

Abstract
Chronic inflammation is a major risk factor underlying aging and the associated diseases of aging; of particular interest is insulin resistance during aging. Chronic inflammation impairs normal lipid accumulation, adipose tissue function, mitochondrial function, and causes endoplasmic reticulum (ER) stress, which lead to insulin resistance. However, some studies show that insulin resistance itself amplifies chronic inflammation. The activity of the insulin-dependent Akt signaling pathway is highlighted because of its decrease in insulin-sensitive organs, like liver and muscle, which may underlie insulin resistance and hyperinsulinemia, and its increased levels in non-metabolic organs, such as kidney and aorta. In that the prevalence of obesity has increased substantially for all age groups in recent years, our review summarizes the data showing the involvement of chronic inflammation in obesity-induced insulin resistance, which perpetuates reciprocal interactions between the chronic inflammatory process and increased adiposity, thereby accelerating the aging process.
Keywords: Insulin resistance, Molecular inflammation, Aging
Introduction
Chronic, low-grade, systemic inflammation is wildly accepted as a significant risk factor underlying aging and major diseases (Yu and Chung 2006). Inflammation, as part of the key innate immune defense system, is an essential protective mechanism that fights against not only invading pathogens and other exogenous harmful materials, but also endogenously produced substances (Mogensen 2009) that are tightly regulated under normal conditions. However, unresolved chronic inflammation is known to exacerbate and underlie age-related diseases such as cancer, type 2 diabetes, cardiovascular diseases, arthritis, osteoporosis, and Alzheimer’s disease (Howcroft et al. 2013).
Strong evidence was presented on a plausible relationship between inflammation and obesity-related diseases (Gregor and Hotamisligil 2011). It was shown that an age-related increase in visceral adiposity results in high circulating levels of pro-inflammatory cytokines that interfere with insulin signaling during the aging process (Sepe et al. 2011). Pro-inflammatory macrophages, a major source of cytokines, are present in much higher numbers in the adipose tissue of obese subjects than lean subjects (Heilbronn and Campbell 2008).
Pro-inflammatory cytokines were shown to act in an autocrine or paracrine manner to induce insulin resistance in peripheral tissues and macrophages (Schenk et al. 2008; Tilg and Moschen 2008). The effect of pro-inflammatory cytokines that induces insulin resistance begins by the ameliorated tyrosine phosphorylation of insulin receptor substrate (IRS)-1.
In this review, we summarize recent work on the close association between chronic inflammation and insulin resistance during aging process.
Molecular insights into the inflammatory process during aging
In chronic inflammation, the inflammatory response is driven by the versatile transcription factor, nuclear factor-κB (NF-κB) during aging. NF-κB is activated by the IκB kinase (IKK)/NF-κB inducing kinase (NIK) and the mitogen activated protein kinase (MAPK) pathways (Fig. 1). Once activated, NF-κB promotes the expression of several other pro-inflammatory cellular mediators, like prostaglandins (PGs), inducible nitric oxide synthase (iNOS), and inflammation-related cytokines. PG metabolites are produced by the action of the enzyme, cyclooxygenase (COX). These are important pro-inflammatory mediators that are known to increase reactive oxygen species (ROS) production, which leads to DNA and tissue damage (Yu and Chung 2001). PGs-induced ROS generation steadily increases in peripheral tissue during aging (Chung et al. 2011). Several studies using COX-2 deficient mice reveal impaired inflammatory responses through the attenuation of PGs generation (Morham et al. 1995; Dinchuk et al. 1995). Not only ROS but also pro-inflammatory cytokines such as interleukin (IL)-1β and IL-6 are enhanced by PGs (Aoki and Narumiya 2012). Nitric oxide (NO) is one of the most physiologically important radical species known to induce the chronic inflammatory response. NO is synthesized by iNOS and can cause pathogenesis and destruction of various tissues, similar to that seen in chronic inflammatory (Zamora et al. 2000).
Fig. 1.
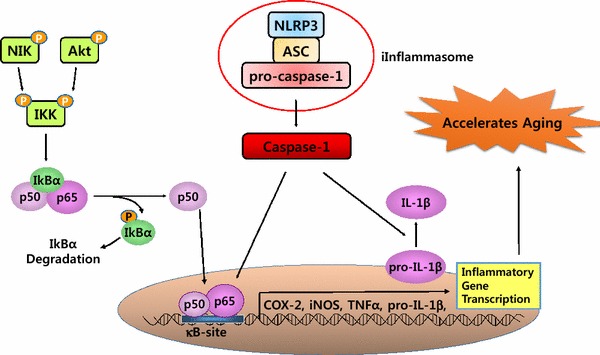
Signaling pathways downstream of NIK and Akt activation. Extracellular receptors bind to their ligands and signal via NF-κB inducing kinase (NIK) or Akt molecules. This leads to phosphorylation of the IκB kinase (IKK) subunits and the subsequent phosphorylation of IκBα, which leads to its ubiquitination and proteasomal degradation. Inflammasome activation also triggers signaling through nuclear factor-κB (NF-κB) and Interleukin-1β (IL-1β). NF-κB is then released into the nucleus where it acts as a transcription factor and stimulates inflammatory responses
The recently established inflammasome, an essential component of the innate immune response, plays a key role in the inflammatory process by the secretion of the cytokines, IL-1β and IL-18 (Agostini et al. 2004). The close cooperative actions between the inflammasome and NF-κB undoubtedly exacerbates the chronic inflammatory status during the aging process (Howcroft et al. 2013). The most studied NLRP3 inflammasome is an intracellular protein complex composed of NOD-like receptor (NLR) proteins, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1, which result in caspase-1 activation. The formation of the inflammasome induces auto-processing and activation of caspase-1 leading to the processing of cellular substrates, namely IL-1β and IL-18. Activation of the well-studied NLRP3 inflammasome has a diverse sensing ability reported to participate in aging-related inflammatory processes. It is worth noting the recent important report by Youm et al in which the authors describe that the suppression of the NLRP3 inflammasome extended the healthy lifespan of mice by attenuating age-related degenerative changes, including cognitive decline. Based on their findings, the authors proposed that suppression of aberrant NLRP3 activity during aging may well attenuate age-related diseases by reducing chronic inflammation (Youm et al. 2013).
Insulin resistance and altered PI3K/Akt signaling pathway
Through mechanistic exploration, numerous studies have produced various interesting data, but to date, there seems to be no unifying consensus, implying the complex nature of the subject (Muoio and Newgard 2008; Kahn et al. 2006; Saini 2010). One interesting observations is that insulin resistance, at cellular and molecular levels, is characterized by dysfunction in the IRS/phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, which results in impaired glucose uptake in hepatocytes and adipocytes. The process involved in glucose uptake has a series of sequential events beginning at the insulin receptor (IR), which is a heterotetramer consisting of two α subunits and two β subunits that are linked by disulfide bonds. Insulin binds to the α subunit of the IR and activates the tyrosine kinase in the β subunits to initiate insulin signaling. Once the tyrosine kinase of the IR is activated, it promotes auto-phosphorylation of the β subunit, where phosphorylation of tyrosine residues and a conformational change are required to amplify the kinase activity. The activated receptor then phosphorylates adaptor molecules, such as IRS-1 and IRS-2, which permit the recruitment and activation of phosphatidylinositol 3-kinase (PI3-kinase). Activated PI3-kinase produces 3-phosphoinositides, phosphatidyl-inositol-3,4-bisphosphate (PIP2) and phosphatidyl-inositil-3,4,5-trisphosphate (PIP3) that bind to phosphoinositide-dependent kinase 1 (PDK1). Akt activates glucose transporter 4 (GLUT4), which moves to the cell surface to transport glucose into the cell.
Possible causes underlying insulin resistance through inflammation activation during aging
Lipid accumulation
Lipids have a wide variety of roles in biological systems. These roles are a consequence of their chemical and physical properties. However, excess lipid accumulation induces insulin resistance during aging. Recently studies have shown that inflammatory responses disrupt normal lipid accumulation. For example, pro-inflammatory cytokines such as IL-6 and tumor necrosis factor-α (TNFα) impair the lipid accumulation by promoting WNT signaling (Gustafson and Smith 2006). Indeed, inflammation leads to lipid accumulation through disruption of the expression of proteins related to lipid metabolism, such as sterol regulatory element binding protein (SREBP), a basic helix-loop-helix transcription factor that controls the expression of genes required for cholesterol, fatty acids, triglycerides and phospholipids synthesis (Ferre and Foufelle 2007).
A recent cDNA microarray study from our laboratory (Park et al. 2013) documented that the genes upregulated during aging contain several major inflammatory genes. The upregulated genes included NF-κB1alpha and a 90 kDa ribosomal protein, S6 kinase, which is related to mammalian target of rapamycin (mTOR) in aged rats (Park et al. 2013). In the aging transcriptome, genes that are related to both the immune response and to inflammation were upregulated with age, particularly those related to cytokine–cytokine receptor interaction, natural killer cell mediated cytotoxicity, and primary immunodeficiency (Anderson et al. 2010). Down-regulated genes, such as peroxisome proliferator-activated receptors (PPARs) associated with lipid metabolism, were also observed with aging according to the gene ontology data (Hong et al. 2010). Similarly, energy metabolism, insulin signaling, and PPAR signaling genes were down-regulated, as shown by the analysis of the protein–protein interaction network that was composed of genes differentially expressed during aging (Park et al. 2013). The involvement of inflammation was also shown by the reduction of ABCA1-mediated cholesterol efflux that was regulated by PPARs in acceleration of foam cell formation through disrupting LDL receptors-mediated negative feedback regulation in vascular smooth muscle cells and mesangial cells (Ma et al. 2008). These data further support that metabolic disorders associated with lipid accumulation are closely related to chronic inflammation at the molecular level during aging.
Dysregulated adipose tissue
White adipose tissue (WAT) not only is involved in energy storage in the body, but also secretes a variety of bioactive molecules including adiponectin, IL-1β, IL-6, and TNFα, which enable the adipose tissue to communicate with other tissues and organs, such as the liver, skeletal muscle, and central nervous system. Through this interaction network, WAT participates in regulating important biological processes, such as food intake, energy homeostasis, and insulin resistance. Adipose tissue is at the nexus of mechanisms and signaling pathways involved in metabolic disorder, inflammation, aging and age-related diseases. Obesity and aging are affected by increasing both the size and number of adipocytes. Adipogenesis can lead to a large number of new adipocytes (i.e., hyperplasia), which produce more adiponectin and fewer pro-inflammatory adipokines. In contrast, the prevalence of hypertrophied adipocytes in adipose tissue encourages increased macrophage infiltration (Despres 2006; Goossens 2008). The recruitment of macrophages into adipose tissue is the initial event leading to obesity-induced inflammation and insulin resistance (Fig. 2).
Fig. 2.
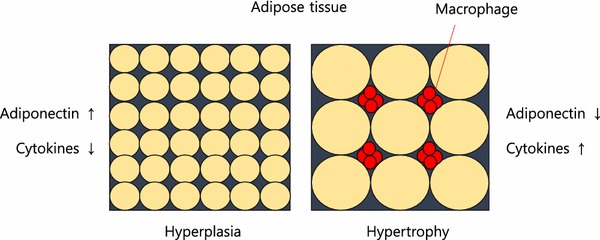
Obesity is determined by increasing both, the size and number of adipocytes. Adipogenesis can lead to a large number of new adipocytes (hyperplasia) which produce more adiponectin and less inflammatory adipokines. Conversely, hypertrophied adipocytes produce less adiponectin and more inflammatory adipokines. The prevalence of hypertrophied adipocytes in adipose tissue leads to a reduction in blood flow with subsequent hypoxia and macrophage infiltration. In addition, cytokines produced by macrophages inhibit adipogenesis
In general, over-nutrition and aging cause adipocytes to secrete chemokines, such as pro-inflammatory monocyte chemotactic protein-1 (MCP-1) and others that provide a chemotactic gradient that attracts monocytes to the adipose tissue where they become adipose tissue macrophages (ATMs). Once pro-inflammatory ATMs migrate into adipose tissue, they also secrete their own chemokines, which attract additional macrophages creating a feed-forward inflammatory process (Olefsky and Glass 2010). Thus, ATMs perpetuate inflammation, by inducing local and systemic increases of pro-inflammatory molecules like IFNγ, IL-1β, IL-6, and TNFα, while decreasing other adipokines, such as adiponectin.
Hypertrophied adipocytes, which are up to 150–200 μm in diameter are larger than the normal distance that O2 can diffuse, which is 100–200 μm (Skurk et al. 2007; Brahimi-Horn and Pouyssegur 2007), which leads to a hypoxic condition. Hypoxia in obese adipose tissue has been well characterized by many researchers, and so has the induction of a key regulator, hypoxia-inducible factor-1 (HIF-1) (Hosogai et al. 2007; Wang 2007). HIF-1 is a transcription factor that accumulates during hypoxia and increases the mRNA expression of a wide variety of genes that stimulate erythropoiesis, angiogenesis, and glycolysis (Semenza 2000). Thus, it provides a major link to the pathogenesis of insulin resistance (Olefsky and Glass 2010).
Visceral adipose tissue refers to intra-abdominal fat around the intestines and it correlates with liver fat. Visceral adipose tissue has metabolic characteristics that differ from those of subcutaneous fat. It is more metabolically active with a high free fatty acid (FFA) turnover, and the increased flux of FFAs promotes insulin resistance at a cellular level and increases hepatic very low density lipoprotein (VLDL) production. Indeed, excess visceral adiposity has been shown to accompany chronic low-grade inflammation (Giorgino et al. 2005). Studies on the visceral adipose tissue from lean or obese subjects demonstrate that macrophage specific markers and chemokines involved in monocyte chemotaxis, as well as their receptors, are elevated in obese visceral fat compared with lean controls (Huber et al. 2008).
Mitochondrial dysfunction
For many years, it has been known that mitochondrial dysfunction is associated with type 2 diabetes mellitus (T2DM) and age-related insulin resistance (Stump et al. 2003; Petersen et al. 2003). The mitochondrial oxidative phosphorylation capacity is defined as the maximal ADP-stimulated oxidative phosphorylation elicited by a high ADP:ATP ratio that reflects the maximal energy demands of the cell at saturating concentrations of substrates (Chance et al. 2006; Prompers et al. 2006). Mitochondrial oxidative phosphorylation and unlimited oxygen supply is very important for the function of the cell. However, subjects with T2DM, often have mitochondrial dysfunctions such as reduced insulin-stimulated ATP production, lower protein expression of respiratory chain subunits, decreased mitochondrial DNA, and decreased mitochondrial size and density (Ritov et al. 2005; Heilbronn et al. 2007; Szendroedi et al. 2007; Hwang et al. 2010).
In addition, several studies have shown that the oxidative phosphorylation capacity is impaired in muscle biopsy samples from patients with T2DM compared with healthy individuals (Phielix et al. 2008; Boushel et al. 2007). Moreover, the expression of genes for mitochondrial oxidative phosphorylation is related to insulin resistance (Petersen et al. 2003). Mitochondrial dysfunction results in the accumulation of fatty acid metabolites, diacylglycerol (DAG), and long chain fatty acyl-CoA (Itani et al. 2002) causes decreased fatty acid oxidation. Intracellular accumulation of DAG activates protein kinase C (PKC), which in turn activates IKK and JNK leading to increased serine phosphorylation of IRS, and consequently attenuation of insulin signaling (Griffin et al. 1999; Yu et al. 2002). Indeed, with aging, mutations and deletions occur in mtDNA, leading to impaired function of the respiratory chain and enhanced ROS production (Chomyn and Attardi 2003). Thus, mitochondrial dysfunction is highly likely one of the major causes of the age-related chronic inflammation.
ER stress
Endoplasmic reticulum (ER) is an organelle that is comprised of a reticular membranous network that extends throughout the cytoplasm and is contiguous with the nuclear envelope. This organelle is involved in several important cellular functions that affect the homeostasis of the whole-organism. The ER is also the cellular site where all secretory and integral membrane proteins are folded and post-translationally modified in an ATP dependent process mediated by chaperones, which allows the cell to respond to misfolded proteins within the ER. These pathways are collectively known as the unfolded protein response (UPR) and are important for normal cellular homeostasis and organismal development, and may play key roles in the pathogenesis of many diseases. In addition, the ER is also a site of Ca2+ storage and biosynthesis for steroids, cholesterol, and lipids (van Meer et al. 2008; Ron and Walter 2007).
Recently studies demonstrated that fatty acid overload rapidly induces ER stress in pancreatic β cells and hepatocytes that leads to impaired insulin secretion and lucose uptake, respectively (Borradaile et al. 2006; Kharroubi et al. 2004). Recent studies also suggest that ER stress plays an important role in the onset of obesity, insulin resistance, and T2DM (Wellen and Hotamisligil 2005; Eizirik et al. 2008; Ozawa et al. 2005). ER stress in hepatocytes and adipocytes contributes to insulin resistance, in part through IRE1-dependent, JNK mediated inhibition of IRS-1 tyrosine phosphorylation, and increased serine phosphorylation (Ozawa et al. 2005; Hirosumi et al. 2002). Several chaperones and folding enzymes such as glucose regulated proteins 78 (GRP78; which is also known as immunoglobulin binding protein [BiP]), protein disulfide isomerase (PDI), calnexin, and calreticulin participate in protein folding in the ER. These chaperones and foldases decrease with age (Naidoo et al. 2005; Nuss et al. 2008; Rabek et al. 2003).
ER stress has been demonstrated to trigger activation of JNK as well as IKK by increasing IRE1 and leads to inducing NF-κB activation (Brown et al. 2008). Increased JNK and NF-κB signaling would then induce the expression of pro-inflammatory cytokines (Hirosumi et al. 2002). ER stress is also a major source of ROS generation due to Ca2+release. This increases the concentration of cytosolic Ca2+ and subsequently stimulates mitochondria metabolism to produces more ROS. Thus, the ER may be a proximal site that senses over-nutrition and translates it into metabolic and age-related inflammatory responses.
Interdependency between inflammation and insulin resistance during aging
Although many studies have focused on the effects of inflammatory responses on insulin resistance, only few studies have specifically investigated the effects of insulin resistance on inflammatory responses. Exposure to excessive amounts of nutrients and energy can impair the insulin signaling in metabolically important tissues such as the liver, adipose tissue, and skeletal muscle. Insulin resistance is therefore closely associated with chronic inflammation, likely due to increased levels of pro-inflammatory cytokines.
In fact, type 2 diabetes induced by insulin resistance has recently been suggested as a cause of accelerated aging (Mokdad et al. 2001). In an insulin resistant state, serum insulin levels are increased to a hyperinsulinemia state that activates the Akt/IKK signaling pathway, and thus activates NF-κB, a core transcription factor involved in the inflammatory response in non-metabolic organs (Fig. 3). Over recent years, the incidence of NF-κB activation in human kidney has risen dramatically, primarily because of the increasing prevalence of both obesity and insulin resistance (El-Atat et al. 2004). Obesity and insulin resistance both make substantial contributions to renal disease by up-regulating Akt activity. Many studies suggest that chronic insulin resistance, accompanied by hyperinsulinemia, contributes to several pathologies, such as atherosclerosis, through augmenting the effects of pro-inflammatory cytokines (Lele 2007; Goldschmidt-Clermont et al. 2005). In practice, high levels of the pro-inflammatory cytokines have been found in the adipose tissue of a diabetic rat model. They activate JNK and IKK/NF-κB signaling pathways, resulting in or from the up-regulation of potential mediators of chronic inflammation, which perpetuates reciprocal interactions between the chronic inflammation and adiposity, leading to accelerated aging.
Fig. 3.
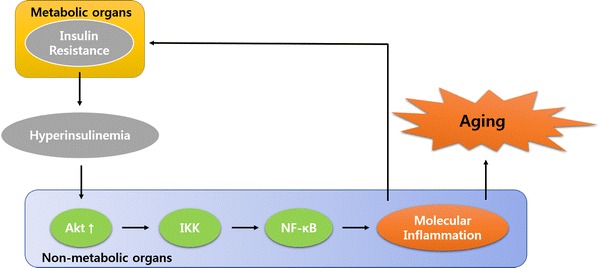
Crosstalk between insulin resistance in metabolic organs and molecular inflammation in non-metabolic organs. The serum insulin concentration is up regulated in an insulin resistance state. Insulin resistance-induced hyperinsulinemia stimulates inflammatory responses in non-metabolic organs such as kidney by activating the Akt/IκB kinase (IKK) signaling pathway
Therefore, it is possible that therapeutic drugs for insulin resistance, such as AMPK activator or PPARα/γ dual agonist, have the potential to become anti-aging agents by ameliorating insulin resistance. Actually, metformin, an AMPK activator, extends lifespan in C. elegansand mice (Cabreiro et al. 2013; Martin-Montalvo et al. 2013). Indeed, it has been shown that insulin-induced FOXO1 is a master protein that turns on the expression of another key inflammatory cytokine, IL-1β that also inhibits insulin signaling (Su et al. 2009). Thus, FOXO1 inhibitors may have the potential to develop into anti-aging agents through their ability to inhibit IL-1β production.
It is still unclear whether insulin resistance or inflammation occurs first. It is clear however, that there is a vicious cycle between insulin resistance and inflammation that occurs during aging and contributes to accelerated aging. Also, over-nutrition intake, such as with a western diet, could accelerate aging by increasing both inflammation and insulin resistance.
Conclusion
Many aging hypotheses have been proposed over the past several decades, including the recent molecular inflammation hypothesis of aging. This hypothesis of molecular inflammation is based on activities of the key NF-κB signaling pathway, which plays a crucial role in activating the systemic inflammatory response during the aging process when there is increased adiposity. Chronic inflammation due to obesity-induced ectopic tissue, such as liver and muscle lipid accumulation further exacerbates insulin resistance. A state of chronic inflammation perpetuates an insulin resistant state, and the interactions between a chronic inflammatory response and increased adiposity likely accelerate the aging process.
Acknowledgments
This work was supported for two years by Pusan National University Research Grant.
References
- Agostini L, Martinon F, Burns K, Mcdermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/S1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Progress in retinal and eye research. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends in Pharmacological Sciences. 2012;33:304–311. doi: 10.1016/j.tips.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ET, Ory DS, Schaffer JE. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Molecular Biology of the Cell. 2006;17:770–778. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahimi-Horn MC, Pouyssegur J. Oxygen, a source of life and stress. FEBS Letters. 2007;581:3582–3591. doi: 10.1016/j.febslet.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Brown MR, Clark KD, Gulia M, Zhao Z, Garczynski SF, Crim JW, Suderman RJ, Strand MR. An insulin-like peptide regulates egg maturation and metabolism in the mosquito Aedes aegypti. Proceedings of the National Academy of Sciences USA. 2008;105:5716–5721. doi: 10.1073/pnas.0800478105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cocheme HM, Noori T, Weinkove D, Schuster E, Greene ND, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Im J, Nioka S, Kushmerick M. Skeletal muscle energetics with PNMR: Personal views and historic perspectives. NMR in Biomedicine. 2006;19:904–926. doi: 10.1002/nbm.1109. [DOI] [PubMed] [Google Scholar]
- Chomyn A, Attardi G. MtDNA mutations in aging and apoptosis. Biochemical and biophysical research communications. 2003;304:519–529. doi: 10.1016/S0006-291X(03)00625-9. [DOI] [PubMed] [Google Scholar]
- Chung HY, Lee EK, Choi YJ, Kim JM, Kim DH, Zou Y, Kim CH, Lee J, Kim HS, Kim ND, Jung JH, Yu BP. Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. Journal of Dental Research. 2011;90:830–840. doi: 10.1177/0022034510387794. [DOI] [PubMed] [Google Scholar]
- Despres JP. Is visceral obesity the cause of the metabolic syndrome? Annals of Medicine. 2006;38:52–63. doi: 10.1080/07853890500383895. [DOI] [PubMed] [Google Scholar]
- Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- El-Atat FA, Stas SN, Mcfarlane SI, Sowers JR. The relationship between hyperinsulinemia, hypertension and progressive renal disease. Journal of the American Society of Nephrology. 2004;15:2816–2827. doi: 10.1097/01.ASN.0000133698.80390.37. [DOI] [PubMed] [Google Scholar]
- Ferre P, Foufelle F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Hormone Research. 2007;68:72–82. doi: 10.1159/000100426. [DOI] [PubMed] [Google Scholar]
- Giorgino F, Laviola L, Eriksson JW. Regional differences of insulin action in adipose tissue: Insights from in vivo and in vitro studies. Acta Physiologica Scandinavica. 2005;183:13–30. doi: 10.1111/j.1365-201X.2004.01385.x. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont PJ, Creager MA, Losordo DW, Lam GK, Wassef M, Dzau VJ. Atherosclerosis 2005: Recent discoveries and novel hypotheses. Circulation. 2005;112:3348–3353. doi: 10.1161/CIRCULATIONAHA.105.577460. [DOI] [PubMed] [Google Scholar]
- Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiology & Behavior. 2008;94:206–218. doi: 10.1016/j.physbeh.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- Gustafson B, Smith U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3-L1 preadipocytes. Journal of Biological Chemistry. 2006;281:9507–9516. doi: 10.1074/jbc.M512077200. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Current Pharmaceutical Design. 2008;14:1225–1230. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- Heilbronn LK, Gan SK, Turner N, Campbell LV, Chisholm DJ. Markers of mitochondrial biogenesis and metabolism are lower in overweight and obese insulin-resistant subjects. Journal of Clinical Endocrinology and Metabolism. 2007;92:1467–1473. doi: 10.1210/jc.2006-2210. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Hong SE, Heo HS, Kim DH, Kim MS, Kim CH, Lee J, Yoo MA, Yu BP, Leeuwenburgh C, Chung HY. Revealing system-level correlations between aging and calorie restriction using a mouse transcriptome. Age (Dordr) 2010;32:15–30. doi: 10.1007/s11357-009-9106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- Howcroft TK, Campisi J, Louis GB, Smith MT, Wise B, Wyss-Coray T, Augustine AD, Mcelhaney JE, Kohanski R, Sierra F. The role of inflammation in age-related disease. Aging (Albany NY) 2013;5:84–93. doi: 10.18632/aging.100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, Zlabinger GJ, Stulnig TM, Chemokine CC. chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. Journal of Clinical Endocrinology and Metabolism. 2008;93:3215–3221. doi: 10.1210/jc.2007-2630. [DOI] [PubMed] [Google Scholar]
- Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, Smoke CC, Meyer C, Hojlund K, Yi Z, Mandarino LJ. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes. 2010;59:33–42. doi: 10.2337/db09-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology. 2004;145:5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- Lele RD. Causation, prevention and reversal of vascular endothelial dysfunction. Journal of the Association of Physicians of India. 2007;55:643–651. [PubMed] [Google Scholar]
- Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology. 2008;48:770–781. doi: 10.1002/hep.22423. [DOI] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, De Cabo R. Metformin improves healthspan and lifespan in mice. Nature communications. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clinical microbiology reviews. 2009;22:240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nature Reviews Molecular Cell Biology. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Giang W, Galante RJ, Pack AI. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. Journal of Neurochemistry. 2005;92:1150–1157. doi: 10.1111/j.1471-4159.2004.02952.x. [DOI] [PubMed] [Google Scholar]
- Nuss JE, Choksi KB, Deford JH, Papaconstantinou J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochemical and Biophysical Research Communication. 2008;365:355–361. doi: 10.1016/j.bbrc.2007.10.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual Review of Physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y, Ogawa S. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes. 2005;54:657–663. doi: 10.2337/diabetes.54.3.657. [DOI] [PubMed] [Google Scholar]
- Park D, Lee EK, Jang EJ, Jeong HO, Kim BC, Ha YM, Hong SE, Yu BP, Chung HY. Identification of the dichotomous role of age-related LCK in calorie restriction revealed by integrative analysis of cDNA microarray and interactome. Age (Dordr) 2013;35:1045–1060. doi: 10.1007/s11357-012-9426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, Dipietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phielix E, Schrauwen-Hinderling VB, Mensink M, Lenaers E, Meex R, Hoeks J, Kooi ME, Moonen-Kornips E, Sels JP, Hesselink MK, Schrauwen P. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57:2943–2949. doi: 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prompers JJ, Jeneson JA, Drost MR, Oomens CC, Strijkers GJ, Nicolay K. Dynamic MRS and MRI of skeletal muscle function and biomechanics. NMR in Biomedicine. 2006;19:927–953. doi: 10.1002/nbm.1095. [DOI] [PubMed] [Google Scholar]
- Rabek JP, Boylston WH, 3rd, Papaconstantinou J. Carbonylation of ER chaperone proteins in aged mouse liver. Biochemical and Biophysical Research Communication. 2003;305:566–572. doi: 10.1016/S0006-291X(03)00826-X. [DOI] [PubMed] [Google Scholar]
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Saini V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J Diabetes. 2010;1:68–75. doi: 10.4239/wjd.v1.i3.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: Modulation by nutrients and inflammation. The Journal of Clinical Investigation. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. Journal of Applied Physiology. 2000;1985(88):1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Sepe A, Tchkonia T, Thomou T, Zamboni M, Kirkland JL. Aging and regional differences in fat cell progenitors—A mini-review. Gerontology. 2011;57:66–75. doi: 10.1159/000279755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. Journal of Clinical Endocrinology and Metabolism. 2007;92:1023–1033. doi: 10.1210/jc.2006-1055. [DOI] [PubMed] [Google Scholar]
- Stump CS, Short KR, Bigelow ML, Schimke JM, Nair KS. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proceedings of the National Academy of Sciences. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D, Coudriet GM, Hyun Kim D, Lu Y, Perdomo G, Qu S, Slusher S, Tse HM, Piganelli J, Giannoukakis N, Zhang J, Dong HH. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes. 2009;58:2624–2633. doi: 10.2337/db09-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szendroedi J, Schmid AI, Chmelik M, Toth C, Brehm A, Krssak M, Nowotny P, Wolzt M, Waldhausl W, Roden M. Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Medicine. 2007;4:e154. doi: 10.1371/journal.pmed.0040154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Molecular Medicine. 2008;14:222–231. doi: 10.2119/2007-00119.Tilg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nature Reviews Molecular Cell Biology. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY. Hypoxia/Reoxygenation induces nitric oxide and TNF-alpha release from cultured microglia but not astrocytes of the rat. Chinese Journal of Physiology. 2007;50:127–134. [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. Journal of Clinical Investigation. 2005;115:1111–1119. doi: 10.1172/JCI200525102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, Mccabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Munzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metabolism. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BP, Chung HY. Oxidative stress and vascular aging. Diabetes Research and Clinical Practice. 2001;54(Suppl 2):S73–S80. doi: 10.1016/S0168-8227(01)00338-2. [DOI] [PubMed] [Google Scholar]
- Yu BP, Chung HY. Adaptive mechanisms to oxidative stress during aging. Mechanisms of Ageing and Development. 2006;127:436–443. doi: 10.1016/j.mad.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. Journal of Biological Chemistry. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Molecular Medicine. 2000;6:347–373. [PMC free article] [PubMed] [Google Scholar]