

Significance
The heart contracts more forcefully in response to fear, stress, or exercise through the fight-or-flight response. This physiological process is mediated by β-adrenergic receptors acting through adenylyl cyclase, cAMP, and cAMP-dependent protein kinase (PKA), which phosphorylates the cardiac calcium channel CaV1.2 and increases its activity. We show that mutation of a single amino acid residue, Ser-1700, in a PKA phosphorylation site at the interface between the distal and proximal C-terminal domains substantially disrupts this regulatory mechanism in mice. Basal and β-adrenergic–stimulated calcium currents, myocyte contractility, and stress-induced exercise capacity are all reduced. Moreover, these mice develop cardiac hypertrophy, an indication of failure of cardiac homeostasis in vivo. Evidently, phosphorylation of Ser-1700 is a primary event in cardiovascular regulation.
Keywords: β-adrenergic, calcium channel, protein kinase, cAMP, cardiac hypertrophy
Abstract
L-type calcium (Ca2+) currents conducted by voltage-gated Ca2+ channel CaV1.2 initiate excitation–contraction coupling in cardiomyocytes. Upon activation of β-adrenergic receptors, phosphorylation of CaV1.2 channels by cAMP-dependent protein kinase (PKA) increases channel activity, thereby allowing more Ca2+ entry into the cell, which leads to more forceful contraction. In vitro reconstitution studies and in vivo proteomics analysis have revealed that Ser-1700 is a key site of phosphorylation mediating this effect, but the functional role of this amino acid residue in regulation in vivo has remained uncertain. Here we have studied the regulation of calcium current and cell contraction of cardiomyocytes in vitro and cardiac function and homeostasis in vivo in a mouse line expressing the mutation Ser-1700–Ala in the CaV1.2 channel. We found that preventing phosphorylation at this site decreased the basal L-type CaV1.2 current in both neonatal and adult cardiomyocytes. In addition, the incremental increase elicited by isoproterenol was abolished in neonatal cardiomyocytes and was substantially reduced in young adult myocytes. In contrast, cellular contractility was only moderately reduced compared with wild type, suggesting a greater reserve of contractile function and/or recruitment of compensatory mechanisms. Mutant mice develop cardiac hypertrophy by the age of 3–4 mo, and maximal stress-induced exercise tolerance is reduced, indicating impaired physiological regulation in the fight-or-flight response. Our results demonstrate that phosphorylation at Ser-1700 alone is essential to maintain basal Ca2+ current and regulation by β-adrenergic activation. As a consequence, blocking PKA phosphorylation at this site impairs cardiovascular physiology in vivo, leading to reduced exercise capacity in the fight-or-flight response and development of cardiac hypertrophy.
Upon membrane depolarization, CaV1.2 channels conduct L-type calcium (Ca2+) current into cardiomyocytes and initiate excitation–contraction coupling (1, 2). Ca2+ influx through Cav1.2 channels activates Ca2+ release from the sarcoplasmic reticulum, which leads to contraction of myofilaments. As the initiator of excitation–contraction coupling, Ca2+ influx via CaV1.2 channels is tightly regulated. Under conditions of fear, stress, and exercise, the sympathetic nervous system activates the fight-or-flight response, in which the marked increase in contractile force of the heart is caused by epinephrine and norepinephrine acting through β-adrenergic receptors, activation of adenylyl cyclase, increased cAMP, activation of cAMP-dependent protein kinase (PKA), and phosphorylation of the CaV1.2 channel (1, 3). Phosphorylation of the CaV1.2 channel leads to a threefold to fourfold increase in peak current amplitude in mammalian cardiomyocytes. Regulation of the CaV1.2 channel by the cAMP signaling pathway is altered in cardiac hypertrophy and heart failure (4–6). Under those pathological conditions, responsiveness of CaV1.2 channel activity to β-adrenergic receptors and PKA activation is severely blunted, resulting in diminished contractile reserve and impaired fight-or-flight response (6, 7). Enormous effort has been devoted to understanding how β-adrenergic regulation of the CaV1.2 channel is achieved, but the exact molecular mechanisms remain unresolved.
CaV1.2 channels contain multiple subunits, including a pore-forming α11.2 subunit (also designated α1C), β and α2δ subunits that modulate expression of CaV1.2 at the cell surface, and possibly γ subunits (8). The closely related CaV1.1 and CaV1.2 channels in skeletal and cardiac muscle, respectively, are both proteolytically processed near the center of their large C-terminal domains (9, 10), and the distal C terminus (dCT) remains associated noncovalently with the proximal C terminus (pCT) and serves as a potent autoinhibitor (11, 12). Regulation of CaV1.2 channels by PKA was reconstituted in nonmuscle cells with a dynamic range of threefold to fourfold similar to native cardiomyocytes by building the autoinhibitory CaV1.2 complex through cotransfection of each of its components (13). Successful reconstitution required an A Kinase Anchoring Protein (AKAP), which recruits PKA to the dCT (13–15). Deletion of the dCT in vivo results in loss of regulation of the L-type Ca2+ current by the β-adrenergic pathway and embryonic death from heart failure (16, 17). These results suggest that the autoinhibited CaV1.2 signaling complex serves as the substrate for β-adrenergic regulation, and disruption of this complex leads to heart failure.
PKA is responsible for phosphorylation of the CaV1.2 channel in response to β-adrenergic stimulation in cardiac myocytes (18–22). Although multiple PKA sites have been identified in α1 subunits by in vitro phosphorylation (10, 23), none of these sites is required for regulation of CaV1.2 channels in vivo. For example, PKA-dependent phosphorylation of S1928 is prominent in transfected cells and cardiomyocytes (10, 24), but its phosphorylation has little or no effect on β-adrenergic up-regulation of cardiac CaV1.2 channel activity in transfected cells or cardiomyocytes (13, 25, 26). Two sites in the C terminus of the skeletal muscle CaV1.1 channel are phosphorylated in vivo as assessed by mass spectrometry (S1575 and T1579), and phosphorylation of S1575 is increased by β-adrenergic stimulation (27). These sites are conserved in cardiac CaV1.2 channels as S1700 and T1704, and phosphoproteomics analysis revealed β-adrenergic–stimulated phosphorylation of S1700 by PKA (28). S1700 and T1704 reside at the interface between the pCT and dCT. In studies of the CaV1.2 signaling complex reconstituted in nonmuscle cells, phosphorylation of both sites was required for normal basal channel activity, whereas only S1700 was essential for PKA stimulation (13). Mutation of S1700 and T1704 to Ala in STAA mice reduced basal activity and CaV1.2 channel regulation by the β-adrenergic pathway in cardiomyocytes (29). To further dissect the contribution of S1700, we studied a mutant mouse line expressing CaV1.2 channel with the S1700A mutation (SA mice). Our results demonstrate that this single phosphorylation site is required for normal regulation of CaV1.2 channels, contraction of cardiac myocytes, exercise capacity, and cardiac homeostasis.
Results
S1700A Mutation in CaV1.2 Channels in Mice.
S1700 and T1704 are located on the same side of an alpha helix, strategically positioned in the interaction face between the dCT and pCT (Fig. 1A). Standard gene-targeting procedures were used to generate mice in which the CaV1.2 channel harbors the S1700A mutation (Fig. 1B). SA mice are viable and show no overtly distinguishable phenotypes before postnatal day 60. Examination of expression of mutant CaV1.2 channels by immunoblotting extracts from whole heart revealed a trend toward reduced channel expression that did not reach significance (Fig. 1C). More importantly, in acutely dissociated ventricular myocytes, immunocytochemical staining of CaV1.2 channels with an antibody recognizing the intracellular linker connecting domains II and III of the α11.2 subunit (30, 31) showed levels of specific staining in integrated Z-stack images that were comparable to WT (Fig. 1C). Moreover, line-scan analysis from surface and middle planes of Z-stack images revealed that the cellular distribution of mutant CaV1.2 channels is similar to WT (Fig. S1), with only a small reduction in staining intensity at the surface of the cells. No significant difference in specific immunostaining was observed with the anti-CH2 antibody, which recognizes the dCT (Fig. 1D), suggesting an unchanged ratio of dCT to pCT in SA cardiomyocytes, in contrast to the increase in dCT observed in STAA cardiomyocytes (29). Using the phosphospecific antibody anti-CH3P against phospho-S1928, we observed similar basal immunostaining, and 100 nM isoproterenol (Iso) increased phosphorylation to a similar degree in WT and SA myocytes (Fig. 1E). Thus, overall responsiveness of β-adrenergic signaling is preserved in myocytes expressing CaV1.2/S1700A. Together, these results indicate that Ala substitution for S1700 alone does not substantially affect CaV1.2 channel expression or distribution, the stoichiometry of the dCT and pCT in the autoinhibitory complex, or the overall β-adrenergic signaling to CaV1.2 channels as judged from phosphorylation of S1928.
Fig. 1.

Alanine substitution of S1700 in Cav1.2 channels. (A) Molecular model of S1700 and T1704 at the interface between the dCT and pCT. (B) Mutations indicated by an asterisk (*) were introduced into exon 41 of CaV1.2 genomic DNA by homologous recombination to generate a knock-in mouse line expressing Ala at S1700 (SA). The point mutation is immediately followed by the Neo cassette. (C) (Top) Representative immunoblot of heart extracts of WT (left two lanes) and SA (right two lanes) animals using an antibody targeting the intracellular II–III loop of CaV1.2 channels (anti-CNC1). (Middle) Representative immunostaining using anti-CNC1 antibody. (Bottom) Relative expression of CaV1.2 channel in WT and SA hearts was quantified with immunoblot (Left) and fluorescence immunostaining (Right). (D, Upper) Representative immunostaining of cardiomyocytes from WT and SA mice using an antibody recognizing the dCT (anti-CH2) under control conditions and 10 min after 100 nM Iso stimulation. (Lower) Relative fluorescence intensity was quantified. (E, Upper) Representative immunostaining of cardiomyocytes from WT and SA using an antibody recognizing phosphorylated S1928 under basal conditions and in response to 100 nM Iso stimulation. (Lower) The fluorescence intensity was quantified. ***P < 0.001 (two-way ANOVA, Iso vs. control).
Reduced Basal Ba2+ Currents and Blunted Response to β-Adrenergic Stimulation in SA Neonatal Cardiomyocytes.
Using Ba2+ as charge carrier, we examined basal CaV1.2 channel function in neonatal cardiomyocytes dissociated on postnatal days 0–2, before the hypertrophy that develops later in life for SA mice (see below) could adversely affect CaV1.2 regulation. Basal CaV1.2 channel activity was significantly reduced in SA myocytes. The peak L-type current at +10 mV was 419 ± 55 pA in WT and 268 ± 38 pA in SA cardiomyocytes (P < 0.001). We tested responsiveness to β-adrenergic stimulation by comparing L-type current before and 3 min after addition of 1 µM Iso (Fig. 2A). A statistically significant increase was observed in WT but not in SA cardiomyocytes (Fig. 2B). These results indicated that preventing phosphorylation of S1700 reduces both basal L-type Ca2+ channel activity and its up-regulation by the β-adrenergic/PKA pathway. These effects of the S1700A mutation on CaV1.2 channel function in neonatal myocytes suggest that the changes we observe in channel regulation are intrinsic to the phosphorylation state of this site, rather than being secondary to the resulting development of cellular pathology.
Fig. 2.
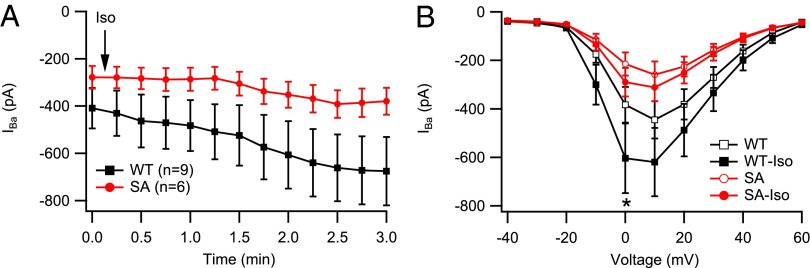
Reduced basal Ba2+ current and impaired response to β-adrenergic activation by 1 μM Iso in SA neonatal cardiomyocytes. (A) Time course of IBa following application of Iso (arrow). Basal current was stable for 2 min before zero time. (B) Current–voltage relationship in unstimulated cardiomyocytes and after stimulation by 1 μM Iso. Statistical significance was determined by two-way ANOVA with a Bonferroni posttest. *P < 0.05 (Iso vs. basal).
Response to β-Adrenergic Stimulation in Young Adult SA Myocytes.
We also tested β-adrenergic responsiveness in myocytes isolated from young-adult animals when β-adrenergic signaling was fully mature. Basal currents in myocytes from SA mutant mice were small relative to currents in WT myocytes (Fig. 3 A–C, red). Similarly, the increments in Ca2+ current amplitude induced by treatment for 5 min with 3, 10, and 100 nM Iso were also smaller for SA myocytes than WT (Fig. 3 A–C). The shape of the current–voltage relationships for WT cells treated with 0, 3, 10, or 100 nM Iso (Fig. 3 A–C, black) reveals the well-known negative shift of the Ca2+ current after Iso treatment. The smaller current amplitudes in SA myocytes (Fig. 3 A–C, red) prevented a quantitative assessment of differences in the negative shift in the current–voltage relationship, but the negative shift seemed less prominent at 100 nM Iso in SA myocytes compared with WT. The most prominent effect of the SA mutation is the marked reduction in peak amplitude of the Iso-stimulated CaV1.2 current over this concentration range in comparison with WT myocytes (Fig. 3D).
Fig. 3.

Reduced basal Ca2+ current and impaired response to β-adrenergic activation by Iso in adult cardiomyocytes. (A–C) ICa was recorded under basal conditions and 5 min after application of 3 nM (A), 10 nM (B), and 100 nM (C) Iso in WT (n = 8–18) and SA (n = 11–19) cardiomyocytes. Basal current was stable for 2 min before addition of Iso. (D) Mean absolute magnitudes of peak current plotted vs. Iso concentration. (E) Baseline subtracted Iso-induced increment in ICa density plotted against Iso concentration. *P < 0.05 (SA vs. STAA; ref. 29); ***P < 0.001 (WT vs. SA).
It is typical to assess the β-adrenergic stimulation of CaV1.2 channel activity by comparing the ratio of current observed before and after treatment with Iso. However, this metric is misleading in SA myocytes because both the denominator (basal current) and the numerator (Iso-stimulated current) are substantially reduced, and the reduction in basal current in the denominator artifactually increases the stimulation ratio. Our dissociated SA cardiomyocytes have approximately the same density and distribution of CaV1.2 channels as WT (Fig. 1B and Fig. S1). Therefore, to quantify the extent to which current could be increased through these channels, basal CaV1.2 current was subtracted, and the increments in CaV1.2 current induced by Iso were plotted vs. concentration. The Iso-induced increment in peak CaV1.2 current in SA myocytes (Fig. 3E, red) showed significant reduction at low concentrations of Iso and was greatly reduced in response to full activation of the β-adrenergic pathway with 100 nM Iso in comparison with WT myocytes (19.4 ± 1.2 pA/pf for WT vs. 6.5 ± 0.2 pA/pf for SA; P < 0.001; 33% of WT; Fig. 3E, black).
It is of interest to compare the concentration–response curves for myocytes from SA and STAA mice (Fig. 3E, light gray). The results for WT myocytes in these two datasets are nearly identical (Fig. 3E, black and gray). In contrast, the form of the concentration–response curves is noticeably different in STAA and SA myocytes. The double mutation of S1700 and T1704 has a greater effect at low Iso concentrations, shifting the concentration–response curve toward higher concentrations (Fig. 3E, gray; ref. 29), whereas the single S1700A mutation has less effect at low levels of Iso stimulation but a similar effect at 100 nM Iso (Fig. 3E, red). Possible interpretations of this difference in concentration dependence are considered in Discussion.
Impaired β-Adrenergic Regulation of Contraction in SA Myocytes.
Ca2+ entry via the CaV1.2 channel is the first step in excitation–contraction coupling, which induces Ca2+ release from the sarcoplasmic reticulum and myocyte contraction in a nonlinear manner. To determine the degree to which reduced Ca2+ entry in the SA myocytes affects regulation of contraction, we examined the response of cellular contractility to Iso stimulation. Myocytes were stimulated at 1 Hz in the absence and presence of a range of concentrations of Iso, and cell shortening was measured by microscopy. As in WT, SA myocytes contracted in response to stimulation (Fig. 4A), and the extent of contraction increased in response to Iso (Fig. 4B). Surprisingly, despite the substantial decrease in CaV1.2 current, contraction was not significantly different from WT, except at 10 nM Iso, where it was decreased by 30%. Thus, contraction and its regulation by Iso are better preserved than Ca2+ current and its regulation.
Fig. 4.

In vitro cell shortening in response to Iso stimulation. (A) A representative trace of cell shortening in response to field stimulation at 1 Hz. (B) Cell shortening was quantified at basal levels and in response to 3, 10, and 100 nM Iso. **P < 0.01 (WT vs. SA).
In our previous work, we found that the phenotype of CaV1.2 STAA mutant mice included frequent episodes of Iso-induced asynchronous contractions and contraction failures (29). These phenotypes were not observed for myocytes from the SA mice studied here, suggesting a milder deficit in control of contractility compared with STAA mice.
Reduced Exercise Capacity in SA Mice.
To assess the importance of phosphorylation at S1700 in the fight-or-flight response in vivo, we forced WT and SA mice to run uphill on a treadmill to escape a foot shock at the bottom of the slope until exhaustion. SA mice were able to run uphill and escape the shock for ∼30% less distance than WT mice (Fig. 5A). This deficit was similar to that of STAA mice (29), indicating a substantial reduction in contractile reserve and impairment of the fight-or-flight response in vivo for both mouse lines.
Fig. 5.

Exercise tolerance and cardiac hypertrophy. (A) Total distance run on a treadmill. (B and C) Cardiac hypertrophy measured as heart/body-weight ratio (B) or membrane capacitance measured in whole-cell patch-clamp experiments (C). *P < 0.05. n = 7 WT, 5 SA (B); 45 WT, 46 SA (C).
Cardiac Hypertrophy in SA Mice.
Disruption of Ca2+ homeostasis has severe consequences in the heart (2, 4, 32). Our results show that blocking phosphorylation at S1700 reduces basal and Iso-stimulated L-type Ca2+ currents and cellular contractility in SA myocytes. In our previous studies of STAA mice (29), this deficit was associated with a striking increase in heart to body weight ratio and cellular hypertrophy. Similar phenotypes were observed in the SA mice studied here, with a 26% increase in heart/body-weight ratio (Fig. 5B) and a 15% increase in cellular hypertrophy measured as cell-surface capacitance (Fig. 5C) compared with WT. These results indicate that the lack of phosphorylation of S1700 and the resulting reduction in basal and β-adrenergic–stimulated Ca2+ current lead to cardiac hypertrophy in vivo.
Discussion
This report describes the effects of a single serine-to-alanine mutation in a key cAMP-dependent phosphorylation site in the CaV1.2 channel on regulation of Ca2+ current and contraction in dissociated cardiomyocytes and on cardiac performance and homeostasis in vivo. Mutation of this PKA phosphorylation site reduced basal CaV1.2 current in cardiac myocytes to ∼37% of its WT value, and maximal stimulation with 100 nM Iso increased CaV1.2 current to only ∼33% of the WT value. Importantly, these effects occurred despite similar levels of expression and subcellular distribution of CaV1.2 protein in SA and WT cardiomyocytes. Thus, phosphorylation of S1700 is essential for setting both the level of basal CaV1.2 activity and the β-adrenergic stimulation of Ca2+ current. In contrast to the effects on Ca2+ current, overall β-adrenergic signaling was normal, as assessed by phosphorylation of S1928 in the C-terminal domain of CaV1.2 in response to Iso. The effect of the S1700A mutation on cell contraction was less than on the CaV1.2 current itself. Therefore, the primary physiological deficits at the cellular level were the size of the Cav1.2 current and the stimulation of peak current by Iso. Despite normal β-adrenergic signaling and the limited effect on cell contraction, the altered regulation of CaV1.2 was associated with striking cardiac hypertrophy and substantial reduction in maximal stress-induced exercise capacity in vivo. These are remarkable physiological consequences from the mutation of a single amino acid residue in a protein phosphorylation site. The significance of these results for CaV1.2 channel regulation and cardiovascular function is considered in more detail below.
Phosphorylation of S1700 Is Required for Basal Activity of CaV1.2 Channels.
The basal L-type Ca2+ current is regulated by PKA as well as other protein kinases (13). In transfected nonmuscle cells, Ala substitution for both S1700 and T1704 reduced basal L-type Ca2+ currents, indicating that these two phosphorylation sites are both involved in control of basal CaV1.2 channel activity (13). T1704 is a substrate for casein kinase II, whereas S1700 is phosphorylated by PKA and Ca2+/calmodulin-dependent protein kinase II (13, 27). Our results showing that the single S1700A mutation reduced basal Ca2+ current to ∼37% of normal demonstrates the importance of phosphorylation of this site for the basal level of Ca2+ current in cardiac myocytes. We observed a similar reduction in basal current amplitude for the STAA mutant, in which both S1700 and T1704 are mutated (29). The simplest explanation for those results is that the SA mutation is responsible for nearly all of the reduction in current observed for the combined STAA construct. However, it is also possible that phosphorylation at both sites is necessary to sustain normal regulation of the basal level of CaV1.2 current in cardiomyocytes, and therefore mutation of either site would give a reduction similar to mutating both. In any case, it is clear from the results presented here that phosphorylation of S1700 is a primary determinant of the level of CaV1.2 current at basal levels of protein phosphorylation.
Phosphorylation of S1700 Is Required for Normal Stimulation of CaV1.2 Current in the Fight-or-Flight Response.
In transfected nonmuscle cells, mutation of S1700 prevented up-regulation of CaV1.2 channel activity in response to treatment with Iso (13). The S1700A mutation resulted in greatly reduced peak Ca2+ current in response to 3, 10, and 100 nM Iso in myocytes from SA mice. This loss of regulation was not caused by alteration in β-adrenergic signaling per se, because phosphorylation of S1928 was unchanged from WT. Moreover, the remaining stimulation of CaV1.2 current by Iso had similar concentration dependence to WT. Altogether, our results in transfected nonmuscle cells and SA mice provide strong evidence that S1700 is the primary site of PKA phosphorylation mediating the fight-or-flight response in cardiac myocytes.
Contrasting results were reported from studies in which endogenous CaV1.2 channels in cardiomyocytes were blocked with a dihydropyridine antagonist and exogenous WT or S1700A/T1704A CaV1.2 channels harboring a mutation that prevents dihydropyridine block were expressed from a transgene (33). In this experimental system, the response of the CaV1.2 channels to 200 nM Iso was not significantly reduced by combined S1700A and T1704A mutations. As we have shown in transfected cells, regulation of CaV1.2 channels by the PKA pathway requires a stoichiometric complex of channel subunits, AKAP, and PKA (13). One plausible reason for the difference in results is the possibility that exogenously expressed CaV1.2 channels do not achieve the correct subcellular location or the correct protein composition and stoichiometry of the CaV1.2 signaling complex.
Role of T1704 Phosphorylation in Regulation of Basal CaV1.2 Activity.
T1704 is predicted to be a substrate for casein kinase II (13, 27), which is constitutively active in cardiac myocytes (34, 35). Thus, basal phosphorylation of this site may be high and not strongly regulated by Iso. T1704 is present in SA, but not in STAA myocytes. S1700 and T1704 are located at the interface where the dCT and pCT interact (Fig. 1A). Despite this difference in available phosphorylation sites at this key interface, we found little difference in basal Ca2+ current between SA and STAA myocytes. Experiments on CaV1.2 channels in transfected cells showed that combining the T1704A mutation with S1700A further decreased the level of basal current (13), suggesting an important role for basal phosphorylation of T1704. In STAA mice, other compensatory changes in regulation of basal phosphorylation and dephosphorylation or other modulators of channel activity may have partially restored basal Ca2+ current despite the mutation of T1704.
Concentration Dependence of Iso Stimulation in STAA and SA Myocytes.
In STAA myocytes, the IC50 for Iso stimulation of CaV1.2 activity is shifted ∼4.6-fold to a higher concentration compared with WT (27). In contrast, the concentration dependence for Iso stimulation is similar for SA and WT mice (Fig. 3E). Thus, phosphorylation at T1704 may destabilize the autoinhibitory interaction between the pCT and dCT and thereby enhance the ability of phosphorylation at S1700 to disinhibit the channel, even though phospho-T1704 alone is not able to relieve autoinhibition in cardiomyocytes. Because the availability of T1704 for phosphorylation is the only known difference between SA and STAA myocytes, the 4.6-fold shift in the concentration–response relationship toward higher Iso concentrations in STAA mice suggests either that phosphorylation of T1704 increases the availability or efficacy of S1700 phosphorylation or that, in the absence of T1704 phosphorylation, an additional unknown site is engaged at higher levels of stimulation of the PKA pathway.
Remaining β-Adrenergic Regulation of CaV1.2 Channels in SA and STAA Myocytes.
Despite the large reduction in basal CaV1.2 current in SA and STAA myocytes, CaV1.2 channels are substantially up-regulated by β-adrenergic/PKA signaling, and the increment in current induced by treatment with 100 nM Iso is approximately one-third of WT (Fig. 3E). This finding implicates one or more PKA sites other than S1700 in the regulation of CaV1.2 channels. A well-characterized site that is phosphorylated in an Iso- and PKA-dependent fashion is S1928 (10, 23, 24); however, no evidence has emerged for a strong effect of phosphorylation of this site in the regulation of cardiac CaV1.2 channels in transfected cells (13) or in cardiomyocytes dissociated from mutant mice (25, 26). It is possible that regulation via phosphorylation of S1928 is unmasked in vivo by mutation of S1700 or that additional, unidentified PKA phosphorylation site(s) are responsible for the remaining regulation of CaV1.2 channels in STAA and SA mice. These additional phosphorylation sites might reside in either the α1 or β subunits.
Block of Phosphorylation of S1700 Has a Smaller Effect on Contraction.
Ca2+ entry through CaV1.2 channels initiates excitation–contraction coupling, but the relationship between Ca2+ entry and contraction is plastic and nonlinear (32, 36). For that reason, submaximal reduction of Ca2+ entry often causes a proportionally smaller reduction in contractile force. This smaller reduction is particularly striking in the SA mutant. Basal CaV1.2 current was reduced to 37% of WT, and the increment in current induced by treatment with 100 nM Iso was only 33% of WT, yet the effects of the SA mutation on myocyte contraction was much smaller by comparison. Contraction increased as a function of Iso concentration but was noticeably less forceful at each Iso concentration, with reductions ranging from 40% at baseline to 12% at 100 nM. However, because of the variability in contraction among single cells, the reduction relative to WT was only significant at 10 nM Iso (30%), which was far less than the 67% decrease in CaV1.2 current. Thus, the effectiveness of coupling between Ca2+ entry and subsequent contraction is greatly increased in SA myocytes, such that much less initial Ca2+ entry triggers levels of contraction that are close to WT values. It is likely that the efficacy of Ca2+ entry in initiating excitation–contraction coupling is increased by physiological compensation for the reduced basal and stimulated Ca2+ entry.
In STAA mice, we observed arrhythmic contractions and contraction failure in 20–25% of dissociated myocytes at 10 and 100 nM Iso (29). Surprisingly, we did not observe a similar incidence of arrhythmia or contractile failure in myocytes from SA mice, despite their similar reduction in basal and stimulated CaV1.2 current. This finding suggests that block of phosphorylation of T1704 might be responsible for triggering such arrhythmic behavior.
Reduced Exercise Capacity and Cardiac Hypertrophy in SA Mice.
Despite the modest reduction in cellular contractility in dissociated SA myocytes, we observed substantially reduced maximal exercise capacity and striking cardiac hypertrophy in SA mice. Our test of exercise capacity engages the fight-or-flight response, because the mice run up the treadmill continuously to escape a foot shock until they reach exhaustion. The reduction in exercise capacity is as large as we observed with STAA mice, again indicating that phosphorylation of S1700 is a primary event in the fight-or-flight response. This reduced exercise capacity of SA mice, plus the changes in intracellular Ca2+ signaling that result from reduced basal and stimulated CaV1.2 current, likely trigger cardiac hypertrophy in SA mice. Previous studies have shown that many changes in Ca-dependent signaling pathways can induce hypertrophy in experimental animals, including changes in Ca2+/calmodulin-dependent protein kinase II activity, calcineurin, level of expression of CaV1.2 channels, and regulation of CaV1.2 channels (4, 5, 37, 38). Remarkably, simply deleting the dCT of CaV1.2 channels is sufficient to cause prenatal cardiac hypertrophy and heart failure (16, 17). Moreover, both increased and decreased expression of CaV1.2 channels can cause hypertrophy and heart failure (37, 38), suggesting a precise balance point for healthy cardiac function. Our results reveal that mutation of a single amino acid residue that impairs basal and β-adrenergic regulation of CaV1.2 channels is sufficient to induce cardiac hypertrophy. Evidently, tight regulation of CaV1.2 channel activity by PKA phosphorylation of S1700 is absolutely essential for normal cardiovascular function and homeostasis in vivo. The role of dysregulation of CaV1.2 channels as a pathogenic effect and potential therapeutic target in heart failure deserves further examination.
Materials and Methods
A mouse line carrying the S1700A mutation was generated by Ingenious Targeting Laboratory using standard methods (SI Materials and Methods). Neonatal cardiomyocytes were isolated as described (16) at postnatal day 0–2. L-type Ba2+ currents in neonatal cardiomyocytes were recorded as described (16). Adult ventricular myocytes were isolated by using a protocol adapted from ref. 39. The whole-cell configuration of the patch-clamp technique was used to record Ca2+ currents from adult ventricular myocytes within 1–6 h of isolation (24). For measurement of cell contraction, freshly isolated ventricular myocytes were perfused with Tyrode’s solution in the absence and presence of Iso. Cells were field-stimulated at 1 Hz. Cell length was measured from video frames and expressed as percent of original length (29). To measure exercise tolerance, mice ran up a 5% incline on a treadmill to escape a mild foot shock, and the distance they ran was recorded (29). Immunocytochemical studies of the distribution of CaV1.2 channels in ventricular myocytes were carried out as described (24). Polyclonal anti-CaV1.2 (anti-CNC1) was generated against amino acid sequences in the intracellular loop between domains II and III of CaV1.2 and characterized (30, 31). Polyclonal anti-CH3P recognizes phosphorylated S1928 and anti-CH2 antibody recognizes a site in dCT. All of the data are reported as the means ± SE. One-way ANOVA with a Bonferroni posttest (GraphPad Prism; Version 5.0) was used to compare the averages from multiple groups. See SI Materials and Methods for additional experimental details.
Supplementary Material
Acknowledgments
This work was supported by American Heart Association Award 12SDG12080084 (to Y.F.) and National Heart, Lung, and Blood Institute Award R01HL085372 (to W.A.C.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1419129111/-/DCSupplemental.
References
- 1.Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301(5901):569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 3.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74(2):365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 4.Richard S, et al. ‘Ca2+-induced Ca2+ entry’ or how the L-type Ca2+ channel remodels its own signalling pathway in cardiac cells. Prog Biophys Mol Biol. 2006;90(1-3):118–135. doi: 10.1016/j.pbiomolbio.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 6.Pleger ST, Boucher M, Most P, Koch WJ. Targeting myocardial β-adrenergic receptor signaling and calcium cycling for heart failure gene therapy. J Card Fail. 2007;13(5):401–414. doi: 10.1016/j.cardfail.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 7.El-Armouche A, Eschenhagen T. β-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev. 2009;14(4):225–241. doi: 10.1007/s10741-008-9132-8. [DOI] [PubMed] [Google Scholar]
- 8.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 9.De Jongh KS, Warner C, Colvin AA, Catterall WA. Characterization of the two size forms of the α1 subunit of skeletal muscle L-type calcium channels. Proc Natl Acad Sci USA. 1991;88(23):10778–10782. doi: 10.1073/pnas.88.23.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Jongh KS, et al. Specific phosphorylation of a site in the full-length form of the α1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry. 1996;35(32):10392–10402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- 11.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576(Pt 1):87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hulme JT, et al. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. Proc Natl Acad Sci USA. 2005;102(14):5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010;3(141):ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hulme JT, Ahn M, Hauschka SD, Scheuer T, Catterall WA. A novel leucine zipper targets AKAP15 and cyclic AMP-dependent protein kinase to the C terminus of the skeletal muscle Ca2+ channel and modulates its function. J Biol Chem. 2002;277(6):4079–4087. doi: 10.1074/jbc.M109814200. [DOI] [PubMed] [Google Scholar]
- 15.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. β-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci USA. 2003;100(22):13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Y, et al. Deletion of the distal C terminus of CaV1.2 channels leads to loss of β-adrenergic regulation and heart failure in vivo. J Biol Chem. 2011;286(14):12617–12626. doi: 10.1074/jbc.M110.175307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domes K, et al. Truncation of murine CaV1.2 at Asp-1904 results in heart failure after birth. J Biol Chem. 2011;286(39):33863–33871. doi: 10.1074/jbc.M111.252312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsien RW, Giles W, Greengard P. Cyclic AMP mediates the effects of adrenaline on cardiac purkinje fibres. Nat New Biol. 1972;240(101):181–183. doi: 10.1038/newbio240181a0. [DOI] [PubMed] [Google Scholar]
- 19.Tsien RW. Calcium channels in excitable cell membranes. Annu Rev Physiol. 1983;45:341–358. doi: 10.1146/annurev.ph.45.030183.002013. [DOI] [PubMed] [Google Scholar]
- 20.Reuter H, Cachelin AB, De Peyer JE, Kokubun S. Modulation of calcium channels in cultured cardiac cells by isoproterenol and 8-bromo-cAMP. Cold Spring Harb Symp Quant Biol. 1983;48(Pt 1):193–200. doi: 10.1101/sqb.1983.048.01.022. [DOI] [PubMed] [Google Scholar]
- 21.Kameyama M, Hescheler J, Hofmann F, Trautwein W. Modulation of Ca current during the phosphorylation cycle in the guinea pig heart. Pflugers Arch. 1986;407(2):123–128. doi: 10.1007/BF00580662. [DOI] [PubMed] [Google Scholar]
- 22.Trautwein W, Hescheler J. Regulation of cardiac L-type calcium current by phosphorylation and G proteins. Annu Rev Physiol. 1990;52:257–274. doi: 10.1146/annurev.ph.52.030190.001353. [DOI] [PubMed] [Google Scholar]
- 23.Mitterdorfer J, et al. Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel α1 subunits. Biochemistry. 1996;35(29):9400–9406. doi: 10.1021/bi960683o. [DOI] [PubMed] [Google Scholar]
- 24.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during β1-adrenergic regulation. Proc Natl Acad Sci USA. 2006;103(44):16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ganesan AN, Maack C, Johns DC, Sidor A, O’Rourke B. β-adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of α1C but not serine 1928. Circ Res. 2006;98(2):e11–e18. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemke T, et al. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Ca v 1.2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008;283(50):34738–34744. doi: 10.1074/jbc.M804981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emrick MA, Sadilek M, Konoki K, Catterall WA. β-adrenergic-regulated phosphorylation of the skeletal muscle CaV1.1 channel in the fight-or-flight response. Proc Natl Acad Sci USA. 2010;107(43):18712–18717. doi: 10.1073/pnas.1012384107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundby A, et al. In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci Signal. 2013;6(278):rs11. doi: 10.1126/scisignal.2003506. [DOI] [PubMed] [Google Scholar]
- 29.Fu Y, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation sites required for regulation of cardiac calcium channels in the fight-or-flight response. Proc Natl Acad Sci USA. 2013;110(48):19621–19626. doi: 10.1073/pnas.1319421110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hell JW, et al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J Cell Biol. 1993;123(4):949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hell JW, et al. Differential phosphorylation of two size forms of the neuronal class C L-type calcium channel α1 subunit. J Biol Chem. 1993;268(26):19451–19457. [PubMed] [Google Scholar]
- 32.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87(4):275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, et al. β-adrenergic regulation of the L-type Ca2+ channel does not require phosphorylation of α1C Ser1700. Circ Res. 2013;113(7):871–880. doi: 10.1161/CIRCRESAHA.113.301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McFarland TP, Sleiman NH, Yaeger DB, Cala SE. The cytosolic protein kinase CK2 phosphorylates cardiac calsequestrin in intact cells. Mol Cell Biochem. 2011;353(1-2):81–91. doi: 10.1007/s11010-011-0777-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Issinger OG. Casein kinases: pleiotropic mediators of cellular regulation. Pharmacol Ther. 1993;59(1):1–30. doi: 10.1016/0163-7258(93)90039-g. [DOI] [PubMed] [Google Scholar]
- 36.Ginsburg KS, Bers DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004;556(Pt 2):463–480. doi: 10.1113/jphysiol.2003.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goonasekera SA, et al. Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122(1):280–290. doi: 10.1172/JCI58227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, et al. Dilated cardiomyopathy with increased SR Ca2+ loading preceded by a hypercontractile state and diastolic failure in the α(1C)TG mouse. PLoS ONE. 2009;4(1):e4133. doi: 10.1371/journal.pone.0004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.