

Abstract
Bone-derived fibroblast growth factor-23 (FGF23) plays an important role in systemic phosphate turnover. Increased FGF23 activity results in hypophosphatemic, while reduced activity is linked to hyperphosphatemic disorders. FGF23, together with klotho as co-factor, can activate FGF-receptors in its target tissues to exert its functions. However, molecular regulation of FGF23 synthesis is not clearly defined, and recent studies have found that PTH can activate the nuclear receptor-associated protein-1 (Nurr1) to induce FGF23 transcription in bone cells.
Keywords: Kidney, Bone, transcription, regulation
Commentary
Optimal phosphate balance is important for many physiological functions from cell signaling to energy metabolism to skeletal mineralization. Inadequate phosphate balance disrupts a multitude of physiological processes, and can cause or exacerbate age-associated disorders, cardiovascular calcification and bone mineralization defects 1. Physiologic phosphate balance is maintained by interactions among the intestine, kidney, parathyroid gland and bone. Phosphate absorption takes place mostly in the small intestine, and the sodium-dependent phosphate (NaPi) co-transporter, NaPi-2b, facilitates such absorption. Of relevance, the intestinal NaPi-2b activity is influenced by 1,25-dihydroxyvitamin D, which can increase the expression of intestinal NaPi-2b protein to augment intestinal phosphate uptake by the enterocytes. Likewise, renal phosphate reabsorption is mostly accomplished by the NaPi-2a and NaPi-2c co-transporters. Parathyroid hormone (PTH) increases urinary phosphate excretion by reducing NaPi-dependent phosphate uptake in the proximal tubular epithelial cells. Recent studies have shown that bone-derived fibroblast growth factor 23 (FGF23) and kidney-derived klotho can also directly suppress sodium phosphate co-transporter activities 2. In addition to intestine and kidney, bone also plays a major role in maintaining phosphate balance. When serum phosphate levels are low, the bone releases additional phosphate to maintain the homeostatic balance by increasing resorption, a process that is mostly influenced by the activity of PTH and 1,25-dihydroxyvitamin D. PTH alters receptor activator of nuclear factor kappa-B ligand-osteoprotegerin (Rankl/Opg) balance, by acting on the stromal cells to stimulate Rankl expression, and also by reducing the expression of Opg and vitamin D. On the other hand, PTH stimulates osteoclast differentiation and activity, resulting in increased bone resorption. Furthermore, PTH and 1,25-dihydroxyvitamin D can induce skeletal FGF23 production to regulate serum phosphate levels and thereby can influence bone resorption.
FGF23 is a ∼30 kDa protein that is proteolytically processed to smaller N-terminal (∼18 kDa) and C-terminal tail (∼12 kDa) fragments. Structural analysis of FGF23 protein found a FGF receptor (FGFR)-binding domain at the N-terminal and a potential klotho-interacting site at the C-terminal tail 3. Recent studies have shown that the C-terminal tail determines the functionality of FGF23 protein. For instance, when the C-terminal tail of the FGF2 protein was replaced with the C-terminal tail of FGF23, the chimeric protein containing the N-terminal of FGF2 and C-terminal tail of FGF23 could act as a phosphatonin, and could also reduce renal 1α(OH) synthesis 4. It is worth mentioning that the presence of the C-terminal tail of FGF23 in the chimeric protein paved the way for the klotho interaction. Recently, family with sequence similarity 20, member C (Fam20C) has shown to phosphorylate FGF23 on a Ser-x-Glu motif, and such phosphorylation promotes FGF23 proteolysis by furin through blocking O-glycosylation by polypeptide N-acetylgalactosaminyltransferase 3 (GalNAc-T3). This observation suggests that interplay between phosphorylation and O-glycosylation of FGF23 may be a critical posttranslational mechanism by which the activity of secreted FGF23 protein is determined 5.
Once secreted as a bioactive protein, FGF23, in presence of klotho, can induce downstream signaling molecules, as demonstrated by the activation of early growth response element-1 (Egr-1) and the phosphorylation of FGF receptor substrate-2a, extracellular signal-regulated kinase (ERK), p38, Jun N-terminal kinase (JNK), and AKT. Of relevance, these signaling phosphoproteins were detected only when cells were challenged with both FGF23 and klotho, and not in cells treated with FGF23 without klotho. In accord with these in vitro observations, in vivo studies have shown that bioactive FGF23 protein could significantly reduce serum phosphate level in wild-type and Fgf23 knockout mice, but failed to exert such phosphate lowering effects in Fgf23/klotho double knockout mice, again suggesting that without klotho, FGF23 loses its phosphate regulating abilities. Moreover, the FGF23-induced hypophosphatemic phenotype of hyp mutant mice was reversed to hyperphosphatemia in the hyp/klotho double mutant mice, despite significantly higher serum FGF23 levels in double mutants 6. In a similar line of observation, an inactivating mutation in the human Klotho gene resulted in severe hyperphosphatemia in a tumoral calcinosis patient, despite high serum FGF23 levels 7. Summarizing these above-mentioned observations, an indispensable role of klotho in FGF23-mediated urinary phosphate excretion is obvious. One of the possible mechanisms of FGF23-induced urinary phosphate excretion is that it suppresses NaPi-2a and NaPi-2c co-transporters, either directly or through influencing PTH activity.
PTH, an 84 amino acid protein, is produced in response to low levels of serum calcium and secreted PTH acts on the bone and kidney to increase serum calcium level. Low serum calcium levels reduce calcium-sensor receptor (CaR) signaling and allow active PTH to be secreted, which then binds to the PTH receptor 1, a seven transmembrane G-protein coupled receptor, to activate the PKA, PKC, and MAPK pathways in kidney and bone. In addition to serum calcium, vitamin D can also suppress PTH expression and parathyroid hyperplasia. It is believed that FGF23 and PTH mutually regulate each other in a negative feedback loop, where PTH stimulates FGF23 production and FGF23 in turn suppresses PTH synthesis. When PTH was genetically ablated from Fgf23 knockout mice, serum calcium levels were normalized in double mutant (Fgf23-/-/Pth-/-) mice, despite high serum 1,25-dihydroxyvitamin D and high serum phosphate levels, suggesting that some of the biochemical changes documented in Fgf23-/- mice are mediated by PTH 8. It is a well-known fact that continuous administration of PTH for a prolonged period can reduce bone mass. However, when Fgf23-/- mice were challenged with constant PTH infusion through osmotic minipumps for 3 weeks, PTH-induced bone loss was more severe, suggesting that FGF23 might exert a protective effect against the long-term catabolic effects of PTH on bone 8.
In a mouse model of hyperparathyroidism, FGF23 levels positively correlated with those of PTH, but inversely with serum phosphate levels 9. Presence of both klotho and FGF receptors in the parathyroid glands raised the possibility that parathyroid gland might be a target organ for FGF23 activities. Using bovine parathyroid cells, FGF23 was shown to suppress PTH production 10. On the other hand, other studies have claimed that the activation of the PTH receptor in bone via the PKA signaling pathway suppresses the Wnt inhibitor sclerostin, thereby allowing Wnt signaling to increase FGF23 synthesis. It is, however, worth mentioning that reducing Wnt signaling in vivo, by genetically inactivating its co-receptor, low-density lipoprotein receptor-related protein 6 (Lrp6), did not affect FGF23-induced hypophosphatemia in Hyp mice, as shown in Hyp/Lrp6 double mutant mice. Furthermore, injection of bioactive FGF23 protein into Lrp6 mutant mice reduced serum phosphate levels to a similar degree as FGF23 injection into wild-type mice, providing a genetic and pharmacological evidence for a WNT-independent function of FGF23 in the regulation of phosphate homeostasis 11.
Meir et al. 12 (this issue) claimed that PTH, by activating nuclear orphan receptor (Nurr1), can increase the transcription of FGF23 in bone cells. The transcription factor Nurr1 has been shown to be important for neuronal development. Structural analysis has found that Nurr1 protein is lacking a ligand-binding cavity, and therefore may act as a ligand-independent transcription factor. In the FGF23 promoter region, the presence of Nurr1 response elements raises the possibility of its role in FGF23 synthesis. In a cell-based system, through over-expression and knock down of Nurr1, an association between PTH and FGF23 is suggested 12. Moreover, in a rat model of chronic kidney disease (CKD), increased Nurr1 mRNA and protein levels were associated with increased FGF23 mRNA expression, Calcimimetic treatment of these CKD animals reduced PTH and FGF23 levels, along with decreased calvarial Nurr1 mRNA and protein expression 12. Despite the presence of Nurr1 responsive elements in FGF23 promoter regions, the functionality of Nurr1 responsive elements in FGF23 synthesis is not yet defined, and without mutagenesis studies, whether increased expression of Nurr1 and FGF23 is a mutual regulation or merely an epiphenomenon, could not be established. Moreover, to provide in vivo direct evidence, further studies will be needed to show that inactivating PTH signaling, by targeting its receptors, can block PTH induced Nurr1 and FGF23 expression in long bones. It is also worth mentioning that PTH induced FGF23 synthesis is a cell-line specific phenomenon. For instance, while PTH can induce FGF23 in UMR106 cell lines, no such response of PTH on FGF23 is noted in ROS16/2.8 cells.
Despite a better understanding of FGF23 biology in systemic regulation of phosphate turnover 1, factors inducing its skeletal expression are not yet fully documented. 1,25-dihydroxyvitamin D, phosphate, calcium, iron, leptin, acidosis, secreted klotho and PTH are the factors currently known to induce FGF23 production (Fig. 1). It is a well-accepted fact that PTH can induce the synthesis of 1,25-dihydroxyvitamin D in the kidney, and that, in turn, 1,25-dihydroxyvitamin D can inhibit PTH secretion by the parathyroid glands. Recent studies have claimed that 1,25-dihydroxyvitamin D can stimulate FGF23 production in bone and that FGF23 can suppress 1,25-dihydroxyvitamin D production and PTH secretion. The ability of PTH to directly stimulate FGF23 expression forms an endocrine regulatory feedback loop to control mineral ion metabolism. Studies, as the one highlighted here 12, will help us to understand molecular regulation of FGF23 synthesis and identify its yet to be documented functions.
Figure 1.
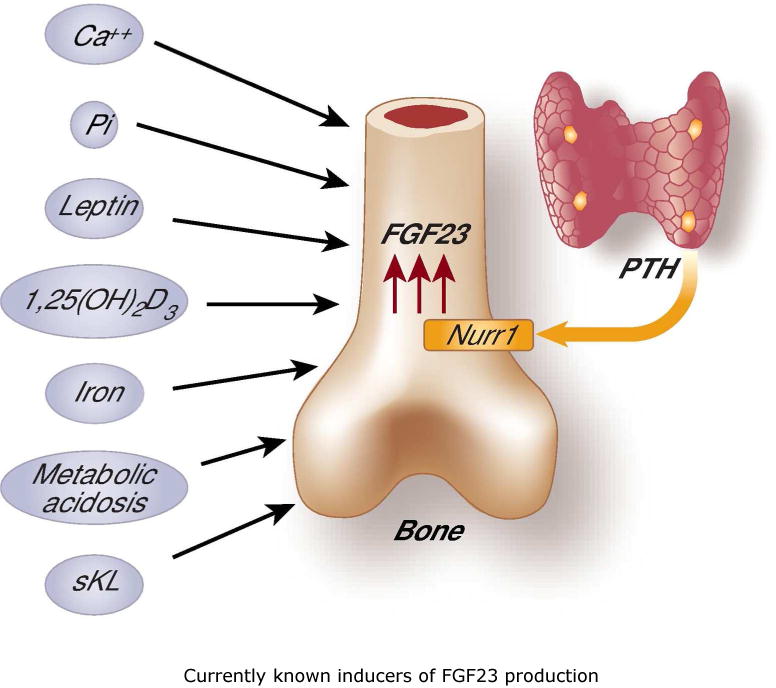
Schematic representation of currently known inducers of FGF23 production. (left) calcium (Ca++), phosphate (P), active vitamin D [1,25(OH)2D3], leptin, secreted Klotho (sKL), iron, and metabolic acidosis; (right) proposed direct stimulation of FGF23 by PTH via the Nurr1 transcription factor.
Acknowledgments
Dr. Lanske is funded by NIH- DK097105.
Footnotes
Disclosures: None
References
- 1.Razzaque MS. The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5:611–619. doi: 10.1038/nrendo.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24:3438–3450. doi: 10.1096/fj.10-154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goetz R, Nakada Y, Hu MC, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A. 2010;107:407–412. doi: 10.1073/pnas.0902006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goetz R, Ohnishi M, Kir S, et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J Biol Chem. 2012;287:29134–29146. doi: 10.1074/jbc.M112.342980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tagliabracci VS, Engel JL, Wiley SE, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014;111:5520–5525. doi: 10.1073/pnas.1402218111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakatani T, Ohnishi M, Razzaque MS. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009;23:3702–3711. doi: 10.1096/fj.08-123992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Q, Sitara D, Sato T, et al. PTH ablation ameliorates the anomalies of Fgf23-deficient mice by suppressing the elevated vitamin D and calcium levels. Endocrinology. 2011;152:4053–4061. doi: 10.1210/en.2011-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawata T, Imanishi Y, Kobayashi K, et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–2688. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 10.Krajisnik T, Bjorklund P, Marsell R, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125–131. doi: 10.1677/JOE-07-0267. [DOI] [PubMed] [Google Scholar]
- 11.Uchihashi K, Nakatani T, Goetz R, et al. FGF23-induced hypophosphatemia persists in Hyp mice deficient in the WNT coreceptor Lrp6. Contrib Nephrol. 2013;180:124–137. doi: 10.1159/000346792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meir T, Durlacher K, Amir G, et al. PTH activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. kidney Int. 2014 doi: 10.1038/ki.2014.215. [DOI] [PubMed] [Google Scholar]