and see Fig. 2A below). In contrast, migration of the endogenous ADAM8 proform in two ERα-positive cell lines was unaffected by treatment with PNGase F (Fig. 1E). This did not seem to be due to a general defect in the glycosylation machinery in these cells, as the endogenous E-cadherin appeared to be N-glycosylated normally as judged by the effects of PNGase F treatment of WCEs from MCF-7 cells (Fig. 1F), consistent with the findings of other groups showing N-glycosylation in these cells (22, 32, 33). Thus, endogenous ADAM8 is subjected to N-glycosylation in ERα-negative but not in ERα-positive breast cancer cells.
FIGURE 2.

ADAM8 induction by hypoxia and dimerization are differentially regulated in ERα-negative versus -positive breast cancer cells. A and B, indicated ERα-negative (A) or ERα-positive (B) breast cancer lines were cultured under normoxic (Normox.) or hypoxic (Hypox.) (1% O2) conditions for 24 h. WCEs were isolated and samples before (−) and after (+) treatment with PNGase F subjected to Western blotting for ADAM8, as above. C, samples of WCEs (50 μg) from MCF-7 and MDA-MB-231 cells before (−) or following (+) BS3 cross-linking were compared using Western blotting for ADAM8 and for β-tubulin. The white dashed line indicates the migratory position of the proform in ERα-negative MDA-MB-231 cells. MDA-231, MDA-MB-231.
Recently we demonstrated that when MDA-MB-231 cells were grown under hypoxic conditions, the levels of ADAM8 were induced and its processing was enhanced (7). Thus, we next tested the effects of hypoxia on N-glycosylation and processing of ADAM8 in ERα-negative versus -positive breast cancer lines. Following incubation of cultures under 1% oxygen for 24 h, WCEs were digested with PNGaseF and subjected to Western blotting. Large increases were seen in the levels of the proform, active, and remnant forms of ADAM8 in MDA-MB-231 cells under hypoxic conditions (Fig. 2A, left panel), consistent with our previous findings (7). Similarly, hypoxia resulted in substantial increases in ADAM8 proform and active form in the SUM-149 line (Fig. 2A, right panel). Notably, all of the ADAM8 forms were hydrolyzed with PNGase F in the two ERα-negative cell lines. In contrast, hypoxia failed to induce either the level of expression, or the glycosylation and processing of ADAM8 in the two ERα-positive cell lines tested here (Fig. 2B). Thus, hypoxia induces the expression, N-glycosylation, and processing of ADAM8 selectively in ERα-negative lines.
ADAM8 Dimers Are Not Detected on the Surface of ERα-positive Breast Cancer Cells
As the active form of ADAM8 results from autocatalytic clipping by the proform after its dimerization, we used chemical cross-linking to investigate whether the absence of processed ADAM8 forms in ERα-positive breast cancer cells could be correlated with an inability to dimerize. To this end, MCF-7 and MDA-MB-231 cells were subjected to treatment with the non-cell permeable cross-linking reagent BS3. Total protein extracts were separated under reducing conditions by SDS-PAGE and analyzed by immunoblotting for ADAM8. As shown in Fig. 2C, BS3 treatment of MDA-MB-231 cells generated an additional molecular species that migrated slightly faster than 250 kDa, which is approximately twice the apparent molecular size of the 120-kDa proform, suggesting that a portion of the ADAM8 protein on the surface of the MDA-MB-231 cells had dimerized thereby permitting autocatalytic processing to the active form. In contrast, no higher molecular species were seen with BS3 treatment of MCF-7 cells. These results are consistent with the model that ADAM8 is present exclusively as a monomer on the surface of MCF-7 cells and that it does not undergo dimerization, and hence is unable to cleave the proform in this ERα-positive line.
ADAM8 Has Both Complex and High Mannose N-Glycans in ERα-negative Breast Cancer Cells
We next sought to characterize the glycosylation modifications of ADAM8 that take place in the ERα-negative cells. The high mannose N-glycosylation of proteins that occurs in the endoplasmic reticulum is further modified in the Golgi with additional N-glycans, leading to loss of EndoH sensitivity. Thus, we evaluated the nature of the glycans on each of the ADAM8 forms in the ERα-negative lines using sensitivity to EndoH versus PNGaseF (Fig. 3A). EndoH digestion resulted in increased mobility of the ADAM8 proform in MDA-MB-231 and SUM-149 cell lysates, whereas the active form was EndoH resistant. As seen above, all of the ADAM8 forms are sensitive to PNGaseF digestion. These findings suggest that the initial carbohydrate structures added to the ADAM8 proform are high mannose oligosaccharides and that these are further modified in the active form to complex-type N-glycans.
FIGURE 3.

ADAM8 has both high mannose and complex N-glycans in ERα-negative breast cancer cells. A, WCEs from MDA-MB-231 and SUM-149 cells before (−) and after (+) digestion with PNGase F (PF) or Endo H (EH) were examined by Western blotting for ADAM8 expression, as in Fig. 1. B, MDA-MB-231 and SUM-149 cells were incubated with 1 μg/ml tunicamycin (TN) or an equivalent volume of vehicle control (−) for 24 h and WCEs prepared. A sample of WCE from control cells were treated with PNGase F (PF) and compared with untreated control or tunicamycin-treated cells by Western blotting. C, samples of WCEs (50 μg) from MCF-7 and MDA-MB-231 cells were compared before (−) or following tunicamycin treatment (+) using Western blotting for ADAM8 and for β-tubulin.
Inhibition of ADAM8 N-Glycosylation by Tunicamycin Prevents Processing
To test the role of N-glycosylation of ADAM8 in ERα-negative breast cancer lines, MDA-MB-231 and SUM-149 cells were treated for 24 h with 1 μg/ml of tunicamycin (Fig. 3B), which inhibits addition of N-linked oligosaccharides to nascent polypeptides (34). Only one band was observed upon tunicamycin treatment, which co-migrated with the faster mobility, unglycosylated proform band seen following PNGaseF treatment. The processed active and remnant forms were not detected following tunicamycin treatment. Furthermore, the level of the proform band was reduced as a result of tunicamycin treatment (Fig. 3B). In contrast, when MCF-7 cells were treated with tunicamycin (Fig. 3C), neither protein stability nor its glycosylation were affected. Together, these results suggest that inhibition of N-glycosylation of ADAM8 in ERα-negative breast cancer lines prevented processing of the proform, and that unglycosylated ADAM8 protein may be less stable.
Processing of ADAM8 Is Reduced in ADAM8 N-Glycosylation Mutants
The consensus sequence for N-glycosylation is Asn-X-Thr/Ser, with X corresponding to any amino acid, except proline. Using the NetNgly program, which takes advantage of artificial neural networks for prediction, we identified four putative strong sites of N-glycosylation, with higher than 0.5 thresholds in human ADAM8 (Fig. 4A). Two of these sites were located in the prodomain (Asn-67 and Asn-91), one within the disintegrin domain (Asn-436) and one between the cysteine-rich and EGF-like domains (Asn-612). A three-dimensional model of ADAM8 (residues 195–647 that includes the MP domain, DI domain, CRD, and the ELD) was generated using Swissmodel (35), and the crystal structure of ADAM22 as a template (36). The figure was generated using Pymol (PyMOL Molecular Graphics System, Schrödinger, LLC), and shows the location of the residues 436 and 612, both of which are predicted from this model to be surface accessible (Fig. 4B). Unfortunately, no suitable template could be found to model the N-terminal region of ADAM8.
FIGURE 4.

All four putative sites in ADAM8 are N-glycosylated. A, schematic representation of ADAM8-Myc/His protein with the different domains and putative N-glycosylation sites indicated. N-Glycosylation sites were individually mutated from Asn to Gln in the pCDNA 3.1/Myc-His (−)Ver B vector, which expresses proteins with a C-terminal Myc/His tag. Pro: prodomain; MP: metalloproteinase domain: DI: disintegrin domain; CRD: cysteine-rich domain; ELD: EGF-like domain; TM: transmembrane domain; CTD: cytoplasmic domain. B, representation of the predicted three-dimensional model of ADAM8 (195–647 residues including MP, DI, CRD, and ELD). N-Glycosylation residues 436 and 612 are highlighted in blue. Figures produced using PyMol. C and D, HEK-293 (C) or MDA-MB-231 (D) cells were transiently transfected with empty vector (EV) DNA, or with vectors expressing Myc-His-tagged WT ADAM8 or N67Q, N91Q, N436Q, or N612Q N-glycosylation mutants. Cells were lysed after 24 h, and samples subjected to immunoblot analysis of ADAM8 expression, as above, using a c-Myc antibody to detect ectopically expressed proteins. The white dashed line indicates the migratory position of the proform expressed by WT ADAM8.
To test whether ADAM8 was indeed N-glycosylated at these positions, site-directed mutagenesis was performed to individually replace the asparagine with glutamine in ADAM8 within the pCDNA 3.1/Myc-His (−) Ver B vector, which expresses Myc tagged proteins. Wild-type (WT) and mutant human ADAM8 (hADAM8) proteins were ectopically expressed in HEK-293 cells, which lack endogenous ADAM8. The ADAM8 proform of each of the mutants migrated more rapidly than the WT ADAM8 proform, suggesting all N-glycosylation sites were occupied consistent with the increased mobility following PNGaseF treatment (Fig. 4C and see below). The processing of the N67Q mutant proform was only slightly reduced compared with WT, whereas the N91Q and N612Q mutants displayed very little proform processing. Consistent with its localization, the active form of the N436Q mutant displayed faster migration than WT proteins, as did the remnant form (Fig. 4C); furthermore, the amounts of active and remnant forms were greatly reduced.
We next expressed the WT and mutant ADAM8 forms in MDA-MB-231 breast cancer cells (Fig. 4D). Again, the proform of all four mutants migrated more rapidly than the WT proform, consistent with N-glycosylation of ADAM8 at all of the sites in these cells. Furthermore, processing of the N612Q and N91Q mutants was greatly reduced, as judged by the very low levels of active and remnant forms. The N67Q and N436Q mutants had less profound effects on processing with lower relative levels of active or remnant forms compared with the proform. Taken together, our data indicate that all four sites of glycosylation are functional in both the MDA-MB-231 breast cancer and HEK-293 cells, and the loss of N-glycosylation at the Asn-91 and Asn-612 positions greatly reduces the processing of ADAM8 in these cells.
To investigate the type of glycans present in the ectopically expressed N-glycosylation mutants, we assessed their susceptibility to PNGase F and Endo H digestion following transfection in HEK-293 cells (Fig. 5, A and B). The proforms of the N67Q and N91Q mutants were digested with EndoH, indicating that both of these two sites are modified by addition of high mannose sugars. Furthermore, the N436Q and N612Q proform mutants retained EndoH sensitivity consistent with the presence of high-mannose N-glycans (Fig. 5B). The active and remnant forms of the N67Q and N436Q mutants processing by PNGaseF was similar to the WT ADAM8, although the amounts of these forms with the N436Q mutant were greatly reduced and their migration substantially faster, consistent with reduced glycosylation. Processing of the N91Q and N612Q ADAM8 mutants was again severely inhibited. Thus, these findings suggest that the preliminary N-glycosylation step at the two sites present in the proform are modified by addition of high mannose glycans, which likely takes place in the endoplasmic reticulum, and that following clipping of ADAM8 to the active form, further processing to complex sugars takes place on the Asn-436 and Asn-612 sites in the Golgi.
FIGURE 5.

Cell surface expression of ADAM8 and its N-glycosylation mutants. A and B, WCEs were isolated from HEK-293 cells transiently transfected with vectors expressing WT, N67Q or N91Q prodomain N-glycosylation mutants (A) or WT, N436Q DI, or N612Q CRD mutants (B). Samples were analyzed before (−) and after digestion with PNGase F (PF) or Endo H (EH) by Western blotting using a c-Myc antibody. The white dashed line indicates the migratory position of the proform expressed by WT ADAM8. C, HEK-293 cells transiently expressing WT ADAM8 and the indicated N-glycosylation ADAM8 mutants, were harvested and analyzed for ADAM8 cell surface expression by FACS. Unpermeabilized cells were stained using a goat anti-ADAM8 ectodomain antibody (AF1031) and a secondary Alexa fluor 488 chicken anti-goat IgG. Propidium iodide was used to exclude dead cells from analysis. Background staining in EV-transfected cells is shown in the first panel. The percentage of FITC-positive cells in R8 is indicated in each histogram.
ADAM8 N91Q and N612Q Mutants Display Altered Cellular Localization and Are Unable to Reach the Cell Surface
To address whether loss of glycosylation alters the ability of ADAM8 to reach the cell surface, we performed FACS analysis of transfected HEK-293 cells using an ADAM8 ectodomain antibody on unpermeabilized cells. HEK-293 cells were transiently transfected with vectors expressing WT or mutant ADAM8 proteins or with empty vector (EV) DNA (Fig. 5C). Consistent with the absence of endogenous ADAM8 in HEK-293 cells, almost no staining was seen with the EV DNA. A substantial increase was observed upon ectopic WT ADAM8 protein expression. The levels of WT ADAM8 protein expression were heterogenous in the mixed transiently transfected cell population, and thus the scan was divided into 4 regions. R5 represents the ADAM8-negative population, and R6-R7 the cell population characterized by low ADAM8 expression. R8 represents those cells in the population with the highest levels of ADAM8 expression on their surface. Approximately 25% of the cells displayed high WT ADAM8 cell surface expression (R8). Substantial numbers of cells were also seen in the R8 population with the N67Q and N436Q mutants (Fig. 5C), although their extents were lower than with the WT ADAM8, consistent with their reduced processing seen above (Fig. 4C). The R8 populations for N67Q and N436Q were 20 and 10%, respectively. In contrast, cells staining high for ADAM8 were almost undetectable with the N91Q and N612Q mutants (∼1 and 3%, respectively), indicating post-translational N-glycosylation at these sites are critical for correct cellular localization of ADAM8 on the cell surface.
To further test the effects of the N-glycosylation mutations on ADAM8 cellular localization, we performed immunofluorescence of ectopically expressed WT and mutant ADAM8 proteins in SUM-149 breast cancer cells using an antibody targeting the Myc-tag fused to ADAM8 (Fig. 6A). SUM-149 breast cancer cells were selected because they express high endogenous levels of E-cadherin that can be easily used as a cell-surface marker. Transfected cultures were co-stained with the ADAM8 ectodomain antibody and an E-cadherin antibody targeting its extracellular part. WT ADAM8 (red) was appropriately processed and observed at the surface of the SUM-149 cells, as shown by the co-localization (yellow) with E-cadherin (green) (Fig. 6A). The localization of the N67Q and N436Q mutant ADAM8 proteins was similar to WT ADAM8. In contrast, the N91Q and N612Q mutant ADAM8 proteins were localized to the perinuclear area and were not detected at the surface of SUM-149 cells, consistent with the findings obtained with FACS analysis.
FIGURE 6.
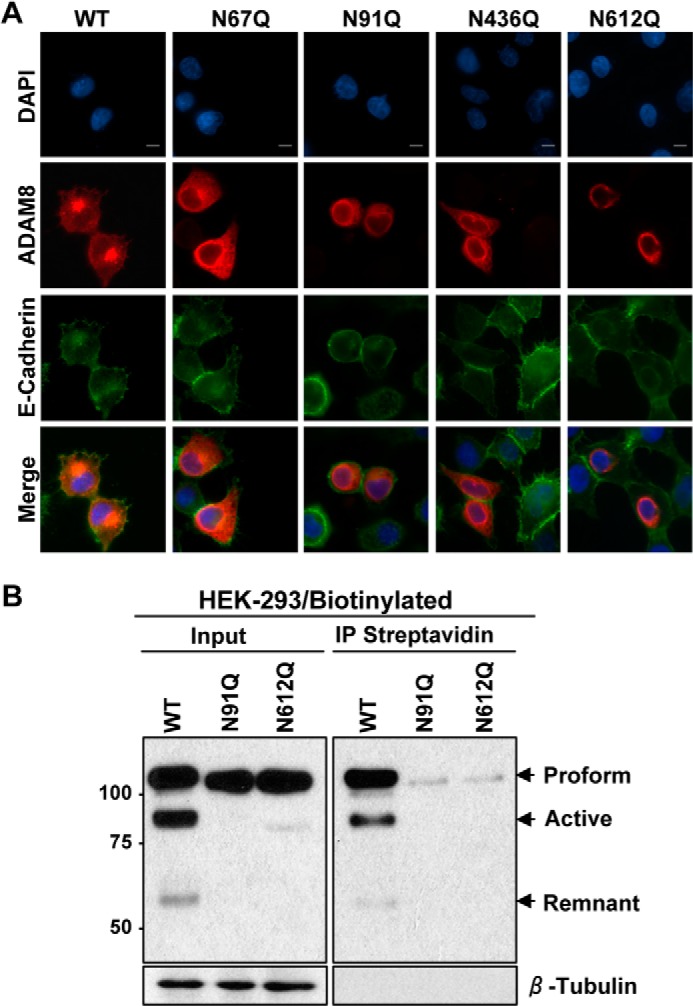
N91Q and N612Q ADAM8 mutants are unable to reach the cell surface. A, SUM-149 cells were transiently transfected with vectors expressing WT ADAM8 or N67Q, N91Q, N436Q, or N612Q N-glycosylation mutants. Cells were fixed, permeabilized, and subjected to immunofluorescent staining for ADAM8 expression using an anti-ADAM8 ectodomain antibody (AF1031) (red), and for the cell surface marker E-cadherin (green), and with DAPI (blue) to label the nuclei. Images were taken using a Nikon 80i Upright Research Fluorescence Scope (40× objective) with an Andoe Clara-E Camera and processed with NIS-Elements Basic software. Scale bar, 10 μm. B, HEK-293 cells were transiently transfected with vectors expressing WT ADAM8 or N-glycosylation mutants N91Q and N612Q. The cell surface proteins were labeled with biotin and pulled down by streptavidin (SA)-conjugated beads. Samples (10%) of extracts (Input) and pulled down biotinylated ADAM8 were detected by Western blotting using a Myc antibody.
To specifically test for surface staining of the N91Q and N612Q ADAM8 proteins, a biotinylation assay was performed. Surface proteins on transfected HEK-293 cells were biotinylated and pulled down using streptavidin-agarose followed by Western blotting (Fig. 6B). The WT ADAM8 proform, active and remnant forms were brought down with Streptavidin indicating they were all on the cell surface. As seen above, only abundant N91Q and N612Q ADAM8 proform proteins were present in the transfected HEK-293 cells (Fig. 6B). Notably, only a very small amount of the N91Q and N612Q proteins were detectable on the cell surface as judged by streptavidin pull-down, consistent with the findings obtained with both the FACS and immunofluorescence analyses. Thus, glycosylation at the Asn-91 and Asn-612 sites is required both for ADAM8 processing and localization at the cell surface.
ADAM8 N91Q Is Retained in the Golgi and ADAM8 N612Q in the Endoplasmic Reticulum
We next investigated the subcellular localization of the N91Q and N612Q mutant ADAM8 proteins using immunofluorescence. Mannose-6-phosphate receptor (M6PR) and phosphor-disulfide isomerase (PDI) were used as markers for the Golgi and the endoplasmic reticulum, respectively. Following transfection into HEK-293 cells, ectopically expressed WT ADAM8 was again visualized at the plasma membrane and in the cytoplasm. Co-staining with M6PR indicated extensive localization of WT ADAM8 in the Golgi (Fig. 7A), whereas only a low level of co-staining was observed within the endoplasmic reticulum (Fig. 7B). As seen above, the N91Q ADAM8 protein localized to punctated vesicles in the perinuclear area and these co-stained with M6PR, indicating this mutant protein was present within the Golgi (Fig. 7A). The N612Q ADAM8 mutant protein, which again appeared perinuclear, co-stained with PDI indicating it is localized in the endoplasmic reticulum (Fig. 7B). Thus, the mutations at Asn-91 and Asn-612 cause ADAM8 to be incorrectly localized within the cell.
FIGURE 7.
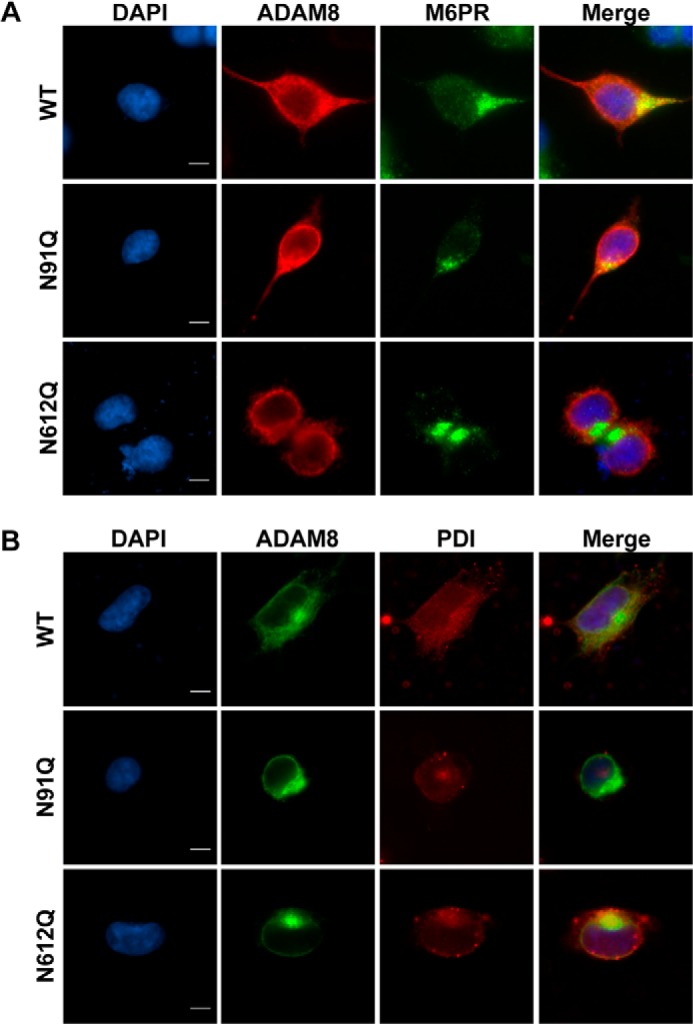
N91Q and N612Q mutant ADAM8 proteins are retained in the Golgi and ER, respectively. A and B, HEK-293 cells were transiently transfected with vectors expressing WT ADAM8 or N-glycosylation mutants N91Q and N612Q. Cells were fixed, permeabilized, and subjected to immunofluorescent staining using (A) ADAM8 LSBio antibody (red), M6PR (Golgi marker) (green), and DAPI (blue) or (B) Myc antibody (green); PDI (ER marker) (red) and DAPI (blue). Images were taken as described in Fig. 6A. Scale bar, 10 μm.
N436Q ADAM8 Displays Reduced Stability
The N436Q ADAM8 mutant appeared to be processed correctly and to localize to the cell surface but the levels of the processed forms observed were substantially reduced. As N-glycosylation has been shown to control ADAM10 stability (37), we asked whether the loss of glycosylation at Asn-436 similarly affected the stability of ADAM8. To test for altered stability, WT ADAM8 and N436Q mutant were overexpressed in HEK-293 cells and cultures treated with cycloheximide for 2 or 6 h to inhibit protein synthesis, in the absence or presence of the lysosomal inhibitor chloroquine (Fig. 8A). Upon treatment with cycloheximide, we observed a modest decrease in the amount of WT ADAM8 proform over the 6-h period. Addition of chloroquine increased the level of the proform as well as the active form, although to a lesser extent, suggesting that both may be subjected to lysosomal degradation. Cycloheximide treatment of cells expressing the N436Q mutant resulted in a larger 2.3-fold decrease in the amount of the proform, and to an almost complete loss of active form, suggesting active N436Q protein is indeed less stable (Fig. 8A). Addition of chloroquine again partially rescued the levels of the proform and the active form, but the amounts were substantially below what was seen with WT ADAM8 protein. To assess directly the localization of these proteins, HEK-293 cells expressing either WT or N436Q ADAM8 were subjected to indirect immunofluorescence for ADAM8 (red) and the lysosomal-associated membrane protein (LAMP) 2 (green). WT ADAM8 co-localized with LAMP 2 as indicated by the yellow staining in the merged images (Fig. 8B). Interestingly, the N436Q protein was also detected in the lysosomal compartment and the staining appeared more intense. The localization of both the WT and N436Q ADAM8 proteins in the lysosome suggest these proteins are turned over in this compartment. Next, we tested whether the turnover of endogenous ADAM8 protein was sensitive to the chloroquine. Notably when the MDA-MB-231 cells were treated with cycloheximide for 6 h in the absence of chloroquine (Fig. 8C, right panel), an average 2.3-fold decrease was observed in three independent replicates. Loss of ADAM8 in MDA-MB-231 cells was partially blocked by the addition of chloroquine, suggesting the endogenous ADAM8 protein also undergoes degradation in the lysosomal compartment. Lastly, we sought to assess the stability of the larger ADAM8 proform protein expressed in MCF-7 cells (Fig. 8C, left panel). Interestingly, the turnover of the ADAM8 protein was similar to that seen in MDA-MB-231 cells (2.9-fold average) and was also partially inhibited in the presence of chloroquine. Together these findings suggest that the N436Q mutant ADAM8 protein is substantially less stable than the WT protein and that ADAM8 degradation occurs, at least in part, in the lysosomal compartment.
FIGURE 8.

N436Q is less stable than WT ADAM8 and degraded in the lysosomal compartment. A, HEK-293 cells were transiently transfected with WT ADAM8 or N436Q as in Fig. 4C. After 24 h, cells were treated with carrier DMSO (−), 25 μg/ml cycloheximide (CHX) in the absence (−) or presence (+) of 50 μm chloroquine (Chloroq) for 6 h. WCEs were analyzed by Western blotting for ADAM8 expression (using a Myc antibody) and for β-tubulin. All lanes were from the same gel, but cut to re-align as indicated by the vertical line. The values below are the amount of normalized ADAM8 Proform relative to the control sample (untreated sample), which was set to 100%, determined as described under “Experimental Procedures.” Results are from one of three representative blots with similar data. B, HEK-293 cells were transiently transfected with vectors expressing WT ADAM8 or its N-glycosylation mutant N436Q. Cells were fixed, permeabilized, and subjected to immunofluorescent staining using ADAM8 LSBio antibody (red); LAMP 2 (Lysosomal marker) (green), and DAPI (blue). Images were taken as described in Fig. 6A. Scale bar, 10 μm. C, MCF-7 and MDA-MB-231 cells were treated for 6 h with DMSO (−), cycloheximide and/or chloroquine, as above. WCEs were run on the same gel and analyzed for expression of endogenous ADAM8 (LSBio antibody) and β-tubulin by Western blotting. The values below are the average amounts of normalized ADAM8 Proform relative to the control sample, which was set to 100%, of three independent replicates. The vertical line indicates that the blots for MCF-7, and MDA-MB-231 cell lysates were exposed for different lengths of time.
Reduced Glycosylation and Processing Decreases ADAM8 Metalloproteinase Activity
Next, we investigated whether the glycosylation mutants of ADAM8 display decreased metalloproteinase activity. To this end, we used one of the well-established ADAM8 substrates CD23 (2). Cleavage of CD23 by the metalloproteinase activity of ADAM8 leads specifically to the release of a 29-kDa soluble fragment. As a negative control for protease activity, an E335Q ADAM8 mutant, which carries an inactivating mutation in the metalloproteinase domain of ADAM8, was used. Vectors expressing WT ADAM8, glycosylation mutants (N67Q, N91Q, N436Q, or N612Q), E335Q mutant, or EV DNA were co-transfected with the C-terminal HA-tagged CD23 (CD23-HA) vector in HEK-293 cells. After 6 h, the medium was replaced with serum-free medium. Sixteen hours later, conditioned medium was harvested and WCEs prepared. Samples of the medium were analyzed by Western blotting for soluble cleaved CD23 (Fig. 9A, upper panel) and the WCEs for CD23 and ADAM8 protein in the lysates (Fig. 9A, upper and lower panels). WT ADAM8 induced the accumulation of cleaved 29-kDa CD23-HA in the supernatant, while essentially no cleavage was detected with the EV or E335Q mutant. Consistently, the relative level of CD23 in the cell lysates with the EV was substantially higher than in the presence of WT ADAM8 and comparable to the E335Q mutant. Mutation of N91Q and N612Q caused a decrease in the amount of shed CD23 to almost undetectable levels, and relatively more CD23 was seen in the lysates. These findings are consistent with the reduction in processing of N91Q and N612Q mutants to active form (Fig. 9, lower panel) and with the lack of their surface expression seen above. Transfection with the N436Q mutant led to substantially reduced production of soluble CD23 consistent with the decrease in amount of processed active ADAM8 observed, suggesting that the Asn-436 active form retained metalloproteinase activity. The N67Q mutation, which had the most modest effects on ADAM8 processing and location, displayed substantial levels of CD23 cleavage, indicating that its processed form retained activity. Overall, mutations N91Q and N612Q, which had the most substantial effects on ADAM8 processing and location, led to the largest decreases in its ability to release CD23 from the cell surface. We also tested the effects of knockdown of the endogenous ADAM8 on CD23 shedding using a MDA-MB-231 clone with stable ADAM8 knockdown resulting from ADAM8 shRNA (shA8). As a control, a MDA-MB-231 clone expressing an shControl RNA (shCtrl) was used. Stable knockdown of ADAM8 decreased the amount of released 29-kDa CD23 fragment compared with the shCtrl cells, consistently more CD23 was detected in the cell lysates (Fig. 8B), confirming the role of ADAM8 in this enzymatic reaction.
FIGURE 9.
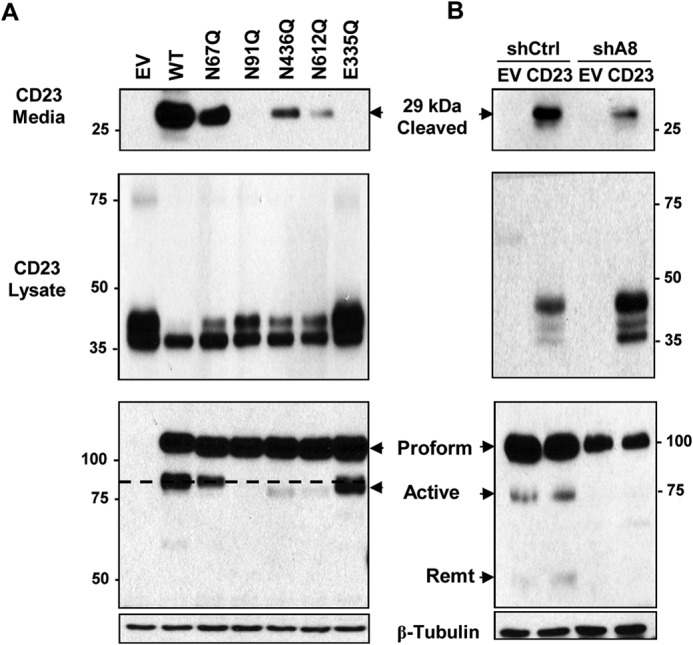
Glycosylation mutants with reduced processing display decreased ADAM8 metalloproteinase activity. A, C-terminal HA-tagged CD23 (CD23-HA) expression vector was co-transfected with EV DNA or with vectors expressing WT ADAM8, the indicated N-glycosylation mutants or metalloproteinase inactive E335Q mutant (EQ) in HEK-293 cells. 6 h later, the medium was replaced with serum-free medium. After 16 h, supernatants, and cells were collected. Medium samples were concentrated and assessed by Western blotting for cleaved CD23 (29 kDa) using an anti-HA antibody (top panel). Alternatively, WCEs were analyzed by Western blotting for CD23 expression using an HA antibody (middle panel) and for ADAM8 expression using a Myc antibody (bottom panels). The black dashed line indicates the migratory position of the active form of WT ADAM8. Note that the ADAM8-processed E335Q protein, while running near the position of active WT protein, has no detectable metalloproteinase activity, suggesting an alternative, non-functional enzymatic processing occurs in these cells. B, stable clones of MDA-MB-231 expressing shA8 RNA or control shRNA (shCtrl) were transfected with C-terminal HA-tagged CD23 (CD23-HA) vector DNA or EV DNA and processed and analyzed as in part A.
DISCUSSION
Here we show for the first time that human ADAM8 activity is tightly regulated by N-glycosylation, which extensively impacts its dimerization and subsequent processing, stability, and ability to localize to the cell surface of ERα-negative breast cancer cells. Site-directed mutagenesis supports the conclusion that human ADAM8 is N-glycosylated at four sites. Mutation of two of these sites: Asn-91 (located in the prodomain) and Asn-612 (located between the CRD and ELD), reduced the processing of ADAM8 and its localization at the cell surface, two events required for ADAM8 to promote breast tumor growth and metastatic dissemination (7). N-Glycosylation at the Asn-91 was required for ADAM8 exit from the Golgi, while an inability to N-glycosylate ADAM8 on Asn-612 resulted in localization of ADAM8 in the endoplasmic reticulum. Glycosylation of Asn-436 affected ADAM8 stability, particularly of the active form, whereas Asn-67 in the prodomain had only modest effects on processing or activity. Moreover, growth of ERα-negative breast cancer cells under hypoxic conditions increased both the level of ADAM8 expression as well as its glycosylation. No O-glycosylation of ADAM8 could be detected in breast cancer cells (data not shown). Interestingly, N-glycosylation and processing of ADAM8 was essentially not detectable in ERα-positive breast cancer cell lines, which are known to be less invasive and metastatic, and this correlated with a lack of extensive dimerization of ADAM8. Thus, the processing, localization, stability, and activity of ADAM8 depend upon its accurate N-glycosylation.
Several members of the ADAM family of proteins, including ADAM9, ADAM10, ADAM12, and ADAM17, have been implicated in the pathogenesis or progression of cancer (38–40). Of these different ADAM proteins, post-translational modifications of ADAM10, ADAM12, and ADAM17 have been shown to play major roles in their activities. ADAM10 is also N-glycosylated at four sites and these post-translational modifications are crucial for its processing, activity, and resistance to proteolysis (37). Interestingly, higher expression of an N-linked glycosylated form of ADAM12 was observed in more aggressive form of various cancer cell lines (41), and differential glycosylation and activity of ADAM17 was observed when human ADAM17 was expressed in mammalian and insect cells (42). Previous studies have shown that increased ADAM8 expression is associated with a more advanced stage of cancer, including non-small cell lung cancer (13, 43), prostate cancer (44), renal cell carcinoma (45), and pancreatic cancer (15). Further, our laboratory has reported ADAM8 is abundantly expressed in aggressive and invasive TNBCs compared with normal mammary tissue or fibroadenomas, which are the most common benign tumors of the breast (7). Further studies would be required to determine whether additional changes in N-glycosylation of ADAM8 occur with tumor progression.
N-linked glycosylation has varying functional consequences on ADAM8. The N67Q mutation did not affect the expression levels of ADAM8 proform and only slightly reduced the levels of its active and remnant forms, suggesting that glycosylation at the Asn-67 site plays a minor role in processing. Also, the sheddase activity of ADAM8 was only modestly decreased in the N67Q mutant. Interestingly, the Asn-67 site, which is located in the prodomain of human ADAM8, is the only site that is absent in Mus musculus ADAM8 protein. In contrast, the three other sites of N-glycosylation (Asn-91, Asn-436, and Asn-612 in the human ADAM8 protein) played critical roles for its processing. Putative counterparts to these sites are detected in mouse ADAM8 (located at Asn-88, Asn-430, and Asn-613); although, it remains to be determined whether these sites are actually functional in murine ADAM8. In human ADAM8, mutation at Asn-91 decreased its processing, ability to exit the Golgi and thereby its cell surface localization. Similarly, mutation of ADAM8 at Asn-612 reduced its ability to localize on the cell surface or to be appropriately processed due to an inability to exit from the endoplasmic reticulum. The reduced staining in post-Golgi compartments in N91Q-expressing cells compared with the WT ADAM8 suggests that glycosylation at the Asn-91 site in the prodomain is the sorting determinant for exiting from the Golgi complex and Asn-612 for the endoplasmic reticulum. Notably the failure of ADAM8 to appropriately localize to the cell surface would decrease its ability to influence the breast tumor microenvironment (7). The Asn-91 and Asn-612 mutations in ADAM8 may have altered its ability to interact with lectin chaperones in the endoplasmic reticulum, and thereby decreased its ability to fold correctly. Interestingly, incorrectly folded glycoproteins are generally aggregated in the endoplasmic reticulum and subject to endoplasmic reticulum associated degradation (ERAD) or transported to the Golgi for lysosomal mediated degradation or alternatively translocated to the cytoplasm for proteasomal degradation (19). Hence, mutation at these sites may result in the failure of ADAM8 to attain a proper conformation. Furthermore, mutation of the Asn-436 glycosylation site (located within the DI domain) resulted in decreased ADAM8 active form stability, in part due to degradation via the lysosome. This likely explains the substantial decrease in the detectable level of ADAM8 in MDA-MB-231 and SUM-149 cells treated with tunicamycin, which inhibits all glycosylation. It remains to be determined whether the proteasome or ERAD also play a role in ADAM8 turnover.
ADAM8 N-glycosylation and processing was not detected in ERα-positive breast cancer cells and could not be induced in these cells by hypoxia. As glycosylation of E-cadherin occurred in these cells, there does not appear to be a general glycosylation defect and thus the reason for this absence is unclear. Interestingly, despite the absence of glycosylation, the ADAM8 proform migrated more slowly in the ERα-positive compared with ERα-negative breast cancer cells. These findings suggest the possibility that some other post-translational modification(s) occurred in the ERα-positive breast cancer cells that contributed to this apparent increased size, e.g. phosphorylation. If so, this modification may be responsible for blocking glycosylation and ADAM8 processing. Studies are in progress to identify and compare all of the post-translational modifications of ADAM8 in ERα-positive versus -negative breast cancer cells.
Acknowledgments
We thank Stephen Ethier and Nader Rahimi for generously providing SUM-149 and HEK-293 cells, respectively and Joerg Bartsch for the hADAM8 clone and MDA-MB-231 stable shA8 and Ctrl cells. We also gratefully acknowledge the assistance of Dr. Nora Mineva in the analysis of the FACS data.
This work was supported, in whole or in part, by grants from the National Institutes of Health (R01 CA129129 and P01 ES01124, to G. E. S.) and the Dept. of Defense (DOD) postdoctoral fellowship W81XWH-10-1-1003 (to M. R.).
- ADAM
- A Disintegrin And Metalloproteinase
- MP
- metalloproteinase
- DI
- disintegrin
- CRD
- cysteine-rich
- ELD
- EGF-like domain
- mCD23
- membrane-bound CD23
- sCD23
- soluble CD23
- TNBC
- Triple-Negative breast cancer
- ER
- estrogen receptor
- PNGase F
- peptide N-glycosidase F
- Endo H
- endoglycosidase H
- BS3
- bis(sulfosuccinimidyl) suberate
- HMEC
- human mammary epithelial cells
- shA8
- ADAM8 shRNA
- shCtrl
- control shRNA
- hADAM8
- human ADAM8
- WCE
- whole-cell extract
- PEI
- polyethylenimine
- EV
- empty vector
- M6PR
- mannose-6-phosphate receptor
- PDI
- phospho-disulfide isomerase
- LAMP
- lysosomal-associated membrane protein
- ERAD
- endoplasmic reticulum associate degradation.
REFERENCES
- 1. Yoshiyama K., Higuchi Y., Kataoka M., Matsuura K., Yamamoto S. (1997) CD156 (human ADAM8): expression, primary amino acid sequence, and gene location. Genomics 41, 56–62 [DOI] [PubMed] [Google Scholar]
- 2. Koller G., Schlomann U., Golfi P., Ferdous T., Naus S., Bartsch J. W. (2009) ADAM8/MS2/CD156, an emerging drug target in the treatment of inflammatory and invasive pathologies. Curr. Pharm. Des. 15, 2272–2281 [DOI] [PubMed] [Google Scholar]
- 3. Bartsch J. W., Wildeboer D., Koller G., Naus S., Rittger A., Moss M. L., Minai Y., Jockusch H. (2010) Tumor necrosis factor-α (TNF-α) regulates shedding of TNF-α receptor 1 by the metalloprotease-disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci. 30, 12210–12218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gómez M. I., Seaghdha M. O., Prince A. S. (2007) Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. EMBO J. 26, 701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naus S., Richter M., Wildeboer D., Moss M., Schachner M., Bartsch J. W. (2004) Ectodomain shedding of the neural recognition molecule CHL1 by the metalloprotease-disintegrin ADAM8 promotes neurite outgrowth and suppresses neuronal cell death. J. Biol. Chem. 279, 16083–16090 [DOI] [PubMed] [Google Scholar]
- 6. Zack M. D., Malfait A. M., Skepner A. P., Yates M. P., Griggs D. W., Hall T., Hills R. L., Alston J. T., Nemirovskiy O. V., Radabaugh M. R., Leone J. W., Arner E. C., Tortorella M. D. (2009) ADAM-8 isolated from human osteoarthritic chondrocytes cleaves fibronectin at Ala(271). Arthritis Rheum. 60, 2704–2713 [DOI] [PubMed] [Google Scholar]
- 7. Romagnoli M., Mineva N. D., Polmear M., Conrad C., Srinivasan S., Loussouarn D., Barillé-Nion S., Georgakoudi I., Dagg Å., McDermott E. W., Duffy M. J., McGowan P. M., Schlomann U., Parsons M., Bartsch J. W., Sonenshein G. E. (2014) ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol. Med. 6, 278–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fourie A. M., Coles F., Moreno V., Karlsson L. (2003) Catalytic activity of ADAM8, ADAM15, and MDC-L (ADAM28) on synthetic peptide substrates and in ectodomain cleavage of CD23. J. Biol. Chem. 278, 30469–30477 [DOI] [PubMed] [Google Scholar]
- 9. Cooper A. M., Hobson P. S., Jutton M. R., Kao M. W., Drung B., Schmidt B., Fear D. J., Beavil A. J., McDonnell J. M., Sutton B. J., Gould H. J. (2012) Soluble CD23 controls IgE synthesis and homeostasis in human B cells. J. Immunol. 188, 3199–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelly K., Hutchinson G., Nebenius-Oosthuizen D., Smith A. J., Bartsch J. W., Horiuchi K., Rittger A., Manova K., Docherty A. J., Blobel C. P. (2005) Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev. Dyn. 232, 221–231 [DOI] [PubMed] [Google Scholar]
- 11. Matsuno O., Ono E., Ueno T., Takenaka R., Nishitake T., Hiroshige S., Miyazaki E., Kumamoto T., Higuchi Y. (2010) Increased serum ADAM8 concentration in patients with drug-induced eosinophilic pneumonia-ADAM8 expression depends on a the allergen route of entry. Respir Med. 104, 34–39 [DOI] [PubMed] [Google Scholar]
- 12. Choi S. J., Han J. H., Roodman G. D. (2001) ADAM8: a novel osteoclast stimulating factor. J. Bone Miner Res. 16, 814–822 [DOI] [PubMed] [Google Scholar]
- 13. Ishikawa N., Daigo Y., Yasui W., Inai K., Nishimura H., Tsuchiya E., Kohno N., Nakamura Y. (2004) ADAM8 as a novel serological and histochemical marker for lung cancer. Clin. Cancer Res. 10, 8363–8370 [DOI] [PubMed] [Google Scholar]
- 14. Hernández I., Moreno J. L., Zandueta C., Montuenga L., Lecanda F. (2010) Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer. Oncogene 29, 3758–3769 [DOI] [PubMed] [Google Scholar]
- 15. Valkovskaya N., Kayed H., Felix K., Hartmann D., Giese N. A., Osinsky S. P., Friess H., Kleeff J. (2007) ADAM8 expression is associated with increased invasiveness and reduced patient survival in pancreatic cancer. J. Cell Mol. Med. 11, 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wildeboer D., Naus S., Amy Sang Q. X., Bartsch J. W., Pagenstecher A. (2006) Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J. Neuropathol. Exp. Neurol. 65, 516–527 [DOI] [PubMed] [Google Scholar]
- 17. Zielinski V., Brunner M., Heiduschka G., Schneider S., Seemann R., Erovic B., Thurnher D. (2012) ADAM8 in squamous cell carcinoma of the head and neck: a retrospective study. BMC Cancer 12, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Helenius A., Aebi M. (2001) Intracellular functions of N-linked glycans. Science 291, 2364–2369 [DOI] [PubMed] [Google Scholar]
- 19. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 20. Kophengnavong T., Michnowicz J. E., Blackwell T. K. (2000) Establishment of distinct MyoD, E2A, and twist DNA binding specificities by different basic region-DNA conformations. Mol. Cell Biol. 20, 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y. Y., Takahashi M., Gu J. G., Miyoshi E., Matsumoto A., Kitazume S., Taniguchi N. (2008) Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99, 1304–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liwosz A., Lei T., Kukuruzinska M. A. (2006) N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem. 281, 23138–23149 [DOI] [PubMed] [Google Scholar]
- 23. Guo N., Liu Y., Masuda Y., Kawagoe M., Ueno Y., Kameda T., Sugiyama T. (2005) Repeated immunization induces the increase in fucose content on antigen-specific IgG N-linked oligosaccharides. Clin. Biochem. 38, 149–153 [DOI] [PubMed] [Google Scholar]
- 24. Ioffe E., Stanley P. (1994) Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl. Acad. Sci. U.S.A. 91, 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nita-Lazar M., Noonan V., Rebustini I., Walker J., Menko A. S., Kukuruzinska M. A. (2009) Overexpression of DPAGT1 leads to aberrant N-glycosylation of E-cadherin and cellular discohesion in oral cancer. Cancer Res. 69, 5673–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Lampe B., Stallmach A., Riecken E. O. (1993) Altered glycosylation of integrin adhesion molecules in colorectal cancer cells and decreased adhesion to the extracellular matrix. Gut. 34, 829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Calle Y., Palomares T., Castro B., del Olmo M., Bilbao P., Alonso-Varona A. (2000) Tunicamycin treatment reduces intracellular glutathione levels: effect on the metastatic potential of the rhabdomyosarcoma cell line S4MH. Chemotherapy 46, 408–428 [DOI] [PubMed] [Google Scholar]
- 28. Schlomann U., Wildeboer D., Webster A., Antropova O., Zeuschner D., Knight C. G., Docherty A. J., Lambert M., Skelton L., Jockusch H., Bartsch J. W. (2002) The metalloprotease disintegrin ADAM8. Processing by autocatalysis is required for proteolytic activity and cell adhesion. J. Biol. Chem. 277, 48210–48219 [DOI] [PubMed] [Google Scholar]
- 29. Forozan F., Veldman R., Ammerman C. A., Parsa N. Z., Kallioniemi A., Kallioniemi O. P., Ethier S. P. (1999) Molecular cytogenetic analysis of 11 new breast cancer cell lines. Br. J. Cancer 81, 1328–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horton R. M., Ho S. N., Pullen J. K., Hunt H. D., Cai Z., Pease L. R. (1993) Gene splicing by overlap extension. Methods Enzymol. 217, 270–279 [DOI] [PubMed] [Google Scholar]
- 31. Romagnoli M., Belguise K., Yu Z., Wang X., Landesman-Bollag E., Seldin D. C., Chalbos D., Barillé-Nion S., Jézéquel P., Seldin M. L., Sonenshein G. E. (2012) Epithelial-to-mesenchymal transition induced by TGF-β1 is mediated by Blimp-1-dependent repression of BMP-5. Cancer Res. 72, 6268–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geng F., Zhu W., Anderson R. A., Leber B., Andrews D. W. (2012) Multiple post-translational modifications regulate E-cadherin transport during apoptosis. J. Cell Sci. 125, 2615–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu W., Leber B., Andrews D. W. (2001) Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J. 20, 5999–6007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Varki A., Kannagi R., Toole B. P. (2009) Glycosylation Changes in Cancer. in: Essentials of Glycobiology, 2nd Ed., Chapter 44, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu H., Shim A. H., He X. (2009) Structural characterization of the ectodomain of a disintegrin and metalloproteinase-22 (ADAM22), a neural adhesion receptor instead of metalloproteinase: insights on ADAM function. J. Biol. Chem. 284, 29077–29086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Escrevente C., Morais V. A., Keller S., Soares C. M., Altevogt P., Costa J. (2008) Functional role of N-glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim. Biophys. Acta 1780, 905–913 [DOI] [PubMed] [Google Scholar]
- 38. Fry J. L., Toker A. (2010) Secreted and membrane-bound isoforms of protease ADAM9 have opposing effects on breast cancer cell migration. Cancer Res. 70, 8187–8198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Duffy M. J., Mullooly M., O'Donovan N., Sukor S., Crown J., Pierce A., McGowan P. M. (2011) The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin. Proteomics 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murphy G. (2008) The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 8, 929–941 [DOI] [PubMed] [Google Scholar]
- 41. Kodama T., Ikeda E., Okada A., Ohtsuka T., Shimoda M., Shiomi T., Yoshida K., Nakada M., Ohuchi E., Okada Y. (2004) ADAM12 is selectively overexpressed in human glioblastomas and is associated with glioblastoma cell proliferation and shedding of heparin-binding epidermal growth factor. Am. J. Pathol. 165, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chavaroche A., Cudic M., Giulianotti M., Houghten R. A., Fields G. B., Minond D. (2014) Glycosylation of a disintegrin and metalloprotease 17 affects its activity and inhibition. Anal. Biochem. 449, 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu G. C., Hu H. C., Shi M. H. (2008) Expression and clinical significance of a disintegrin and metalloprotease 8 (ADAM8) and epidermal growth factor receptor (EGFR) in non-small cell lung cancer. Ai Zheng 27, 874–878 [PubMed] [Google Scholar]
- 44. Fritzsche F. R., Jung M., Xu C., Rabien A., Schicktanz H., Stephan C., Dietel M., Jung K., Kristiansen G. (2006) ADAM8 expression in prostate cancer is associated with parameters of unfavorable prognosis. Virchows Arch. 449, 628–636 [DOI] [PubMed] [Google Scholar]
- 45. Roemer A., Schwettmann L., Jung M., Stephan C., Roigas J., Kristiansen G., Loening S. A., Lichtinghagen R., Jung K. (2004) The membrane proteases adams and hepsin are differentially expressed in renal cell carcinoma. Are they potential tumor markers? J. Urol. 172, 2162–2166 [DOI] [PubMed] [Google Scholar]