

Significance
Adipocytes evolve from preadipocyte progenitors by the process of adipogenesis. This process is governed by the adipogenic master regulator peroxisome proliferator–activator receptor gamma (PPARγ). PPARγ is also involved in metabolic homeostasis and in pathological states such as obesity and diabetes. Furthermore, a common genetic polymorphism of PPARγ, encoding the proline 12 to alanine substitution (Pro12Ala) variant, is clinically important. Investigating PPARγ regulation is therefore vital for understanding these physiological and pathological conditions. We show that the tyrosine kinase Abelson murine leukemia viral oncogene (c-Abl) is an adipogenic key regulator. c-Abl promotes adipogenesis by phosphorylation and subsequent stabilization of PPARγ. Importantly, the reduced binding affinity of the Pro12Ala variant to c-Abl provides an explanation for this genetic variant phenotype.
Keywords: adipogenesis, PPAR gamma, c-Abl, differentiation, Pro12Ala polymorphism
Abstract
Adipocyte differentiation, or adipogenesis, is a complex and highly regulated process. A recent proteomic analysis has predicted that the nonreceptor tyrosine kinase Abelson murine leukemia viral oncogene (c-Abl) is a putative key regulator of adipogenesis, but the underlying mechanism remained obscure. We found that c-Abl was activated during the early phase of mouse 3T3-L1 preadipocyte differentiation. Moreover, c-Abl activity was essential and its inhibition blocked differentiation to mature adipocytes. c-Abl directly controlled the expression and activity of the master adipogenic regulator peroxisome proliferator–activator receptor gamma 2 (PPARγ2). PPARγ2 physically associated with c-Abl and underwent phosphorylation on two tyrosine residues within its regulatory activation function 1 (AF1) domain. We demonstrated that this process positively regulates PPARγ2 stability and adipogenesis. Remarkably, c-Abl binding to PPARγ2 required the Pro12 residue that has a phenotypically well-studied common human genetic proline 12 alanine substitution (Pro12Ala) polymorphism. Our findings establish a critical role for c-Abl in adipocyte differentiation and explain the behavior of the known Pro12Ala polymorphism.
Adipogenesis is a highly regulated process by which preadipocytes mature into adipocytes, and it is one of the most intensively studied models of cellular differentiation. Peroxisome proliferator–activator receptor gamma (PPARγ) is a transcription factor and a pivotal regulator of adipocyte differentiation (1). It contains an N-terminal transactivation domain (activation function 1, AF1), a DNA binding domain (DBD), and a C-terminal ligand binding domain (LBD, which harbors the ligand-dependent transactivation domain AF2). The AF1 domain of PPARγ2 is involved in transactivation and recognition of the protein by transcriptional coactivators (2, 3) and can also influence ligand binding affinity of the LBD (4). PPARγ dimerizes with retinoid X receptor α (RXRα) and then binds to peroxisome proliferator response elements (PPREs) within the promoter of its targeted genes (5). PPARγ interacts with a wide spectrum of natural lipophilic ligands and may also exhibit high affinity to synthetic ligands, like the thiazolidinediones. Alternative splicing and differential promoter use result in two PPARγ isoforms, a ubiquitously expressed PPARγ1 and an adipose restricted PPARγ2 (6), with the latter harboring a 30-residue extension at its N terminus. Several posttranslational modifications of PPARγ are known, most of which are inhibitory, for example sumoylation (7) and phosphorylation by ERK (8).
Interestingly, a proline residue in the PPARγ2 N terminus is commonly mutated to alanine in the general population (9). The Pro12Ala substitution in PPARγ2 is associated with decreased receptor activity, lower body mass index, and improved insulin sensitivity (10). PPARγ2 Pro12Ala demonstrates a reduced interaction with some of its coactivators (11).
The nonreceptor tyrosine kinase Abelson murine leukemia viral oncogene (c-Abl) is known to regulate several cellular processes and to participate in cell fate decisions like induction of apoptosis or cell cycle arrest (12–14). However, c-Abl is involved in differentiation as well (15–18). Interestingly, c-Abl is highly expressed in human adipocytes (19) and activated upon insulin stimulation, where it participates in regulation of metabolic downstream signals (20). Furthermore, a recent proteomic analysis placed c-Abl as a potential key regulator of adipogenesis (21).
Here we report that c-Abl is activated upon adipocyte differentiation induction and supports PPARγ2 accumulation. PPARγ2 is phosphorylated by c-Abl in the AF1 domain, a modification that promotes PPARγ2 stabilization. Remarkably the PPARγ2 polymorph is a poorer c-Abl substrate. Our results place c-Abl as a key positive regulator of adipocyte differentiation and explain the behavior of the PPARγ2 polymorphism.
Results
c-Abl Activation in Adipogenesis.
We followed c-Abl activation in adipocyte differentiation by using an antibody detecting the c-Abl Y245 autophosphorylated form, a hallmark of an active kinase (22). c-Abl was activated on the first day of differentiation induction and remained active up to the middifferentiation stage (Fig. 1A). Next, we inhibited c-Abl kinase activity using the c-Abl inhibitor STI-571 (Imatinib). STI-571 is also an inhibitor of the tyrosine kinases c-kit and platelet-derived growth factor receptor (PDGFR) (23). Although c-kit is not expressed in 3T3-L1 (24), PDGFR is a negative regulator of adipocyte differentiation but is only expressed during the first hours of adipogenesis, after which it rapidly declines to an undetectable level (25). To minimize the potential inhibition of PDGFR and specifically target c-Abl, STI-571 was added 7 h after differentiation onset and treatment was maintained until day 5. Remarkably, even at a low inhibitor concentration, adipogenesis was markedly reduced (Fig. 1B). Similar results were obtained when we depleted c-Abl using a specific lentiviral vector encoding shRNA (Fig. 1C). Under this condition, expression of the major adipogenic regulator, PPARγ, and two of its important transcriptional targets, adipocyte protein 2 (aP2) and lipoprotein lipase (LPL), was markedly reduced (Fig. 1D). Interestingly, although the PPARγ protein level was shown to decrease with c-Abl inhibition, transcription of both PPARγ isoforms was unchanged, whereas transcription of their target genes was markedly reduced (Fig. 1E). These results suggest that c-Abl regulates PPARγ expression at the protein level in normal adipocyte differentiation.
Fig. 1.

c-Abl is required for adipocyte differentiation. (A) c-Abl activity increases during adipogenesis. 3T3-L1 adipoblasts were induced to differentiate with medium containing 5 μg/mL insulin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine (IBMX). Cells were lysed at the indicated times, and protein levels were analyzed by Western blotting. The proteasomal subunit PSMA4 served as a loading control. (B) STI-571 inhibits adipocyte differentiation. 3T3-L1 cells were induced to differentiate with addition of STI-571 at the indicated concentrations or DMSO on days 0–5. Cells were stained with oil red O and photographed at day 8. Relative intracellular lipid content was obtained by dissolving oil red O and measuring optical density (OD) at 520 nm (***P < 0.005). (C) c-Abl is required in adipocyte differentiation. 3T3-L1 expressing shRNA against either GFP or c-Abl were induced to differentiate and stained with oil red O at day 10. Intracellular lipid content was measured as in B (***P < 0.005). (D) c-Abl is required for expression of PPARγ and its target genes during adipogenesis. 3T3-L1 cells expressing shRNA against either GFP or c-Abl were induced to differentiate and lysed at the indicated times. Protein levels were analyzed by Western blotting. (E) c-Abl is required for transcription of PPARγ proadipogenic target genes but not for transcription of PPARγ itself. At day 4 of differentiation, 3T3-L1 cells were analyzed for mRNA expression of the indicated genes by quantitative PCR (***P < 0.005). Sequences of the oligos used are listed in Table S1.
c-Abl Regulates Accumulation of PPARγ2.
Under c-Abl knockdown, the PPARγ2 protein level was reduced (Fig. 1D), and therefore, we reasoned that c-Abl might play a role in PPARγ2 accumulation. To check whether the low level of endogenous PPARγ2 expression in c-Abl–depleted cells is the result of protein destabilization, we treated c-Abl–depleted 3T3-L1 cells with MG-132, a proteasomal inhibitor (Fig. 2A). PPARγ2 protein level markedly increased after proteasomal blockade, suggesting a role for c-Abl in PPARγ2 accumulation during adipocyte differentiation. Next, we measured PPARγ2 protein half-life in the presence or absence of an active form of c-Abl (Δ1–81 c-Abl). Following translation inhibition by cycloheximide, the level of PPARγ2 gradually dropped to a minimal level within 6 h (Fig. 2B; see also Fig. S5). Remarkably, in the presence of a constitutively active c-Abl, PPARγ2 decay was much slower, whereas a kinase-dead c-Abl had only a minor effect. To investigate the possibility that c-Abl supports PPARγ2 stabilization via inhibition of the polyubiquitination of PPARγ2, we coexpressed HA-tagged ubiquitin together with Flag-tagged PPARγ2 with or without active c-Abl and analyzed the pattern of the polyubiquitinated PPARγ2 ladder. PPARγ2 ubiquitination was indeed significantly inhibited by c-Abl in a kinase-dependent manner (Fig. 2C and Fig. S1). These data suggest that active c-Abl promotes the stability of the PPARγ2 protein.
Fig. 2.

c-Abl prolongs PPARγ2 half-life. (A) Proteasomal inhibition in middifferentiation results in PPARγ2 accumulation. 3T3-L1 cells expressing shRNA to GFP (control) or c-Abl were induced to differentiate using insulin, dexamethasone, and IBMX. At day 5, cells were treated with 25 μg MG-132 and lysed after 5 h. Protein levels were analyzed by immunoblotting. (B) c-Abl stabilizes PPARγ2. HEK293 cells were transfected with plasmids expressing HA-PPARγ2 with or without active c-Abl or a kinase-mutated (KM) c-Abl. Twenty-four hours after transfection, cells were treated with 500 μM cycloheximide for different time points. Equal amounts of total protein lysates were analyzed by Western blotting. (C) c-Abl inhibits PPARγ2 polyubiquitination. HEK293 cells were transfected as indicated. Cell extracts were immunoprecipitated using Flag beads and immunoblotted with anti-HA to visualize polyubiquitination. The ratio between immunoprecipitated polyubiquitinated PPARγ2 ladder and PPARγ2 protein level from three independent experiments was quantified (***P < 0.005; NS, nonsignificant). Immunoblot data from the representative experiment are shown in Fig. S1.
c-Abl Interacts with PPARγ2 and Phosphorylates It.
Having demonstrated that c-Abl is a positive regulator of adipogenesis and its kinase activity is essential for this role, we next asked whether PPARγ2 is a direct substrate of c-Abl. First, we measured their possible physical association in transfected HEK293 cells. When Flag-tagged PPARγ2 was immunoprecipitated, a substantial amount of c-Abl was brought down as well (Fig. 3A). In a reciprocal experiment, PPARγ2 was coimmunoprecipitated with c-Abl (Fig. S2A). Similar results were obtained with the endogenous proteins of 3T3-L1 cells at the middifferentiation stage (Fig. 3B). c-Abl binds proteins through its SH3 domain to the PxxP motifs of the target protein (26). Three PxxP motifs are found in PPARγ2, all of which reside within the AF1 domain (Fig. 3C). The first, 9PxxP12, is located in the unique PPARγ2 N-terminal 30 amino acids that are absent in PPARγ1, the less adipogenic isoform (6, 27). To check whether the 9PxxP12 is involved in binding to c-Abl, we generated a four-amino-acid deletion mutant of PPARγ2 (Δ9–12). Remarkably, c-Abl failed to bind this mutant (Fig. 3D).
Fig. 3.
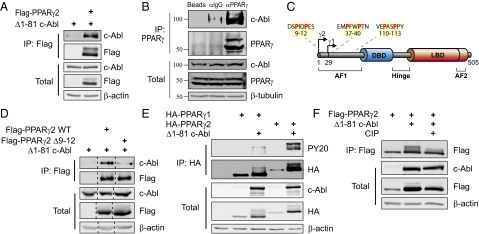
c-Abl binds and phosphorylates PPARγ2. (A) c-Abl binds PPARγ2. HEK293 cells were transfected with the indicated plasmids and analyzed by coimmunoprecipitation. (B) Endogenous c-Abl and PPARγ interact in adipocytes. 3T3-L1 cells were induced to differentiate. At day 5, cells were lysed; immunoprecipitated with either control empty beads, control anti-IgG–coated beads, or anti-PPARγ–coated beads; and protein levels were analyzed by Western blotting. The molecular weight of c-Abl is a bit lower than expected, which may suggest processing as has been previously reported (42, 43). (C) PxxP motifs within PPARγ; arrows denote transcription start sites for PPARγ1 and PPARγ2. The three PxxP motifs in the AF1 domain are highlighted. AF, activation function domain; DBD, DNA binding domain; LBD, ligand binding domain. Image modified from ref. 44. (D) The N-terminal PxxP motif of PPARγ2 is required for interaction with c-Abl. HEK293 cells were transfected with the indicated plasmids and analyzed by immunoprecipitation with anti-Flag beads. (E) c-Abl phosphorylates PPARγ2 but not PPARγ1. HEK293 cells were transfected with the indicated plasmids and analyzed by immunoprecipitation using anti-HA beads. Phosphorylation was detected by probing with an anti-phosphotyrosine antibody (PY20). (F) The mobility shift in the PPARγ2 band represents a phosphorylated form of PPARγ2. HEK293 cells were transfected with the indicated plasmids. Cell lysates were incubated in 37 °C for 30 min in the presence or absence of 150 U/mL calf-intestinal alkaline phosphatase (CIP) and were then analyzed by immunoprecipitation using anti-Flag beads.
Next, we investigated PPARγ2 phosphorylation by c-Abl. We first looked for a difference in degree of phosphorylation by c-Abl between PPARγ2 containing this motif and the PPARγ1 isoform lacking it. Interestingly, when probing with a phosphotyrosine-specific antibody, we found that unlike HA-PPARγ2, HA-PPARγ1 was only poorly phosphorylated by c-Abl (Fig. 3E). Similar results were obtained using Flag-PPARγ2 (Fig. S2B). PPARγ2 phosphorylation was accompanied by an accumulation of the PPARγ2 protein and by a substantial up-shift in the migration pattern on the gel, a fact that may suggest several phosphorylation sites. To verify this possibility, we repeated the experiment in the presence of alkaline phosphatase. This resulted in disappearance of the upshifted form of PPARγ2, implying that the mobility shift is the consequence of phosphorylation (Fig. 3F). These data suggest that c-Abl binds PPARγ2 through the N-terminal PxxP motif and subsequently phosphorylates it.
Phosphorylation of Two Tyrosine Residues of PPARγ2 by c-Abl Increases PPARγ2 Accumulation.
PPARγ is regulated either by ligand binding to its LBD or by other factors, mainly through its AF1 domain (Fig. 3B). To search for potential c-Abl phosphorylation sites on PPARγ2, we investigated the AF1 domain, as important modifications take place in this domain (7, 8). To this end, tyrosine residues in the AF1 domain were mutated into the phosphodead residue phenylalanine. Unlike the wild type, the PPARγ2 Y78F and Y102F mutants were not upshifted in the presence of active c-Abl, but the latter displayed a certain phosphorylation level (Fig. 4A and Fig. S3A). A double Y78F and Y102F mutant (PPARγ2 2YF) was not tyrosine phosphorylated in the presence of active c-Abl (Fig. S3B), delineating these two tyrosine residues as the major c-Abl targets. To check whether these tyrosine residues are phosphorylated by endogenous c-Abl during differentiation, we stably expressed PPARγ2 in NIH 3T3 mouse fibroblasts, which lack endogenous PPARγ2. These cells can be induced to differentiate into adipocytes similar to preadipocytes (28). PPARγ2 that was immunoprecipitated from undifferentiated NIH 3T3 cells was moderately tyrosine phosphorylated (Fig. 4B). Remarkably, at middifferentiation, PPARγ2 but not PPARγ2 2YF was markedly tyrosine phosphorylated. These data suggest that c-Abl phosphorylates PPARγ2 on tyrosine residues 78 and 102, with tyrosine 78 being the major phosphorylation site.
Fig. 4.

Two tyrosine residues in the AF1 domain of PPARγ2 are the phosphorylation targets of c-Abl. (A) Tyr78 and Tyr102 are c-Abl phosphorylation sites in PPARγ2. HEK293 cells were transfected with the indicated plasmids and analyzed by immunoprecipitation. Phosphorylation was detected by probing with an antiphosphotyrosine antibody (PY20). (B) Differentiation induction enhances phosphorylation of Tyr78 and Tyr102 by endogenous c-Abl. NIH 3T3 fibroblasts were infected with control pBabe retroviral vector or with pBabe carrying the indicated PPARγ2 constructs. Cells were induced to differentiate with insulin, dexamethasone, IBMX, and 1 μΜ Rosiglitazone. At day 5, cells were lysed and analyzed by immunoprecipitation. (C) Phosphomimetic mutations on tyrosine residues 78 and 102 result in PPARγ2 stabilization. HEK293 cells were transfected with plasmids expressing the different Flag-PPARγ2 constructs. Twenty-four hours after transfection, cells were treated with 500 μM cycloheximide for different time points. Equal amounts of total protein lysates were analyzed by Western blotting. (D) Phosphomimetic mutations on tyrosine residues 78 and 102 inhibit PPARγ2 polyubiquitination. HEK293 cells were transfected as indicated. Cell extracts were immunoprecipitated using anti-Flag beads and immunoblotted with anti-HA to visualize polyubiquitination. (E) Quantification of the ratio between immunoprecipitated polyubiquitinated PPARγ2 ladder and PPARγ2 protein level from three independent experiments (*P < 0.05).
Next, we wished to check whether tyrosine phosphorylation by c-Abl is a mechanism by which c-Abl promotes PPARγ2 stabilization. To chemically mimic the phosphorylation state of PPARγ2 to uncouple it from the presence of c-Abl, we generated a double Y78E and Y102E mutant (PPARγ2 2YE). We then measured protein half-life of PPARγ2 in cycloheximide-treated cells. Although the protein level of both the wild-type PPARγ2 and the 2YF mutant gradually decreased within 4 h, that of the phosphomimetic PPARγ2 2YE mutant remained constant (Fig. 4C). To investigate whether the increased stabilization of the 2YE mutant is the result of inhibition of its polyubiquitination, we analyzed the different PPARγ2 constructs when coexpressed with an HA-tagged ubiquitin. As expected, PPARγ2 ubiquitination was significantly reduced when the two c-Abl–targeted tyrosines were phosphomimetically mutated, compared with the wild-type or phosphodead mutated PPARγ2 (Fig. 4 D and E). These data suggest that c-Abl modifies PPARγ2 to escape degradation.
PPARγ2 Is Activated by c-Abl.
To investigate the role of c-Abl in inducing PPARγ2 activity, we used the PPRE3 reporter construct containing three copies of the PPARγ-responsive element sequence (29). In the presence of active c-Abl, PPARγ2-induced activation of the promoter was doubled compared with induction with PPARγ2 alone (Fig. 5A). A c-Abl kinase-dead mutant failed to do so (Fig. S4A), suggesting that c-Abl in a kinase-dependent manner supports PPARγ2 transcription activity.
Fig. 5.

c-Abl potentiates PPARγ2 activity. (A) PPARγ2 transcriptional activity is enhanced by c-Abl. The designated plasmids were cotransfected with a PPRE3-Luciferase reporter plasmid into HEK293 cells. After 24 h, cells were lysed and luciferase activity was measured and compared with vector control transfected cells. (B) PPARγ2 transcriptional activity is enhanced by phosphomimetic mutations on tyrosine residues 78 and 102. The designated plasmids were cotransfected with a PPRE3-Luciferase reporter plasmid into HEK293 cells. RXRα was cotransfected with all PPARγ2 constructs, and Rosiglitazone (1 μM) was added 6 h after transfection. After 24 h, cells were analyzed as in A. (C) Tyr78 and Tyr102 phosphorylation promotes adipocyte differentiation. NIH 3T3 fibroblasts were infected with control pBabe retroviral vector or with pBabe carrying the indicated PPARγ2 constructs. Cells were cultured to confluence and then induced to differentiate with medium containing 1 μΜ Rosiglitazone. At day 10, cells were fixed and stained with oil red. Relative intracellular lipid content was obtained by dissolving oil red O and measuring OD at 520 nm (***P < 0.005). (D) Tyr78 and Tyr102 phosphorylation promotes PPARγ target gene induction during adipogenesis. NIH 3T3 fibroblasts were transduced with the indicated PPARγ2 constructs and were induced to differentiate as in C. At day 4, NIH 3T3 cells expressing the indicated PPARγ constructs were analyzed for mRNA expression of the indicated genes by quantitative PCR (***P < 0.005).
Next, we investigated PPARγ2 mutated at the tyrosine residues undergoing phosphorylation by c-Abl in transcription. Compared with either wild-type PPARγ2 or the phosphodead mutants, the Y78E and Y102E phosphomimetic mutants were more active (Fig. 5B). Interestingly, the PPARγ2 2YE double mutant was most active in this assay and in a real-time bioluminescence recording assay (Fig. S4 B and C). These data support a model whereby Y78 and Y102 phosphorylation potentiate PPARγ2 activity.
We next asked whether these PPARγ2 tyrosine residues are involved in adipocyte differentiation. To this end, we overexpressed either wild-type PPARγ2 or the phosphomimetic 2YE mutant in NIH 3T3 mouse fibroblasts. The wild-type PPARγ2 cells displayed a moderate adipogenic phenotype as assessed by oil red staining (Fig. 5C). Remarkably, overexpression of the PPARγ2 2YE phosphomimetic mutant resulted in a substantial increase in differentiation level, both at the level of oil red staining (Fig. 5C) and the PPARγ target genes aP2 and LPL (Fig. 5D). The single phosphomimetic mutants were also more active than the wild type (Fig. S4 D and E). These data suggest that tyrosine phosphorylation of PPARγ2 at the residues targeted by c-Abl plays a role in adipogenesis.
c-Abl Binding Site on PPARγ2 Is a Functional Genetic Polymorphism Site.
As mentioned above, c-Abl was found to bind PPARγ2 through the N-terminal 9PxxP12 sequence. Interestingly, Proline 12 is polymorphic and some individuals carry alanine at this position, the common Pro12Ala variant (9). We therefore wished to check whether polymorphism may influence regulation of PPARγ2 by c-Abl. Remarkably, the Pro12Ala variant was inefficient in binding to c-Abl and in undergoing phosphorylation by the kinase (Fig. 6A). Furthermore, this variant did not accumulate with the expression of active c-Abl (Fig. 6B and Fig. S5).
Fig. 6.

c-Abl binds PPARγ through a genetic polymorphism site. (A) The Pro12Ala (P12A) mutation of PPARγ2 interferes with c-Abl potential to bind and phosphorylate PPARγ2. HEK293 cells were transfected with the indicated plasmids and analyzed by immunoprecipitation using anti-Flag beads. Phosphorylation was detected by probing with an antiphosphotyrosine antibody (PY20). (B) The P12A mutation of PPARγ2 inhibits stabilization by c-Abl. HEK293 cells were transfected with plasmids expressing P12A mutant PPARγ2 with or without active c-Abl. After 24 h, cells were treated with 500 μM cycloheximide for different time points. Equal amounts of total protein lysates were analyzed by Western blotting. (C) Phosphorylation by c-Abl favors binding of PPARγ2 to PGC-1α. HEK293 cells were transfected with the indicated plasmids and analyzed by coimmunoprecipitation.
In a recent work, the Pro12Ala variant was found to poorly bind coactivators like PPAR gamma coactivator 1 alpha (PGC-1α) (11), which is an important component in metabolism regulation by PPARγ (30). To check if c-Abl may affect interaction of PPARγ2 with PGC-1α, we expressed both proteins in HEK293 cells and coimmunoprecipitated PGC-1α in the presence or absence of active c-Abl. Although c-Abl enhanced binding of PGC-1α to wild-type PPARγ2, the phosphodead mutant 2YF was significantly less efficient in PGC-1α binding in the presence of c-Abl (Fig. 6C). Because the expression level of the wild-type and mutant PPARγ in this experiment was the same, this suggests that tyrosine phosphorylation of PPARγ has a more direct role in regulating interaction with cofactors, in addition to the effect on PPARγ stability. These data suggest that reduced affinity of the PPARγ2 Pro12Ala variant to its coactivators may be explained by impairment in c-Abl binding and tyrosine phosphorylation.
Discussion
Here we report a role of c-Abl as a positive mediator regulating adipocyte differentiation. We demonstrate that adipogenic stimuli activate c-Abl. c-Abl binds to the N-terminal PxxP motif of PPARγ2 and phosphorylates two tyrosine residues within the AF1 domain. We also show that this process gives rise to PPARγ2 accumulation, possibly by escaping ubiquitin-dependent proteasomal degradation (Fig. 7). In addition, the tyrosine phosphorylated PPARγ2 appears to form a functional complex with coactivating proteins and ligands, a critical process in inducing the adipogenic target genes and adipogenesis promotion. Interestingly, the effect of c-Abl on PPARγ2 is comparable to that exerted on another important substrate, p73 (13, 31). In both cases, c-Abl participates in cell fate determination by stabilizing a key transcription factor—cell cycle arrest and apoptosis in the case of p73, and cellular differentiation in the case of PPARγ2. Moreover, as c-Abl is known to be engaged in several nuclear processes (32), we believe that its regulation on PPARγ2 also takes place in the nucleus.
Fig. 7.

Regulation of adipogenesis by c-Abl, a model. Under conditions by which c-Abl is not active, PPARγ2 is unstable. Normal adipogenesis is accompanied by c-Abl activation, resulting in PPARγ2 phosphorylation at tyrosine residues 78 and 102, and its subsequent stabilization and binding to transcriptional coactivators. DBD, DNA binding domain; L, PPARγ2 ligand (in red) or RXRα ligand (in gray); LBD, ligand binding domain; P, phosphorylated tyrosine residues; Ub, ubiquitin.
We report here that c-Abl is activated once preadipocytes are induced to differentiate, but the underlying mechanisms remain to be clarified. Upon SH3 domain binding to the PxxP motif, c-Abl might undergo modulation (32), however because c-Abl activation precedes PPARγ2 accumulation, there must be an alternative means of c-Abl activation. c-Abl is known to be activated physiologically by DNA damage response elements like ataxia telangiectasia mutant (ATM) protein (33). Interestingly, both ATM and c-Abl were found to be activated not only by DNA damage induction but also by insulin treatment (20, 34), suggesting a possible mechanism of c-Abl activation by adipogenic induction through ATM. It was also demonstrated that genotoxic drugs or reactive oxygen species can promote adipocyte differentiation of cultured mesenchymal progenitors of adipocytes (35, 36). An intriguing possibility that should be further investigated is that controlled DNA damage is being used during normal adipogenesis to activate key regulators like c-Abl.
As mentioned above, c-Abl was found to be involved in differentiation of diverse cell types (15–18), whereas a recent proteomic analysis showed that it may be a novel adipogenic regulator (21). As we demonstrated, c-Abl inhibition caused a dramatic adipogenic abrogation, underlying its role as a key regulator of this process. Specifically, c-Abl depletion resulted in increased proteasomal degradation of PPARγ2, whereas active c-Abl increased PPARγ2 protein half-life, findings that point to PPARγ2 stabilization as a mechanism by which c-Abl may promote adipocyte differentiation.
A connection between c-Abl and the Pro12Ala polymorphism of PPARγ2 was found in this study, as the proline residue that is mutated in this genetic variant was found to be a part of a c-Abl binding PxxP motif. This fact may be the molecular basis of the reduced activity of the Pro12Ala variant of PPARγ2 and may explain its phenotype. Our data showed that phosphorylation by c-Abl promotes interaction of PPARγ2 with PGC-1α. This result is in line with the finding that the Pro12Ala variant poorly binds coactivators like PGC-1α (11).
Although this work establishes an important regulatory function of c-Abl in adipogenesis in vitro, the role of c-Abl in adipogenesis or in adipose tissue regulation in vivo is yet unclear. c-Abl knockout mice usually die before birth, with surviving mice dying shortly after because of defects in the proper development of the immune system (37). STI-571 (Imatinib) was shown to promote adipocytic differentiation of mesenchymal stromal cells (38) and STI-571 treatment in chronic myelogenous leukemia (CML) patients was associated with fat accumulation and improved insulin sensitivity (39). These data imply that STI-571 positively regulates PPARγ and thus seemingly contradict the observations in our study. However, a recent study has found that these STI-571 effects on adipose and insulin homeostasis are attributed to its inhibition of PDFGR (40). We showed in this study that specific inhibition of c-Abl, either by RNA interference or by the timely administration of STI-571, has an antiadipogenic effect and interferes with PPARγ activity. In the future, drugs that specifically target c-Abl may be developed and thus avoid unwanted effects on other kinases. As PPARγ was shown to promote high fat diet-induced adipocyte hypertrophy (41), a drug that inhibits c-Abl could be potentially useful in treating obesity. In summary, this study establishes an important positive regulatory role for c-Abl in the differentiation of adipocytes and delineates its interplay with the master adipogenic regulator PPARγ2.
Materials and Methods
Cells and Cell Culture.
The cell lines used were human embryonic kidney cells HEK293 and HEK293T, NIH 3T3 mouse fibroblasts, and the mouse preadipocyte cell 3T3-L1. Culture conditions, reagents, plasmids, transfections, mRNA analysis, and immunoblot and coimmunoprecipitation studies are described in detail in SI Materials and Methods.
Reporter Gene Assays.
HEK293 cells were transfected with plasmids expressing the tested constructs along with a promoter-containing firefly luciferase reporter plasmid. Cell lysates were analyzed as detailed in SI Materials and Methods.
Statistical Analysis.
All values presented in graphs represent the average of at least three independent experiments. To estimate distribution of values, SE was calculated. The two-tailed Student t test was used to verify statistical significance in the difference between relevant values.
Supplementary Material
Acknowledgments
We thank M. Rubinstein for providing anti-PPARγ, anti-aP2, and anti-LPL antibodies; C. Kahana for anti-PSMA4 antibody; J. Bar-Tana for the PPRE3 luciferase reporter plasmid; G. Asher for his assistance in real-time bioluminescence recording; and Novartis for STI-571. This work was supported by grants from the Israel Science Foundation (551/11) and from the Minerva Foundation, with funding from the Federal German Ministry for Education and Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411086111/-/DCSupplemental.
References
- 1.Rosen ED. The molecular control of adipogenesis, with special reference to lymphatic pathology. Ann N Y Acad Sci. 2002;979:143–158, discussion 188–196. doi: 10.1111/j.1749-6632.2002.tb04875.x. [DOI] [PubMed] [Google Scholar]
- 2.Castillo G, et al. An adipogenic cofactor bound by the differentiation domain of PPARgamma. EMBO J. 1999;18(13):3676–3687. doi: 10.1093/emboj/18.13.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bugge A, Grontved L, Aagaard MM, Borup R, Mandrup S. The PPARgamma2 A/B-domain plays a gene-specific role in transactivation and cofactor recruitment. Mol Endocrinol. 2009;23(6):794–808. doi: 10.1210/me.2008-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shao D, et al. Interdomain communication regulating ligand binding by PPAR-gamma. Nature. 1998;396(6709):377–380. doi: 10.1038/24634. [DOI] [PubMed] [Google Scholar]
- 5.Chambon P. The nuclear receptor superfamily: A personal retrospect on the first two decades. Mol Endocrinol. 2005;19(6):1418–1428. doi: 10.1210/me.2005-0125. [DOI] [PubMed] [Google Scholar]
- 6.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8(10):1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 7.Ohshima T, Koga H, Shimotohno K. Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. J Biol Chem. 2004;279(28):29551–29557. doi: 10.1074/jbc.M403866200. [DOI] [PubMed] [Google Scholar]
- 8.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274(5295):2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 9.Yen CJ, et al. Molecular scanning of the human peroxisome proliferator activated receptor gamma (hPPAR gamma) gene in diabetic Caucasians: Identification of a Pro12Ala PPAR gamma 2 missense mutation. Biochem Biophys Res Commun. 1997;241(2):270–274. doi: 10.1006/bbrc.1997.7798. [DOI] [PubMed] [Google Scholar]
- 10.Deeb SS, et al. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1998;20(3):284–287. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 11.Heikkinen S, et al. The Pro12Ala PPARgamma2 variant determines metabolism at the gene-environment interface. Cell Metab. 2009;9(1):88–98. doi: 10.1016/j.cmet.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Yuan ZM, et al. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382(6588):272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- 13.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399(6738):809–813. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 14.Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008;29(3):350–361. doi: 10.1016/j.molcel.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Bennett RL, Hoffmann FM. Increased levels of the Drosophila Abelson tyrosine kinase in nerves and muscles: Subcellular localization and mutant phenotypes imply a role in cell-cell interactions. Development. 1992;116(4):953–966. doi: 10.1242/dev.116.4.953. [DOI] [PubMed] [Google Scholar]
- 16.Daniel R, Wong PM, Chung SW. Isoform-specific functions of c-abl: Type I is necessary for differentiation, and type IV is inhibitory to apoptosis. Cell Growth Differ. 1996;7(9):1141–1148. [PubMed] [Google Scholar]
- 17.Koch A, et al. Direct interaction of nerve growth factor receptor, TrkA, with non-receptor tyrosine kinase, c-Abl, through the activation loop. FEBS Lett. 2000;469(1):72–76. doi: 10.1016/s0014-5793(00)01242-4. [DOI] [PubMed] [Google Scholar]
- 18.Lee YC, et al. Src family kinase/abl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene. 2010;29(22):3196–3207. doi: 10.1038/onc.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Neill AJ, Cotter TG, Russell JM, Gaffney EF. Abl expression in human fetal and adult tissues, tumours, and tumour microvessels. J Pathol. 1997;183(3):325–329. doi: 10.1002/(SICI)1096-9896(199711)183:3<325::AID-PATH941>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 20.Frasca F, et al. Role of c-Abl in directing metabolic versus mitogenic effects in insulin receptor signaling. J Biol Chem. 2007;282(36):26077–26088. doi: 10.1074/jbc.M705008200. [DOI] [PubMed] [Google Scholar]
- 21.Wilson B, Liotta LA, Petricoiniii E. Dynamic protein pathway activation mapping of adipose-derived stem cell differentiation implicates novel regulators of adipocyte differentiation. Mol Cell Proteomics. 2013;12(9):2522–2535. doi: 10.1074/mcp.M112.025346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brasher BB, Van Etten RA. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 2000;275(45):35631–35637. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- 23.Buchdunger E, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295(1):139–145. [PubMed] [Google Scholar]
- 24.Matsubara Y, Suzuki H, Ikeda Y, Murata M. Generation of megakaryocytes and platelets from preadipocyte cell line 3T3-L1, but not the parent cell line 3T3, in vitro. Biochem Biophys Res Commun. 2010;402(4):796–800. doi: 10.1016/j.bbrc.2010.10.120. [DOI] [PubMed] [Google Scholar]
- 25.Vaziri C, Faller DV. Down-regulation of platelet-derived growth factor receptor expression during terminal differentiation of 3T3-L1 pre-adipocyte fibroblasts. J Biol Chem. 1996;271(23):13642–13648. doi: 10.1074/jbc.271.23.13642. [DOI] [PubMed] [Google Scholar]
- 26.Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259(5098):1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 27.Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS. PPARgamma knockdown by engineered transcription factors: Exogenous PPARgamma2 but not PPARgamma1 reactivates adipogenesis. Genes Dev. 2002;16(1):27–32. doi: 10.1101/gad.953802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, et al. Identification of a peroxisome proliferator-responsive element upstream of the gene encoding rat peroxisomal enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase. Proc Natl Acad Sci USA. 1992;89(16):7541–7545. doi: 10.1073/pnas.89.16.7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puigserver P, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 31.Tsai KK, Yuan ZM. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 2003;63(12):3418–3424. [PubMed] [Google Scholar]
- 32.Shaul Y. c-Abl: Activation and nuclear targets. Cell Death Differ. 2000;7(1):10–16. doi: 10.1038/sj.cdd.4400626. [DOI] [PubMed] [Google Scholar]
- 33.Baskaran R, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387(6632):516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 34.Yang DQ, Kastan MB. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol. 2000;2(12):893–898. doi: 10.1038/35046542. [DOI] [PubMed] [Google Scholar]
- 35.Altanerova V, Horvathova E, Matuskova M, Kucerova L, Altaner C. Genotoxic damage of human adipose-tissue derived mesenchymal stem cells triggers their terminal differentiation. Neoplasma. 2009;56(6):542–547. doi: 10.4149/neo_2009_06_542. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Lee YJ, Choi H, Ko EH, Kim JW. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. J Biol Chem. 2009;284(16):10601–10609. doi: 10.1074/jbc.M808742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65(7):1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 38.Fitter S, et al. Long-term imatinib therapy promotes bone formation in CML patients. Blood. 2008;111(5):2538–2547. doi: 10.1182/blood-2007-07-104281. [DOI] [PubMed] [Google Scholar]
- 39.Fitter S, et al. Plasma adiponectin levels are markedly elevated in imatinib-treated chronic myeloid leukemia (CML) patients: A mechanism for improved insulin sensitivity in type 2 diabetic CML patients? J Clin Endocrinol Metab. 2010;95(8):3763–3767. doi: 10.1210/jc.2010-0086. [DOI] [PubMed] [Google Scholar]
- 40.Fitter S, Vandyke K, Gronthos S, Zannettino AC. Suppression of PDGF-induced PI3 kinase activity by imatinib promotes adipogenesis and adiponectin secretion. J Mol Endocrinol. 2012;48(3):229–240. doi: 10.1530/JME-12-0003. [DOI] [PubMed] [Google Scholar]
- 41.Kubota N, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4(4):597–609. doi: 10.1016/s1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- 42.Machuy N, Rajalingam K, Rudel T. Requirement of caspase-mediated cleavage of c-Abl during stress-induced apoptosis. Cell Death Differ. 2004;11(3):290–300. doi: 10.1038/sj.cdd.4401336. [DOI] [PubMed] [Google Scholar]
- 43.Yu D, et al. Phosphorylation of DNA topoisomerase I by the c-Abl tyrosine kinase confers camptothecin sensitivity. J Biol Chem. 2004;279(50):51851–51861. doi: 10.1074/jbc.M404396200. [DOI] [PubMed] [Google Scholar]
- 44.Tontonoz P, Spiegelman BM. Fat and beyond: The diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.