

Abstract
Background
Metarhizium anisopliae is an entomopathogenic fungus used in the biological control of some agricultural insect pests, and efforts are underway to use this fungus in the control of insect-borne human diseases. A large repertoire of proteins must be secreted by M. anisopliae to cope with the various available nutrients as this fungus switches through different lifestyles, i.e., from a saprophytic, to an infectious, to a plant endophytic stage. To further evaluate the predicted secretome of M. anisopliae, we employed genomic and transcriptomic analyses, coupled with phylogenomic analysis, focusing on the identification and characterization of secreted proteins.
Results
We determined the M. anisopliae E6 genome sequence and compared this sequence to other entomopathogenic fungi genomes. A robust pipeline was generated to evaluate the predicted secretomes of M. anisopliae and 15 other filamentous fungi, leading to the identification of a core of secreted proteins. Transcriptomic analysis using the tick Rhipicephalus microplus cuticle as an infection model during two periods of infection (48 and 144 h) allowed the identification of several differentially expressed genes. This analysis concluded that a large proportion of the predicted secretome coding genes contained altered transcript levels in the conditions analyzed in this study. In addition, some specific secreted proteins from Metarhizium have an evolutionary history similar to orthologs found in Beauveria/Cordyceps. This similarity suggests that a set of secreted proteins has evolved to participate in entomopathogenicity.
Conclusions
The data presented represents an important step to the characterization of the role of secreted proteins in the virulence and pathogenicity of M. anisopliae.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-822) contains supplementary material, which is available to authorized users.
Keywords: Genome sequence, Entomopathogenic fungi, Secretome, Phylogenomics
Background
It is estimated that over 600,000 species of fungi exist, and it is assumed that these species can be found in almost all habitats on Earth. However, only a few of these species have been described [1]. Most fungal species have developed saprophytic interactions in soil and in water or in association with mycorrhizal plants, as either arbuscular mycorrhizae or ectomycorrhizae. Moreover, fungal species are known to cause disease in several hosts, including mammals, arthropods, and plants [2]. To adapt to such a large variety of habitats, fungi have developed a prolific capability to export proteins to the extracellular space as an important mechanism to acquire nutrients [3]. Therefore, secretomes, which are defined as the global set of proteins produced by a cell and exported to the extracellular space in a determined time and condition, represent an important target for understanding the mechanisms of fungal adaptation. For instance, both saprophytic and pathogenic fungi must quickly adapt to variations in carbon and nitrogen availability. Because fungi generally obtain nutrients from the digestion of extracellular polymers, such as cellulose and chitin, fungi must produce copious amounts of extracellular enzymes to allow for the efficient hydrolysis of biopolymers during the infection process or from their natural environment [3].
A diverse group of fungi is associated with arthropods, the largest class of eukaryotic species on Earth, and plays a role in controlling their populations, in particular of insects [4]. The most well known insect-associated fungi are entomopathogens, which are necrotrophic fungi that actively penetrate the host exoskeleton and proliferate in the hemocoel until all internal tissues have been degraded. The infection process of entomopathogenic fungi depends on the secretion of a plethora of enzymes and toxins, which serve to penetrate and kill the host, as well as to provide nutrients through the action of biopolymer-degrading enzymes [5–10]. The best-characterized example of a relation between an entomopathogenic fungus and its hosts is the genus Metarhizium. Several lines of evidence suggest that the infection cycle of Metarhizium can be schematically divided into the following steps: (i) conidia adherence to the host cuticle through hydrophobic interactions and thin mucilaginous material; (ii) conidia germination and development; (iii) germ-tube differentiation into appressoria; (iv) cuticle penetration; (v) hyphae differentiation into blastospores/hyphal bodies in the hemolymph; (vi) host colonization; (vii) extrusion to the host cadaver surface; and (viii) conidiophore formation and conidia production [11]. The participation of many proteins, including secreted proteins, has been described for the infection process (reviewed in [12]). More recently, the existence of alternative mechanisms has been suggested during the control of Aedes aegypti, the mosquito vector of dengue and yellow fever [13]. Because different Metarhizium species can infect and kill more than 200 species from 50 insect and arthropod families [14], some isolates have been widely used as bioagents to control a wide variety of pests [15]. Indeed, almost 50 different formulations employing Metarhizium are commercially available [16].
In fact, the Metarhizium species generally regarded as M. anisopliae is composed of nine different species, which can be most frequently isolated from either soil or insects [17]. The genomes of the M. anisopliae ARSEF 23, which are currently classified as M. robertsii[17], a broad-spectrum insect pathogen, and of the acridid-specific M. acridum CQMa 102, were characterized [18]. The sequence analysis of these genomes revealed that they are highly syntenic and possess many genes that allow for the different lifestyles of Metarhizium spp. In addition, a phylogenomic analysis showed that M. robertsii and M. acridum are more related to plant endophytes and pathogens than to animal pathogens. Moreover, this analysis showed that the sequenced genome was from M. robertsii, which had been misclassified as M. anisopliae[18]. Further information concerning the evolution of entomopathogenic fungi originates from the characterization of the entomopathogen Beauveria bassiana genome, which contributed to the identification of a common set of gene families potentially associated with fungal entomopathogenicity [19].
The large collection of fungal genomes sequenced, including entomopathogens, plant pathogens, mycopathogens, and mammal pathogens, allows the shared and exclusive genes present in the predicted secretomes of fungal species to be identified. To analyze the importance of secreted proteins in the virulence of fungal pathogens, we sequenced the genome of M. anisopliae strain E6 and performed a comparative study of this genome, emphasizing the predicted secretome among distinct fungal species.
Results and discussion
General features and comparative analyses of the M. anisopliaeE6 genome
The genome sequence of Metarhizium anisopliae E6 was obtained using a 454-based pyrosequencing approach, with 19-fold genome coverage. The assembly performed using Newbler 2.8 and WGS-CA 7.0 software resulted in 191 and 516 scaffolds with 38,326,054 and 38,454,426 bp, respectively (Table 1). The longest scaffold identified using Newbler software was 1,756,362 bp, whereas the longest scaffold identified using WGS-CA software was 638,367 bp. Using Newbler and WGS-CA software, the N50 scaffold size was calculated as 622.80 kb and as 167.52 kb and the N50 contig size was measured as 157.96 kb and as 125.69 kb, respectively. Both assemblies were combined using the assembly tool Minimus2, and the total M. anisopliae genome size was 38.5 Mb (Table 1). In comparison to other entomopathogenic fungal genomes, M. anisopliae E6 has a genome size similar to M. robertsii ARSEF 23 and to M. acridum CQMa 102 and is larger than the Cordyceps militaris CM01 and B. bassiana ARSEF 2860 genomes (Table 2). The GC content of M. anisopliae E6 is 51%, which is similar to the values reported for M. robertsii, M. acridum, C. militaris and B. bassiana, which are fungi traditionally compared with M. anisopliae. The M. anisopliae E6 gene density is 280 genes per Mbp, which is extremely similar to the values found for the fungi previously cited, with an average of 2.7 exons per gene. The number of transfer RNAs (tRNAs) identified in M. anisopliae E6 was 181, which is higher than those tRNAs found in other fungi (Table 2). This finding may reflect the use of different methodologies for analyzing other fungal genomes [18, 19], which were assessed using a combination of different predictors, including tRNAScan-SE [20] and Infernal [21].
Table 1.
General information concerning the M. anisopliae E6 genome assembly
| Software | Newbler 2.8 | WGS-CA 7.0 | Minimus2/Consed |
|---|---|---|---|
| Scaffolds | 191 | 516 | - |
| Total scaffold size (bp) | 38,326,054 | 38,454,426 | - |
| Contigs | 677 | 688 | 376* |
| Total contig size (bp) | 38,369,953 | 38,434,596 | 38,478,534 |
| Scaffold N50 | 622,803 | 167,523 | - |
| Contig N50 | 157,963 | 125,686 | 319,537 |
| Longest scaffold | 1,756,362 | 638,367 | - |
| Longest contig | 735,175 | 638,367 | 1,044,648 |
| Coverage | 19 X | 19.6 X | - |
| Singlets | 3,552 | 39,616 | - |
*A total of 366 contigs larger than 200 base pairs were deposited in NCBI with the accession number JNNZ00000000.
Table 2.
Comparison of the primary genome features between M. anisopliae and other entomopathogenic fungi
| Feature | M. anisopliae | M. robertsii | M. acridum | C. militaris | B. bassiana |
|---|---|---|---|---|---|
| Strain | E6 | ARSEF23 | CQMa102 | CM01 | ARSEF2860 |
| Host-range | Broad | Broad | Locust | Broad | Broad |
| Sequencing plataform | 454 | Solexa | Solexa | 454/Illumina | 454/Illumina |
| Size (Mbp) | 38.5 | 39.0 | 38.1 | 32.2 | 33.7 |
| Coverage | 19 X | 100 X | 107 X | 147 X | 76.7 X |
| GC content (%) | 51.0 | 51.5 | 50.0 | 51.4 | 51.5 |
| Protein-coding genes | 10,817 | 10,582 | 9,849 | 9,684 | 10,366 |
| Gene density* | 280 | 271 | 259 | 301 | 308 |
| Exons/gene | 2.6 | 2.8 | 2.7 | 3.0 | 2.7 |
| tRNA | 181 | 141 | 122 | 136 | 113 |
The genome was predicted to have 10,817 protein-coding genes, a value slightly higher than those values found for M. robertsii (10,582), M. acridum (9,849) and C. militaris (9,684) (Table 2). There are 3,820 hypothetical proteins in M. anisopliae E6, which account for 35.3% of its genome. Based on comparisons with previously cited fungi, M. anisopliae E6 has 690 (6.4%) exclusive protein coding genes (Figure 1A). Both host-generalist fungi M. anisopliae E6 and M. robertsii show a similar number of exclusive (species-specific) genes. However, the specialist fungus M. acridum has a significantly higher number of exclusive genes (875), and C. militaris, the teleomorphic species, has the highest number (2,245) of exclusive genes (Figure 1A) among these entomopathogens. Considering the four fungi, 62.64% (26,224/41,859) of all proteins are shared. As expected, M. anisopliae shares more proteins exclusively with M. robertsii (1,878) than with M. acridum (352) or with C. militaris (141). By excluding all paralogous genes present in each fungal genome, we could analyze the distribution of orthologous genes among the four fungi that presented 1:1 ortholog genes, representing 86% (22,488/26,224) of the shared genes (Figure 1B). BLAST and OrthoMCL analyses were conducted to evaluate the presence of ortholog sequences in Metarhizium spp. and showed that 94.2% of the M. anisopliae E6 coding sequences display matches in M. robertsii and 90.8% display matches in M. acridum. A broader comparison with the sequences of 16 additional fungal genomes revealed that 592 sequences are unique to M. anisopliae E6. Thus, increasing the number of fungal genomes from 4 to 16 from the different lifestyles does not significantly diminish the number of exclusive proteins, which are largely classified as hypothetical (97.5%), suggesting that these proteins may develop specific roles in M. anisopliae that differ from counterparts M. acridum and M. robertsii.
Figure 1.

Comparative genomics analyses of entomopathogenic fungi. Venn diagram comparing the complete proteomes (A) or the ortholog sequences alone (B). M. anisopliae (M. anisopliae E6), M. robertsii (M. anisopliae ARSEF 23), M. acridum (M. acridum CQMa 102) and C. militaris (C. militaris CM01) were used for comparisons. The diagram in B was constructed using best bidirectional BLAST hits, >60% positive amino acid alignments, >60% subject coverage and a cut-off e-value of <1e-05, without paralogs.
Comparative analysis of genes involved in pathogen-host interactions
To evaluate the presence of genes known and suggested to be involved in pathogenic and virulence pathways, the Pathogen-Host Interaction (PHI) database was used to search for orthologous proteins in the M. anisopliae E6 genome. Comparisons with the predicted proteome of M. anisopliae E6 and the PHI-database were conducted employing Blastp analysis and the results were filtered according a stringent criteria (coverage ≥ 50% and e-value ≤ 10-5). Of the 10,817 protein-coding genes, 2,396 (22.1%) exhibited matches with proteins from the PHI database. Similar percentages of M. robertsii and M. acridum proteins also exhibited matches with the PHI databases (21.2% and 21.8%, respectively). However, B. bassiana had a lower percentage of PHI database matches (16.9%). Considering only proteins that show over 70% identity with M. anisopliae orthologs, 50 of 94 matches exhibited a “loss of pathogenicity or reduced virulence” as phenotype characteristics in mutant strains. Chitinase Chi2 (MANI02801, ChimaB1 in M. anisopliae E6) is among the classes of pathogenic proteins represented in M. anisopliae for which mutants exhibited reduced virulence against the cotton bug Dysdercus peruvianus[23]. The M. anisopliae protein MANI18860 (ChimaD1 in M. anisopliae E6) was also found to be a putative virulence factor because this protein is homologous to the chitinase BbCHIT1 from B. bassiana (AAN41259). The overexpression of this gene in B. bassiana led to enhanced virulence against the aphid Myzus persicae[24]. Additionally, three chitin synthase coding genes could be identified (loci MANI15599, MANI17339, and MANI112231). M. anisopliae has an orthologous protein (MANI23390) to M. robertsii histidine kinase 1 (mhk1), whose null mutants showed reduced virulence to Tenebrio molitor larvae [25]. Superoxide dismutase (SOD), mitogen-activated protein kinases (MAP kinases), urease, Cytochrome P450 monooxygenase, and others were also proteins from M. anisopliae with matches in the PHI database (Additional file 1). Therefore, such proteins represent putative virulence determinants and should be exploited in future loss of function mutant experiments.
Comparative secretome analyses
We predicted the refined secretome of M. anisopliae E6, and from other 15 fungal genomes available, selected from different lifestyles, such as plant pathogens (Fusarium graminearum, Fusarium oxysporum, Magnaporthe oryzae and Nectria haematococca), human pathogens (Aspergillus fumigatus and Aspergillus niger), entomopathogens (Beauveria bassiana, C. militaris, M. robertsii and M. acridum), and mycopathogens (Trichoderma atroviride and Trichoderma virens), as well as saprophytes (Aspergillus nidulans, Neurospora crassa and Trichoderma reesei). The goal was to compare the secretome functionalities and to search for evolutionary traits. To perform this task, we combined bioinformatic tools (Additional file 2) based on the approach used for the plant pathogen Fusarium graminearum[26], which has shown high transcriptional and proteomic support. This procedure aimed to detect protein sequences encompassing signal peptides (as detected by SignalP and TargetP tools), a lack of or at most one transmembrane domain (TM) if located within the first 60 amino acids at the N-terminus (as detected by TMHMM), and sequences associated with the extracellular face of the plasma membrane via glycosylphosphatidylinositol (as detected by GPI anchors) after a post-translational modification (PredGPI). Additional cellular localization tools (ProtComp and WoLF PSort) were applied to refine the secretome predictions. Sequences lacking an initial methionine or that were smaller than 20 amino acids were excluded. To ensure that sequences known to permanently reside inside the lumen of the endoplasmic reticulum were not present, we scanned for the PROSITE pattern PS00014 (Endoplasmic reticulum targeting sequence). Our analysis relied on the association of different software modalities to improve our prediction specificity because the utilization of single programs would result in more annotation errors. For example, of the sixteen fungal species analyzed, 2.5% of the proteins predicted to have signal peptides by SignalP were not considered secreted proteins by TargetP, whereas 39.7% of the proteins predicted to have extracellular localization by WoLF PSort were rejected by ProtComp. Recent reports have revealed that the non-classical export of proteins to the extracellular space through vesicles is a conserved mechanism in fungi [27–32]. Although we are aware that the secretory pathway analyzed in this study does not represent the entire repertoire of fungal secreted proteins, notably, the classical mechanism of protein secretion is an important and well-studied route. The M. anisopliae E6 refined secretome represented 3.8% of the complete proteome. Similar proportions were found for all species, ranging from 3.1% (M. acridum and C. militaris) to 4.8% (M. oryzae) (Additional file 3). This proportion was much higher in the previously predicted M. robertsii secretome (17.6%); however, this number was based solely on the presence of signal peptides [18], suggesting that its secretory repertoire may have been overestimated. In contrast, our method predicted that the M. robertsii secretome accounts for 3.7% of the complete proteome (Additional file 3).
To evaluate the functional diversity of the secretomes studied, we employed classification based on the KEGG Orthology (KO) database and association of activities with different fungal lifestyles. The amount of predicted secreted sequences associated with functional groups varied from 33.1% (M. oryzae) to 62.6% (A. niger), indicating a considerable number of proteins with unknown functions for all fungi genomes. For example, 72% of M. anisopliae E6 sequences without KO functions were hypothetical proteins. Among the entomopathogens, we found that M. anisopliae E6 presented a higher number of glycoside hydrolase (GH) sequences, which contain both canonical signatures to be secreted and distributed to cellular compartments. GHs (EC 3.2.1.-) are ubiquitous enzymes found in all domains of life. These proteins can be both intra- and extracellular and play fundamental roles in nutrition by degrading a variety of polymeric carbohydrates [33]. Of note, when compared with fungi with different lifestyles, entomopathogens have relatively fewer GHs, secreted or non-secreted. In fungi, one important class of GH enzymes is the chitinases (EC 3.2.1.14). These proteins are classified into the GH 18 family and are assumed to be involved in insect cuticle degradation and fungi cell wall digestion during morphological changes [9, 34]. As expected, our analysis revealed that entomopathogens and mycoparasites displayed a larger set of secreted chitinases. This finding is consistent with a previous report that characterized the presence of distinct GH members in fungal genomes [35]. Although chitin is absent in mammals and in plants, human pathogens and phytopathogens present a modest set of secreted chitinases that may have antifungal roles [36]. In addition, a consistent number of non-secreted chitinases was found most likely because these chitinases have other roles in the fungal life cycle or are able to reach the extracellular space through different secretion mechanisms or by vesicle transport [28].
Comparative profiling of secretome by classification of Gene Ontology Terms was also applied in order to obtain functionalities predominant in entomopathogen secretomes in relationship to other fungal lifestyles. The annotations related to proteolysis (GO 0006508) and related (peptidase activity – GO 0008233; serine-type peptidase activity – GO 0008236) could be found as overrepresented in entomopathogens when compared to human and plant pathogens, mycopathogens as well to saprophytes (Additional file 4). To establish successful infection, entomopathogens secrete a variety of hydrolytic enzymes, such as proteases (EC 3.4.-.-). A higher number of secreted proteases in this lifestyle group was found; in M. anisopliae E6 and in M. robertsii, these secreted proteases were originated from gene family expansion of serine (EC 3.4.21.-) and aspartic endopeptidases (EC 3.4.23.-). Although it has more genes coding for trypsin (27), the M. robertsii genome codes for slightly fewer secreted enzymes of this class (9) than the M. anisopliae E6 genome (10 of 17). Considering the two types of hydrolyzing peptide bonds, the exopeptidase (EC 3.4.11.-/EC 3.4.16.-/EC 3.4.17.-) and the endopeptidase (EC 3.4.21.-/EC 3.4.23.-/EC 3.4.24.-) families are widely distributed in all analyzed secretomes.
Comparing the predicted secretomes of the three Metarhizium species herein analyzed, the acridid-specific M. acridum and the broad host-range M. anisopliae E6 and M. roberstii, fewer genes in almost all enzyme categories analyzed were found in M. acridum (Additional file 3). This finding is consistent with its narrow range of susceptible hosts. In agreement with this assumption, comparative genome hybridization assays [37] and a comparative genome analysis [18] revealed the absence of several genes in M. acridum compared with M. robertsii. When the secretomes of entomopathogenic fungi were compared with those secretomes from human fungal pathogens, A. niger was found to have the largest repertoire of proteins with a predicted PFAM domain.
We reasoned that fungal secretomes could share a common evolutionary trait. To evaluate this hypothesis, we performed a comparison of the predicted secretome employing the 16 genomes previously analyzed. This hypothesis was shown to be inconsistent. A reasonable explanation is obtained from comparisons of homologs present in indifferent secretomes for all 405 M. anisopliae E6 secreted sequences (Figure 2A and B), as well the analysis of gene duplication (≥2 copies) numbers (Figure 2C) for each predicted secretome. The number of homologs is reduced from 384 for M. robertsii to 88 for A. niger, and the duplication rates varied from 5.7% for T. reesei to 24.2% for A. niger. The closest secretomes to M. anisopliae E6 were the entomopathogens Metarhizium spp., B. bassiana and C. militaris, whereas the human pathogens Aspergillus spp. was the least similar. The plant pathogen Nectria haematococca presented slightly more homologs (156) than the saprophyte T. reesei (149), whereas the other Trichoderma spp., T. virens and T. atroviride, presented 173 and 169 homologs, respectively. These latter numbers are consistent with the reduced repertoire of T. reesei secreted protein families [38], including cellulases, hemicellulases and polysaccharide degradation enzymes.
Figure 2.

Conservation of secreted proteins in fungi. A. Comparison of the M. anisopliae E6 secretome and its homologs in fifteen analyzed fungi genomes. B. Prediction of signal peptide in M. anisopliae E6 homologs. C. Comparison of copy numbers of the M. anisopliae E6 secretome homologs D. Signal peptide presence in putative alternative downstream translation initiation sites for methionine.
The inspection of predicted secreted proteins among homolog sequences reveals that any comparative analysis based on simple homology inferences [26] should be conducted with caution. We observed that the speciation process led to differentiation in the orthologous sequences because some direct homologs between two different species differ in the presence of canonical sequences that classify a protein as secreted (Figure 2B, dark gray and black bars). N. haematococca showed the smallest percentage of secreted homologs (49.4%), whereas even the most taxonomically related M. robertsii had a much larger percentage of secreted homologs (75.8%). Considering only secreted homologs (Figure 2A, light gray bars), the mycoparasite T. atroviride exhibits more homologs (112) than the entomopathogens B. bassiana (106) and C. militaris (100). A considerable number of homologs, ranging from 8.1% to 29.5% for F. oxysporum and for T. virens, respectively, had no detectable signal peptide (Figure 2B, black bars). This finding could represent differences between the N-terminal sequences or alternative start codon predictions [39, 40]. Additionally, the amplitude between the total predicted secretome (Figure 2A), the number of secreted proteins with coding-genes presented in copies (Figure 2C), and the total number of homologs reveals an important set of species-specific secreted proteins for each proteome compared with M. anisopliae E6.
The presence of alternative in-frame translation initiation sites (TIS) is a common feature that has been experimentally verified in a wide range of organisms [41–44] and that may directly influence the cellular localization of proteins [44]. Alternative TIS can occur at distances of hundreds of base pairs from the primary start codon site and has been recently predicted to be a phenomenon that occurs in approximately one-tenth of all Saccharomyces cerevisiae proteins [42]. In an attempt to detect signal peptides in a downstream methionine, which could be an alternative TIS, the M. anisopliae E6 predicted proteome and its homolog sequences were split, beginning with all methionines present at the second to the last 30 residues to the end of each protein. Subsequences were screened for the presence of signal peptides by both SignalP and TargetP tools and were subsequently divided into two categories: (i) signal peptides already detected at the first methionine (M1) of the primary sequence and (ii) signal peptides not detected (Figure 2D). From the 10,817 M. anisopliae E6 coding sequences analyzed, 9,388 could be split into subsequences. Of these subsequences, 987 had signal peptides detected at M1 (category (i)). For 112 (11.3%) of these 987 proteins, an additional signal peptide could be detected (Figure 2D, dark gray bars), accounting for 136 subsequences. Approximately 57.3% of these methionines were located within 20 residues of M1, indicating that M1 may be the same motif detected, although 37.5% were located up to 100 amino acid residues from M1. For 886 (10.5%) of the 8,401 proteins from category ii, a signal peptide motif could be identified in a downstream methionine (Figure 2B, black bars), accounting for 1,254 subsequences. In 88.7% of these sequences, an alternative TIS was present over 100 residues downstream of M1. The same analysis was applied to any proteins classified as secretome-related. Of the 2,524 M. anisopliae E6 secretome homologs, 2,056 could be split into subsequences. Then, these sequences could be grouped into categories (i), with 1,787 sequences, and (ii), with 269 sequences. One hundred and twenty-eight sequences (7.2%) of the group with 1,787 proteins (category (i)) showed an additional motif (alternative TIS), resulting in 134 subsequences (Figure 2B, white bars), of which 70.1% most likely accounted for the same signal within 30 residues of M1. Finally, for only 52 (19.3%) of the 269 proteins (category (ii)), a signal peptide was detected downstream of M1 (Figure 2B, light gray bars), adding 57 subsequences to the alternative TIS, with 35.1% within 50 residues from M1. Together, these results indicate that even if alternative TIS is actually occurring in a downstream methionine for these proteins, the majority of homologs without a detectable signal peptide at M1 may not be secreted, similar to M. anisopliae E6. Again, the possibility of alternative secretion mechanisms cannot be excluded. In fact, these results strongly suggest that such mechanisms exist and may account for the secretion of an important number of proteins.
Among the predicted extracellular proteins of M. anisopliae E6, we could identify 80 glycosylphosphatidylinositol-anchored proteins (GPI-Ps). This post-translational modification has been implicated in protein sorting, trafficking and dynamics in different cells [45]. In yeast, GPI lipids are synthesized in the endoplasmic reticulum, and their addition to target proteins is conducted by a pathway that is composed of 12 steps [46]. The number of GPI-Ps in M. anisopliae E6 is higher than that in other Metarhizium spp. (68 for M. robertsii and 63 for M. acridum) and lower than that in the two other entomopathogens (73 for C. militaris and 76 for B. bassiana). The majority (60%) of the GPI-Ps identified represent conserved hypothetical proteins. However, the GPI-Ps of M. anisopliae E6 showed considerable functional diversity, as revealed by the analysis of conserved domains (Additional file 5). Of the 80 predicted GPI-Ps, 11 proteins could be classified as glycoside hydrolases, whose orthologs were characterized in N. crassa[47], in A nidulans[48], as well in M. robertsii[49].
To identify possible GPI-P orthologs shared by the genome sequences of Metarhizium spp., we conducted a BLAST analysis for M. anisopliae E6, M. robertsii and M. acridum. We found that most of the M. anisopliae E6 GPI-Ps had orthologs in both M. robertsii and M. acridum that also had a GPI anchoring signal. However, in some cases, the GPI-Ps from M. anisopliae had orthologs only in either M. robertsii or M. acridum, or even had exclusive proteins (Additional file 6). In addition, despite the clear presence of orthologs among the three Metarhizium spp., a few orthologous proteins to M. anisopliae E6 GPI-Ps differed only in the presence or absence of the GPI anchoring signal in the Metarhizium spp. counterparts, suggesting a possible difference in their cellular location (Additional file 6). These data suggest that there are differences in their protein cell surface profile, despite their phylogenetic proximity. Indeed, the differences observed in their GPI-P profiles may represent differences in fungal survival in the environment, virulence and host specificity.
Because secretory proteins play a fundamental role in fungi physiology and because their evolution is essential for fungal fitness, gene duplication rates could be significantly higher within secretome genes when compared with the remnant proteome. For the fungal pathogen F. graminearum, genes coding for secreted proteins have been preferentially found in chromosomal regions with higher recombination frequencies [26]. Additionally, gene duplication is an important source of new biological functions because mutations in one of the copies can affect protein structure without being deleterious, whereas the other copy can retain functionality [50, 51]. To evaluate the prediction that secretome coding genes are more susceptible to duplication than the proteome coding genes as a whole, a proportion test was conducted. This analysis supported this hypothesis for the two human pathogens A. fumigatus (p-value < 0.001) and A. niger (p-value < 0.001), for the plant pathogen M. oryzae (p-value < 0.001), for the three saprophytes A. nidulans (p-value < 0.001), N. crassa (p-value = 0.002) and T. reesei (p-value = 0.046) and for the two mycoparasites T. atroviride (p-value < 0.001) and T. virens (p-value = 0.002) (Additional file 7). Conversely, for N. haematococca (p-value = 0.024), this proportion was inverted, such that secreted protein coding genes had less duplication. This finding was consistent with the percentage found for predicted secreted proteins and with the higher duplication rate in supernumerary chromosomes [52]. M. anisopliae is adapted to a diverse range of niches [18, 53, 54], and our analysis revealed that the proportions of duplications for the secretome coding genes compared with the proteome coding genes as a whole are statistically equal. This finding suggests that for entomopathogenic fungi, in contrast to human and plant pathogens, successful adaptation to different habitats may be more qualitative. This hypothesis argues that the presence or absence of specific genes, in contrast to gene duplications, is required for the adaptation of Metarhizium spp. to different habitats.
Longer (more complex) duplicated proteins are more likely to be retained because these proteins have a higher probability of generating new biological functions, as previously observed for several fungal species, including saprophytes and human pathogens [51]. Considering the mean protein size for all sixteen analyzed fungi species, we found that duplicated genes are larger than single copy genes for both the secreted and non-secreted sequences (Figure 3) and that longer genes have more copies (with the exception of secreted proteins with four or more copies). Additionally, independent of the copy number, secreted proteins are generally smaller than the average size of the rest of the proteome (All p-values < 0.001 on one-tailed Student’s t-test). Therefore, evolutionary mechanisms for the selection of secreted proteins based on their size must have occurred to allow for their translocation into the extracellular space or to allow for those proteins to function properly in that external environment.
Figure 3.
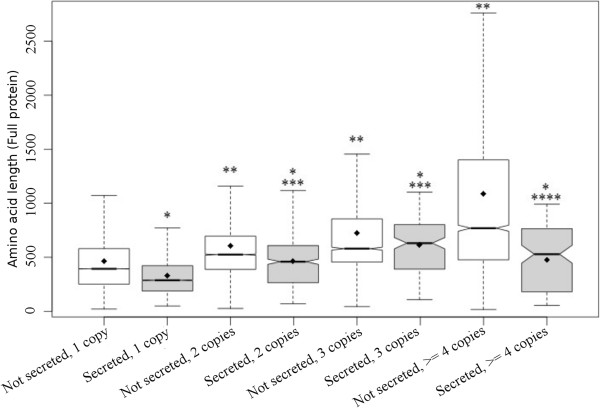
Gene duplication and protein size box-plot for all sequences predicted to be secreted or non-secreted. Mean protein sizes, represented as black dots, were compared with a one-tailed Student’s t-test. *Secreted mean smaller than the non-secreted mean with same copy number, **Non-secreted mean greater than one copy less than the non-secreted mean, ***Secreted mean greater than one copy less than the secreted mean, ****Secreted mean smaller than one copy of the less secreted mean. All p-values < 0.001. Four or more copies were grouped together.
Transcriptome analysis
We conducted the first RNA-Seq experiment to evaluate the differential gene expression profiles of M. anisopliae E6 acting on tick (Rhipicephalus microplus) cuticles to mimic the infection process (as described in the Methods section). These experiments were conducted in three different conditions: (i) a control condition, with spore suspensions cultured in complete medium for 48 hours (C-48 h); (ii) an early cuticle infection condition (I-48 h), with tick cuticles inoculated with spore suspensions and incubated for 48 h in water-agar plates; and (iii) a late cuticle infection condition (I-144 h), with tick cuticles inoculated with spore suspensions and incubated for 144 h in water-agar plates. After mapping reads from all three experimental setups to the genome, approximately 89% of the 10,817 predicted protein coding sequences had at least two uniquely mapped reads (Additional file 8). Notably, the transcriptome results validated our genome annotation since reads that mapped to 415 of the 690 M. anisopliae exclusive genes (60%) could be detected (Additional file 8). The statistical package edgeR [55], which has recently shown better performance considering speed and accuracy than other frequently used tools [56], was used in two pairwise comparisons between the RNA samples, revealing distinct patterns of gene regulation in the tree experimental setups. A principal component analysis (PCA) showed a satisfactory degree of variability among the biological replicates (Additional file 9). The condition mimicking early infection (C-48 h × I-48 h, Figure 4A) triggered a higher amount of up-regulated genes (1,237 genes (FDR ≤ 0.05, log2FC ≥ 1)) compared with down-regulated genes (1,062 genes (FDR ≤ 0.05, log2FC ≥ 1)), with similar proportions of predicted secreted sequences (4.9% and 3.0%, respectively). After an additional 4 days of fungal contact with the host cuticle (I-48 h × I-144 h, Figure 4B), the scenario was inverted, with more genes down-regulated (644) than up-regulated (564), whereas the proportion of the predicted secretome was 8.5% and 1.8%, respectively. Considering secretome genes with more than two mapped reads in any replicate, 55.6%, 59.5% and 47.4% were expressed for setups C-48 h, I-48 h and I-144 h, suggesting that most of these proteins may be constitutively expressed in the condition analyzed in this study.
Figure 4.
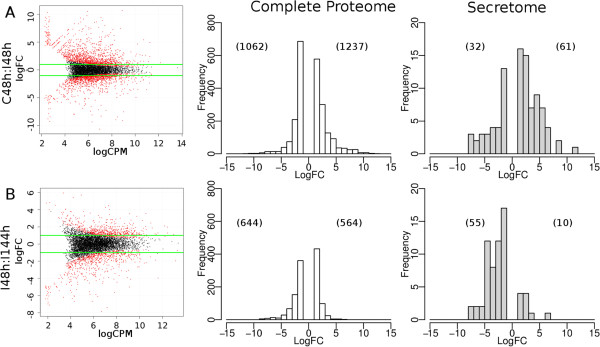
RNA-Seq analysis of differentially expressed genes from M. anisopliae E6. Comparison of the expression profile from C-48 h (Control-48 h) and I-48 h (Infection-48 h) (A) or I-48 h (Infection-48 h) and I-144 h (Infection-144 h) (B) was conducted with the edgeR software package. The overall expression of genes (left panel), the number of differentially expressed genes (middle panel), and the secretome-associated genes (right panel) are shown.
To observe the possible biological roles of differentially expressed genes, superfamily functional categorization [57] was applied (Figure 5). At least 5 different expression profiles could be detected, which encompassed the expression variation of 2,702 genes. This classification is based on the differential expression of functional categories when comparisons between C-48 h and I-48 h (Panels I) and I-48 h and I-144 h (Panels II) were conducted (Figure 5, A to E). The majority of the differentially expressed genes fell into category A (up-regulated in the infection condition compared with the control condition) or E (down-regulated in the infection condition compared with the control condition). Neither the A or E profiles showed differences in their gene expression between the I-48 h and I-144 h conditions; however, these profiles contain differences when C-48 h and I-48 h are compared. In all profiles, genes under constant regulation from the categories “HA: Small molecule binding” (e.g., EC 1.1.1.-/EC 1.3.1.-, binding to NAD or NADP; EC 1.14.13.-, binding to NADH or NADPH; and EC 1.5.3.-, binding to oxygen), “RB: Transferases” (e.g., EC 2.1.1.-, methyltransferases; EC 2.6.1.-, transaminases) and “RC: Other enzymes” were identified. The “RA: Redox” category was also observed in almost all profiles (e.g., EC 1.14.-.-, incorporation or reduction of molecular oxygen). As described for other Metarhizium species [18], the cAMP response element-binding (CREB) protein, which is a major downstream transcription factor in mammals that has not yet been characterized in fungi, was up-regulated (Figure 5A and Additional file 10). A considerable proportion of “G: carbohydrate metabolism and transport” and “OA: protease” categories in profile B is of note because these categories represent gene products involved in the early stages of infection (profile B, up-regulated in I-48 h compared with C-48 h; Figure 5B). Moreover, profile B harbored the highest set of genes (10.5%/37 genes) coding for proteins predicted to be secreted, which is consistent with the expected function of the secretome (Additional file 10). Four subtilisins, which are important enzymes for host cuticle degradation and nutrition [58], were detected in this expression profile (Additional file 10), along with proteins CAS1 and MAS1, whose orthologs in plant pathogens are known to be involved in appressorium formation [59]. Continued contact with the cuticle after 144 hours enhanced the expression of categories “A: RNA binding, metabolism and transport”, “F: Nucleotide metabolism and transport”, “J: Translation” and “RD: Protein interaction” (Figure 5C, profile C, Figure 5C). tRNA synthetases, which are the enzymes responsible for charging the correct amino acid to its cognate tRNA, as well as enzymes acting on ribosome biogenesis and cell cycle progression, were up-regulated [60], indicating that the fungal cells were metabolically active (Additional file 10). As the host cuticle is exhausted, proteases are down-regulated (Figure 5B and D, profiles B and D), and primary metabolism is reduced. Thus, the category “C: energy”, which includes glycolysis and tricarboxylic acid cycle components (e.g., EC 1.2.1.12, Glyceraldehyde 3-phosphate dehydrogenase; EC 5.3.1.1, Triosephosphate isomerase and EC 2.3.3.8, ATP citrate synthase (Additional file 10), was down-regulated, as shown in profile D (Figure 5D).
Figure 5.

Superfamily functional categories of identified expressed genes. The total gene numbers and the percentages of the predicted secretome are for each of the five distinct regulation profiles (A-E). I: edgeR pairwise comparison C-48 h:I-48 h; II: I-48 h:I-144 h. F: Nucleotide m-tr*; G: Carbohydrate m-tr*; HA: Small molecule binding; J: Translation; LA: DNA-binding; O: Protein modification; OA: Proteases; OB: Kinases-phosphatases; P: Ion m-tr*; Q: Secondary metabolism; R: General; RA: Redox; RB: Transferases; RC: Other enzymes. *m-tr: Metabolism and transport. Categories with representativeness < 3% were grouped as Others. The complete classification can be found at: http://supfam.cs.bris.ac.uk/SUPERFAMILY/function.html.
In this study, two trypsin (EC 3.4.21.4) isoforms, which are a class of serine proteases, were found to be up-regulated during early infection, consistent with previous findings [61]. However, one isoform was highly down-regulated (Additional file 10), which could represent differential ambient pH responsiveness [62]. All three isoforms were predicted to be secretome components. Although trypsins are usually more active at an alkaline pH, aspartic endopeptidases (EC 3.4.23.-) are more active at an acidic pH. Five predicted secreted aspartic endopeptidases presented differential expression levels with distinct responses to the environmental conditions. Two were highly down-regulated, whereas the other three were up-regulated during early or late infection (Additional file 10). Additionally, chymotrypsins, which are another class of proteases that were likely originated from horizontal gene transfer from bacteria [63], were also present in two copies in M. anisopliae E6 (MANI06361 and MANI115263, orthologs to M. robertsii MAA_07484). The RNA-Seq analysis revealed that these genes did not present differential transcript levels in the conditions evaluated in this study. Although these enzymes are important for tick cuticle chitin degradation and for fungi cell wall remodeling, no chitinase was up-regulated in the conditions analyzed in this study. One chitin synthase (EC 2.4.1.16) was up-regulated.
The gene that was most down-regulated during the contact period with the host cuticle was a nitrate reductase (EC 1.7.1.3), which is an enzyme that is essential for reducing nitrate to ammonia. This down-regulation can be explained by the physiological condition of nitrogen starvation faced by the fungal cells, which have been shown to be essential for activating distinct virulence functions in plant pathogenic fungi [64]. In accordance, starvation-stress gene A (ssgA), which is a hydrophobin-like protein that leads to decreased fungal sporulation and virulence when deleted [65], was highly up-regulated (Additional file 10).
Infection metabolism induction may produce derivatives of reactive oxygen species (ROS), which are capable of causing damage to diverse cell components, as byproducts. Examples of ROS molecules include hydrogen peroxide (H2O2), superoxide anions (O2-) and nitric oxide (NO). As a defense mechanism against this oxidative stress, superoxide dismutase (EC 1.15.1.1) expression was up-regulated for the conversion of superoxide into O2 and H2O2. Similarly, catalase (EC 1.11.1.6) and catalase-peroxidase (EC 1.11.1.21) expression were up-regulated for H2O2 inactivation, and glutathione S-transferase (EC 2.5.1.18) expression was up-regulated for neutralizing electrophilic substrates. Additionally, a thioredoxin reductase (EC 1.8.1.9) isoform responsible for reducing thioredoxins, which can act as antioxidants by reducing other proteins, was down-regulated. The accumulation of thioredoxin in its oxidized form may also have a protective role because thioredoxin is an effective cysteine oxidant [66] that regulates protein disulfide bond formation in the reducing environment of the cytoplasm, resulting in the downstream regulation of oxidative stress transcriptional factors and chaperones [66]. Peroxiredoxin, which is also an antioxidant enzyme capable of degrading H2O2 (EC 1.11.1.15), was also down-regulated because this enzyme requires the scarce reduced thioredoxin form for its proper function (Additional file 10). For completeness, edgeR statistical analyses are provided (Additional file 11).
Phylogenomic analyses
As the content of the predicted secretomes change according to the fungal species analyzed, homology relations were established among M. anisopliae E6 and a set of fifteen previously cited fungi to evaluate and to compare the evolutionary patterns of the M. anisopliae genome and secretome. In addition a correlation with the transcriptomic data was conducted. All 1:1 ortholog sequences representing different CDSs were concatenated, leading to an aligned file containing 1,947,162 amino acid residues. As expected, the Metarhizium, Trichoderma, Fusarium, and Aspergillus genera formed monophyletic clusters [38, 67–70]. The genera Neurospora and Aspergillus clustered together, with high bootstrap support. Additionally, Metarhizium and Trichoderma formed a statistically well-supported clade in the supermatrix and supertree approaches applied to the shared proteome, which was formed by orthologous proteins only (Figure 6A). When considering the major clades obtained using the maximum likelihood (Figure 6A) and the distance phylogenetic methods (Additional file 12), the trees architectures did not changed significantly.
Figure 6.

Fungal evolutionary history obtained by using a phylogenomic approach. NJ methods were employed to all orthologs identified in the fungal genomes (A). Nearest neighbor interchange method with a neighbor-joining (NJ) tree calculated from average consensus distances were employed to all orthologs identified in the fungal secretomes (B). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates for genomic and 100 for secretomic analysis) is shown next to the branches. The scale bar represents substitutions of amino acids per site.
Some fungal genera, including Trichoderma and Metarhizium, evolved a variety of nutrition strategies. For instance, some Trichoderma species are mycotrophic and, therefore, can grow on living fungi in a process known as mycoparasitism, whereas other species are saprotrophic [71]. Metarhizium species have already been isolated from diverse habitats, primarily from dead arthropod carcasses but also from mycorrhiza and as endophytic [72]. The shared proteome also indicates that the plant pathogens F. graminearum, F. oxysporum and N. haematococca[52, 68, 73] are related to the Metarhizium clade. Wyrebek and coworkers [54] have proposed that Metarhizium species show plant-specific rhizosphere associations within a habitat. Moreover, M. robertsii was also shown to be an endophyte that stimulates root development [53], thus making the direct transfer of nutrients from fungal to plant cells possible [74–76]. Thus, the phylogenomic analysis presented in this study reveals that determining the life-style of M. anisopliae is far more complex than the global set of genes can predict.
The phylogenomic analysis also indicated that M. anisopliae E6 and M. anisopliae ARSEF 23, now identified as M. robertsii[18], share a high number of orthologs. These species formed a statistically well-supported clade, with M. acridum basal to these species, corroborating the results of Bischoff and coworkers [17]. Notably, M. robertsii is morphologically indistinguishable from M. anisopliae. Therefore, phylogenetic analysis is critical for establishing the identity of these fungi.
A phylogenetic analysis was conducted for 212 ortholog-groups representing all proteins that had been identified as putative constituents of the secretome. The evolution of the secretome indicated an important difference when compared with the evolutionary history of these fungi based on their shared proteome. There is a closer evolutionary relation between the Metarhizium clade and that formed by the genera Beauveria and Cordyceps (Figure 6B). Consequently, most proteins secreted by Metarhizium species are similar to those proteins from the Beauveria and Cordyceps genera. These proteins, some of which are important to the infection process, have a pattern of evolution that is extremely similar in fungi with similar hosts.
To identify the proteins responsible for the distinctive evolutionary pattern of the secretome, all individual phylogenies had their topologies compared with the topology of the phylogenomic tree. At least one Metarhizium and one Trichoderma sequence, as well as one Cordyceps or Beauveria sequence, had to be present in a specific secretome ortholog file to be included in the analysis. Finally, 37 secreted proteins were identified as being potentially responsible for the different pattern of evolution of the secretome when compared with the evolution of the organisms represented by the phylogenomic tree. These proteins may explain why fungi with similar hosts cluster together. Thirty four (92%) of those genes are expressed, and 16 (43%) of those genes are differentially expressed during early cuticle infection (I-48 h) and/or during late cuticle infection (I-144 h) (Additional file 13). The biological significance of these findings requires further investigation. Nevertheless, our results show that specific secreted proteins from Metarhizium, Beauveria and Cordyceps species, have an evolutionary history that points for their adaptability to host infection.
Using either proteome or secretome data, M. oryzae and N. crassa form a basal cluster with high bootstrap support. These species belong to the distinct orders Magnaporthales and Sordariales, respectively, and our results corroborate a previous study that noted their evolutionary relation [77]. The Aspergillus genus, which belongs to the Eurotiomycetes order, clusters with the M. oryzae and N. crassa clades and, together, represents the most basal clade in our study. The operational taxonomic units belonging to Hypocreales also form a cluster. These results indicate that the orthologs identified for the proteome and for the secretome have an evolutionary pattern that is consistent with the actual taxonomic classification of fungi.
Conclusions
The M. anisopliae E6 genome and expression profiling analyses provide insights into the molecular mechanisms for adapting to the distinct lifestyles of this entomopathogenic fungus. The comparative analyses presented in this study reveal that Metarhizium spp. genomes harbor a complex set of genes coding for secreted proteins. Such secreted proteins appear to have been selected and maintained in the genome to cope with the distinct lifestyles presented by Metarhizium spp., which can be either entomopathogen-, endophytic- or rhizosphere-associated. The transcriptome profiling of M. anisopliae exposed to infection-mimicking conditions compared with laboratory growth conditions showed many genes that were differentially expressed. Among these genes, many genes coding for secreted proteins could be found, which could represent M. anisopliae virulence determinants. Our results offer selected sequences for further characterization of secreted proteins with potential roles in the M. anisopliae infectious process.
Methods
Sample collection and DNA extraction
M. anisopliae var. anisopliae strain E6 was isolated from Deois flavopicta[11] collected in Espírito Santo State, Brazil. This strain was incubated at 28°C for 48 h in CM (Cove’s Medium) liquid medium [78] for the subsequent DNA isolation, as previously described [23]. The genomic DNA was extracted from the mycelium cultivated in CM and was further purified using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). The quality of the isolated genomic DNA was assessed spectrophotometrically.
Genome sequencing, assembly and annotation
Two shotgun (SG) and one long paired-end (LPE) libraries were constructed using approximately 5 μg of DNA each. The library construction, titration, emulsion PCR and sequencing steps were performed according to the manufacturer’s protocol without modifications. SG libraries were sequenced using GS FLX Titanium chemistry (454-Roche, Brandford, CT, USA). One of the libraries was sequenced in one region of a two-region PicoTiterPlate (PTP) and the other in both regions of a two-region PTP. The LPE library was sequenced using GS FLX standard chemistry (454-Roche, Brandford, CT, USA) in both regions of a two-region PTP.
Replicates [79] software was used to identify and eliminate the artificially replicated sequences produced during the 454-based pyrosequencing. Newbler Assembler version 2.8 and WGS-CA 7.0 software were used to perform the assembly procedures. Minimus2 software [80] was applied to obtain a consensus assembly. The remainder gaps were filled using Consed software [81].
All contig sequences were analyzed and functionally annotated using the System for Automated Bacterial Integrated Annotation (SABIA) [82] altered to annotate eukaryotic genomes by the use of AUGUSTUS [83]. The automatic annotation criteria for assigning an ORF as “valid” included ORFs with BLASTp hits on KEGG, NCBI-nr or UniProtKB/Swiss-Prot databases, respectively; subject and query coverage ≥60%; and positives ≥60%. ORFs with no BLASTp hits found on NCBI-nr, KEGG, UniProtKB/Swiss-Prot, TCDB and Interpro databases or not included in the criteria above were defined as “hypothetical” ORFs.
Selection of refined secretomes and functional analyses
The M. anisopliae strain E6 predicted proteome, as well as those proteomes from fifteen other filamentous fungi (Aspergillus fumigatus Af293, Aspergillus nidulans FGSC A4, Aspergillus niger CBS 513.88, Beauveria bassiana ARSEF 2860, Cordyceps militaris CM01, Fusarium graminearum PH-1, Fusarium oxysporum f. sp. cubense race 1, Metarhizium anisopliae ARSEF 23, Metarhizium acridum CQMa 102, Magnaporthe oryzae 70-15, Neurospora crassa OR74A, Nectria haematococca mpVI 77-13-4, Trichoderma atroviride IMI 206040, Trichoderma reesei QM6a, and Trichoderma virens Gv29-8) were downloaded from the NCBI genome database (http://www.ncbi.nlm.nih.gov/genome/) and considered for in silico secretome analysis.
The prediction of all refined secretomes was based on the procedure described by Brown and coworkers [26] for the plant pathogen Fusarium graminearum. An automatic pipeline was developed using PERL scripts and the MySQL database. Initially, all proteins were screened to remove sequences without an initial methionine and with a mature peptide size of less than 20 amino acids. To detect a signal peptide, proteins with predictions by both SignalP v4.1 [84] (D-score = Y; http://www.cbs.dtu.dk/services/SignalP/) and TargetP v1.1 [85] (LOC = S; http://www.cbs.dtu.dk/services/TargetP/) tools were selected. These proteins were subsequently scanned for the presence of transmembrane regions using the hidden Markov model topology predictor TMHMM [86] (TMHMM v2.0; http://www.cbs.dtu.dk/services/TMHMM/), and we kept those proteins with 0 or 1 TM when a single TM was in the first 60 amino acids in the N-terminal portion. This filtering was necessary as the large majority of secreted proteins spam at the amino terminus of the protein at most 1 TM region, which resembles the signal peptide. PredGPI [87] (FRate ≤ 0.005) was used to predict GPI-anchors (http://gpcr2.biocomp.unibo.it/predgpi/pred.htm). ProtComp v9.1 (with LocDB and PotLocDB, proteins predicted as secreted by both NNets and Integral predictions; http://www.softberry.com) and WoLF PSort v0.2 [88] software were combined to infer the protein localization for the fungi studied (Extr ≥ 17). Finally, a PROSITE scan [89] was used to remove sequences associated with the pattern PS00014 (Endoplasmic reticulum targeting sequence), yielding the refined secretome with GPI-anchored proteins (GPI-Ps).
To assign a predicted function, a BLASTp search (e-value 1e-5) was conducted with selected proteins against the KEGG Orthology [90] database (KO). To avoid spurious domain alignments, we discarded BLAST results for which the alignment size divided by subject size (coverage) was below 50%. Those proteins without significant hits were analyzed with a PFAM-A database using pfam_scan.pl script [91]. The Pathogen-Host Interactions (PHI) database [92] (http://www.phi-base.org/) was used to search for orthologous proteins in M. anisopliae using an e-value of 10-5 and ≥ 50% coverage as criteria. Then, the matches were filtered, and only proteins that shared over 70% identity with M. anisopliae predicted proteins were included. Of these proteins, proteins exhibiting a “loss of pathogenicity or reduced virulence” as phenotype characteristics in the mutant strains were analyzed.
Statistical analyses were conducted using the R statistical package. One-tailed proportion test (prop.test) was used for evaluate secretome gene duplications. Statistical analysis for the comparison of GO enriched terms was conducted with Blast2GO [93].
Transcriptome analysis
Rhipicephalus microplus cuticles were sterilized and used as the sole nutrient source for M. anisopliae E6 growth and development. A spore suspension (5 × 106 spores per ml) was used to inoculate the cuticles by immersion for 30 sec. The inoculated cuticles were disposed over 1% water agar plates and maintained for 48 h and 144 h at 28°C. Each of the two biological replicates consisted of a pool of five plates containing mycelium growth over the cuticles. The comparative control condition was conducted on 100 ml liquid complete medium for 48 h at 28°C. The resulting fungal growth over the host cuticle and on liquid medium was ground to a powder in liquid nitrogen, and the total RNA was extracted using TRIzol® Reagent (Life Technologies, CA, USA) following the manufacturer’s instructions. The total isolated RNA was subjected to DNase treatment using RNase-free DNase I (Thermo Scientific, MA, USA). Large ribosomal RNA molecules were selectively depleted from the total RNA using a RiboMinus™ Eukaryote Kit for RNA-Seq (Life Technologies, CA, USA), and mRNA was concentrated and purified on a RiboMinus™ Concentration Module (Life Technologies, CA, USA).
RNA-Seq was conducted with Ion Torrent technology in an Ion Proton System. FastQC v0.10.0 [http://www.bioinformatics.babraham.ac.uk/projects/fastqc/] software was used for a reads quality check, and the FASTX-Toolkit v0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/) was used for trimming. Reads smaller than 30 nucleotides were discarded. The remaining reads were mapped to the M. anisopliae genome using the spliced read mapper Tophat v2.0.10 [94] with default parameters. HTSeq v0.5.4p5 (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html) was used to count reads aligned to only one position in protein coding regions, and the edgeR package v3.4.0 [55] was used to assess for differential gene expression, with a 5% false discovery rate (FDR ≤ 0.05) and with stringent log fold variation (logFC) ≥ or ≤ -1. Two pairwise comparisons were performed for periods C-48 h × I-48 h and I-48 h × I-144 h. InterProScan was used to assign more general superfamily functional categories v1.73 [57].
Phylogenetic and phylogenomic analyses
OrthoMCL v2.0.8 software [95] was used with default parameters to identify orthologs and paralogs among the complete proteomes of all sixteen studied organisms. A PERL script was developed to select only 1:1 orthologous sequences from the OrthoMCL output such that only a single gene copy was selected from each predicted proteome. The multi-FASTA ortholog files of each protein sequence were used as input for the multiple alignments using CLUSTAL Omega algorithm [96] with default parameters. Subsequently, SCaFos software [97] was used to for the gene concatenation of the 2,684 alignment files. Phylogenies involving the concatenated deduced amino acid sequences from all species were evaluated through distance and probabilistic methods using the PHYLIP package [98], MEGA 5.2 Computing Core [99] and TREE-PUZZLE [100] software.
Initially, multiple 100 bootstrapped data sets were generated by the Seqboot program of the PHYLIP package. Then, these data sets were submitted to ProtDist software analysis to compute a distance matrix under the JTT (Jones-Taylor-Thornton) model of amino acid replacement. The Neighbor software applied the neighbor-joining (NJ) method [101] to the resulting multiple data sets, building trees through the successive clustering of lineages. A consensus tree was obtained using the Consense program of the PHYLIP package. Three different distance matrices (p-distance, Poisson, and JTT) were evaluated using the MEGA 5.2 Computing Core, with the complete deletion and pairwise deletion options for the treatment of gaps. The bootstrap test for phylogeny was performed using 1,000 repetitions.
The quartet-puzzling [102] search algorithm implemented by TREE-PUZZLE was used to reconstruct phylogenetic trees according to the maximum likelihood (ML) approach. The JTT model of amino acid substitution was applied. The quartet-puzzling tree topology was based on 1,000 puzzling steps. The consensus tree was constructed based on a 50% majority rule consensus. The TreeView program [103] and MEGA 5 software were used to visualize and to edit the resulting phylogenies.
In total, 212 ortholog files of the secretome were submitted to phylogenetic analysis using distance methods implemented by MEGA software. The neighbor-joining algorithm, with pairwise deletion of gaps, was applied to the set of data. The p-distance, poisson and JTT matrices were evaluated. The bootstrap test of phylogeny was performed using 1,000 repetitions.
The resulting individual gene phylogenies were submitted to the CLANN software [104] to generate the supertree using a heuristic search of the supertree space for identifying the best tree. The nearest neighbor-interchange and subtree pruning and regrafting methods were tested starting with a neighbor-joining tree calculated from the average consensus distances. A bootstrap analysis was performed using 100 replicates. Then, these individual phylogenies were compared with the phylogenomic tree obtained from the 2,684 alignment files, which was considered to represent the evolutionary history of the analyzed fungi. The aim was to evaluate which secretome proteins have an evolutionary pattern that is not compatible with the organism’s evolution.
During the submission process of this work, the genome sequence from another strain of M. anisopliae (Ma69) was accepted for publication [105]. Altogether, the genomic sequences of Metarhizium spp. will allow a deeper analysis of ancient mechanisms of virulence by entomopathogens.
Sequence submission
The M. anisopliae E6 genome sequence is deposited in NCBI under Accession JNNZ00000000. RNAseq reads were deposited in NCBI SRA under Bioproject accession PRJNA257269.
Electronic supplementary material
Additional file 1: M. anisopliae E6 predicted proteins that exhibit matches in the PHI (Pathogen-Host Interaction) database proteins. (DOCX 34 KB)
Additional file 2: The automated refined secretome prediction pipeline used in this work. (TIFF 188 KB)
Additional file 3: The primary functional categories and PFAM structures of the refined predicted fungal secretomes. (DOC 57 KB)
Additional file 4: Enrichment analysis performed with the GO annotations of the predicted fungal secretomes. Comparisons were conducted with GO annotations of differente lifestyles to entomopathogens employing Fischer enrichment test of the Blast2GO software. (XLSX 46 KB)
Additional file 5: Predicted GPI-Ps coded by the M. anisopliae E6 genome. (DOCX 115 KB)
Additional file 6: Comparative analysis of GPI-Ps coding genes among Metarhizium spp. Each line represents an ortholog, as revealed by the bidirectional best-hit BLAST analysis. Shaded cells represent predicted proteins obtained in the pipeline used in this work to identify secreted proteins. Empty cells represent the lack of an ortholog. (XLSX 16 KB)
Additional file 7: Proportion of gene duplications for sequences predicted as secreted and non-secreted. (DOCX 118 KB)
Additional file 8: RNAseq and alignment analysis statistics. (DOCX 50 KB)
{kind=link}
Additional file 9: Principal component analysis of the expression patterns of the three distinct conditions analyzed. (PNG 96 KB)
Additional file 10: Log fold change (logFC) for selected M. anisopliae E6 loci. C-48 h: Control condition with no infection; I-48 h: host cuticle 48 hours infection and I-144 h: host cuticle 144 hours infection. *no significant variation (FDR < 0.05 and logFC ≥ 1 or ≤ -1). (DOC 52 KB)
Additional file 11: Complete edgeR output for the differential expression analysis. (XLS 2 MB)
Additional file 12: Fungal evolutionary history obtained through a phylogenomic approach using Maximum Likelihood Method. Each internal branch indicates the percentage of times the corresponding cluster was found among the 1,000 intermediate trees. The scale bar represents substitutions of amino acids per site. (TIFF 143 KB)
Additional file 13: Description of the proteins identified as potentially responsible for the differential evolution of the secretome and their expression status based on RNA sequencing. (XLS 30 KB)
Acknowledgments
We would like to thank the staff of LNCC for support. This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Programa de Aperfeiçoamento Pessoal de Nível Superior (CAPES), Fundação de Amparo a Pesquisa do estado do Rio Grande do Sul (FAPERGS) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ).
Footnotes
Competing interests
The authors declare no competing interests.
Authors’ contributions
Conceived and designed the experiments: CCS, MHV, ATRV, and AS. Annotation curation: CCS, AJ, RLMG, CET, GLM, JTB, LGPA, FCA, ALG, NS, RLAP, LB, ML, LS, WOBS, CPS, TRS, EOS, and LK. Comparative analyses: CCS, AJ, RLMG, and CET. Secretome analysis: CCS, AJ, RLMG, and ESO. Transcriptome analysis: CCS, AJ, RLMG, and GLM. Phylogenomic analyses: CET and FCA. Contributed reagents/materials/analysis tools: MHV, ATRV, and AS. Wrote the article: CCS, AJ, RLMG, CET, ESO, and AS. All authors read and approved the final manuscript.
Contributor Information
Charley Christian Staats, Email: staats@cbiot.ufrgs.br.
Ângela Junges, Email: ajunges@cbiot.ufrgs.br.
Rafael Lucas Muniz Guedes, Email: rmguedes@lncc.br.
Claudia Elizabeth Thompson, Email: thompson@cbiot.ufrgs.br.
Guilherme Loss de Morais, Email: guilherme.loss@gmail.com.
Juliano Tomazzoni Boldo, Email: julianoboldo@unipampa.edu.br.
Luiz Gonzaga Paula de Almeida, Email: lgonzaga@lncc.br.
Fábio Carrer Andreis, Email: fabio.andreis@gmail.com.
Alexandra Lehmkuhl Gerber, Email: alegerber@lncc.br.
Nicolau Sbaraini, Email: nicolausbaraini@me.com.
Rana Louise de Andrade da Paixão, Email: ranalouise@cbiot.ufrgs.br.
Leonardo Broetto, Email: lbroetto@gmail.com.
Melissa Landell, Email: melissa.landell@gmail.com.
Lucélia Santi, Email: luca.santi@yahoo.com.br.
Walter Orlando Beys-da-Silva, Email: walterbeys@yahoo.com.br.
Carolina Pereira Silveira, Email: caru.silveira@gmail.com.
Thaiane Rispoli Serrano, Email: thaianerispoli@gmail.com.
Eder Silva de Oliveira, Email: edereto@cbiot.ufrgs.br.
Lívia Kmetzsch, Email: liviak@cbiot.ufrgs.br.
Marilene Henning Vainstein, Email: mhv@cbiot.ufrgs.br.
Ana Tereza Ribeiro de Vasconcelos, Email: atrv@lncc.br.
Augusto Schrank, Email: aschrank@cbiot.ufrgs.br.
References
- 1.Mora C, Tittensor DP, Adl S, Simpson AG, Worm B. How many species are there on Earth and in the ocean? PLoS Biol. 2011;9:e1001127. doi: 10.1371/journal.pbio.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackwell M. The Fungi: 1, 2, 3 … 5.1 million species? Am J Bot. 2011;98:426–438. doi: 10.3732/ajb.1000298. [DOI] [PubMed] [Google Scholar]
- 3.Girard V, Dieryckx C, Job C, Job D. Secretomes: the fungal strike force. Proteomics. 2013;13:597–608. doi: 10.1002/pmic.201200282. [DOI] [PubMed] [Google Scholar]
- 4.Basset Y, Cizek L, Cuénoud P, Didham RK, Guilhaumon F, Missa O, Novotny V, Ødegaard F, Roslin T, Schmidl J, Tishechkin AK, Winchester NN, Roubik DW, Aberlenc H-P, Bail J, Barrios H, Bridle JR, Castaño-Meneses G, Corbara B, Curletti G, da Rocha WD, De Bakker D, Delabie JHC, Dejean A, Fagan LL, Floren A, Kitching RL, Medianero E, Miller SE, de Oliveira EG, et al. Arthropod diversity in a tropical forest. Science. 2012;338:1481–1484. doi: 10.1126/science.1226727. [DOI] [PubMed] [Google Scholar]
- 5.Hasan S, Ahmad A, Purwar A, Khan N, Kundan R, Gupta G. Production of extracellular enzymes in the entomopathogenic fungus Verticillium lecanii. Bioinformation. 2013;9:238–242. doi: 10.6026/97320630009238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.da Silva WO B, Santi L, Correa AP, Silva LA, Bresciani FR, Schrank A, Vainstein MH. The entomopathogen Metarhizium anisopliae can modulate the secretion of lipolytic enzymes in response to different substrates including components of arthropod cuticle. Fungal Biol. 2010;114:911–916. doi: 10.1016/j.funbio.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Santi L, Silva WO, Pinto AF, Schrank A, Vainstein MH. Metarhizium anisopliae host-pathogen interaction: differential immunoproteomics reveals proteins involved in the infection process of arthropods. Fungal Biol. 2010;114:312–319. doi: 10.1016/j.funbio.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Murad AM, Noronha EF, Miller RN, Costa FT, Pereira CD, Mehta A, Caldas RA, Franco OL. Proteomic analysis of Metarhizium anisopliae secretion in the presence of the insect pest Callosobruchus maculatus. Microbiology. 2008;154:3766–3774. doi: 10.1099/mic.0.2008/022913-0. [DOI] [PubMed] [Google Scholar]
- 9.da Silva MV, Santi L, Staats CC, da Costa AM, Colodel EM, Driemeier D, Vainstein MH, Schrank A. Cuticle-induced endo/exoacting chitinase CHIT30 from Metarhizium anisopliae is encoded by an ortholog of the chi3 gene. Res Microbiol. 2005;156:382–392. doi: 10.1016/j.resmic.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Barreto CC, Staats CC, Schrank A, Vainstein MH. Distribution of chitinases in the entomopathogen Metarhizium anisopliae and effect of N-acetylglucosamine in protein secretion. Curr Microbiol. 2004;48:102–107. doi: 10.1007/s00284-003-4063-z. [DOI] [PubMed] [Google Scholar]
- 11.Arruda W, Lubeck I, Schrank A, Vainstein MH. Morphological alterations of Metarhizium anisopliae during penetration of Boophilus microplus ticks. Exp Appl Acarol. 2005;37:231–244. doi: 10.1007/s10493-005-3818-6. [DOI] [PubMed] [Google Scholar]
- 12.Schrank A, Vainstein MH. Metarhizium anisopliae enzymes and toxins. Toxicon. 2010;56:1267–1274. doi: 10.1016/j.toxicon.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 13.Butt TM, Greenfield BP, Greig C, Maffeis TG, Taylor JW, Piasecka J, Dudley E, Abdulla A, Dubovskiy IM, Garrido-Jurado I, Quesada-Moraga E, Penny MW, Eastwood DC. Metarhizium anisopliae pathogenesis of mosquito larvae: a verdict of accidental death. PLoS One. 2013;8:e81686. doi: 10.1371/journal.pone.0081686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freimoser FM, Hu G, St Leger RJ. Variation in gene expression patterns as the insect pathogen Metarhizium anisopliae adapts to different host cuticles or nutrient deprivation in vitro. Microbiology. 2005;151:361–371. doi: 10.1099/mic.0.27560-0. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes EK, Bittencourt VR, Roberts DW. Perspectives on the potential of entomopathogenic fungi in biological control of ticks. Exp Parasitol. 2012;130:300–305. doi: 10.1016/j.exppara.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Faria MR, Wraight SP. Mycoinsecticides and Mycoacaricides: a comprehensive list with worldwide coverage and international classification of formulation types. Biological Control. 2007;43:237–256. doi: 10.1016/j.biocontrol.2007.08.001. [DOI] [Google Scholar]
- 17.Bischoff JF, Rehner SA, Humber RA. A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia. 2009;101:512–530. doi: 10.3852/07-202. [DOI] [PubMed] [Google Scholar]
- 18.Gao Q, Jin K, Ying SH, Zhang Y, Xiao G, Shang Y, Duan Z, Hu X, Xie XQ, Zhou G, Peng G, Luo Z, Huang W, Wang B, Fang W, Wang S, Zhong Y, Ma LJ, St Leger RJ, Zhao GP, Pei Y, Feng MG, Xia Y, Wang C. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011;7:e1001264. doi: 10.1371/journal.pgen.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao G, Ying SH, Zheng P, Wang ZL, Zhang S, Xie XQ, Shang Y, St Leger RJ, Zhao GP, Wang C, Feng MG. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep. 2012;2:483. doi: 10.1038/srep00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32:11–16. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng P, Xia Y, Xiao G, Xiong C, Hu X, Zhang S, Zheng H, Huang Y, Zhou Y, Wang S, Zhao GP, Liu X, St Leger RJ, Wang C. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011;12:R116. doi: 10.1186/gb-2011-12-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boldo JT, Junges A, do Amaral KB, Staats CC, Vainstein MH, Schrank A. Endochitinase CHI2 of the biocontrol fungus Metarhizium anisopliae affects its virulence toward the cotton stainer bug Dysdercus peruvianus. Curr Genet. 2009;55:551–560. doi: 10.1007/s00294-009-0267-5. [DOI] [PubMed] [Google Scholar]
- 24.Fang W, Leng B, Xiao Y, Jin K, Ma J, Fan Y, Feng J, Yang X, Zhang Y, Pei Y. Cloning of Beauveria bassiana chitinase gene Bbchit1 and its application to improve fungal strain virulence. Appl Environ Microbiol. 2005;71:363–370. doi: 10.1128/AEM.71.1.363-370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou G, Wang J, Qiu L, Feng MG. A Group III histidine kinase (mhk1) upstream of high-osmolarity glycerol pathway regulates sporulation, multi-stress tolerance and virulence of Metarhizium robertsii, a fungal entomopathogen. Environ Microbiol. 2012;14:817–829. doi: 10.1111/j.1462-2920.2011.02643.x. [DOI] [PubMed] [Google Scholar]
- 26.Brown NA, Antoniw J, Hammond-Kosack KE. The predicted secretome of the plant pathogenic fungus Fusarium graminearum: a refined comparative analysis. PLoS One. 2012;7:e33731. doi: 10.1371/journal.pone.0033731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oliveira DL, Rizzo J, Joffe LS, Godinho RM, Rodrigues ML. Where do they come from and where do they go: candidates for regulating extracellular vesicle formation in fungi. Int J Mol Sci. 2013;14:9581–9603. doi: 10.3390/ijms14059581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodrigues ML, Nakayasu ES, Almeida IC, Nimrichter L. The impact of proteomics on the understanding of functions and biogenesis of fungal extracellular vesicles. J Proteomics. 2014;97:177–186. doi: 10.1016/j.jprot.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harding CV, Heuser JE, Stahl PD. Exosomes: looking back three decades and into the future. J Cell Biol. 2013;200:367–371. doi: 10.1083/jcb.201212113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vallejo MC, Nakayasu ES, Matsuo AL, Sobreira TJ, Longo LV, Ganiko L, Almeida IC, Puccia R. Vesicle and vesicle-free extracellular proteome of Paracoccidioides brasiliensis: comparative analysis with other pathogenic fungi. J Proteome Res. 2012;11:1676–1685. doi: 10.1021/pr200872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodrigues ML, Nosanchuk JD, Schrank A, Vainstein MH, Casadevall A, Nimrichter L. Vesicular transport systems in fungi. Future Microbiol. 2011;6:1371–1381. doi: 10.2217/fmb.11.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodrigues ML, Djordjevic JT. Unravelling secretion in Cryptococcus neoformans: more than one way to skin a cat. Mycopathologia. 2012;173:407–418. doi: 10.1007/s11046-011-9468-9. [DOI] [PubMed] [Google Scholar]
- 33.Hansen SF, Bettler E, Rinnan A, Engelsen SB, Breton C. Exploring genomes for glycosyltransferases. Mol Biosyst. 2010;6:1773–1781. doi: 10.1039/c000238k. [DOI] [PubMed] [Google Scholar]
- 34.Boldo JT, do Amaral KB, Junges A, Pinto PM, Staats CC, Vainstein MH, Schrank A. Evidence of alternative splicing of the chi2 chitinase gene from Metarhizium anisopliae. Gene. 2010;462:1–7. doi: 10.1016/j.gene.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Gruber S, Seidl-Seiboth V. Self versus non-self: fungal cell wall degradation in Trichoderma. Microbiology. 2012;158:26–34. doi: 10.1099/mic.0.052613-0. [DOI] [PubMed] [Google Scholar]
- 36.Brzezinska MS, Jankiewicz U. Production of antifungal chitinase by Aspergillus niger LOCK 62 and its potential role in the biological control. Curr Microbiol. 2012;65:666–672. doi: 10.1007/s00284-012-0208-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, Leclerque A, Pava-Ripoll M, Fang W, St Leger RJ. Comparative genomics using microarrays reveals divergence and loss of virulence-associated genes in host-specific strains of the insect pathogen Metarhizium anisopliae. Eukaryot Cell. 2009;8:888–898. doi: 10.1128/EC.00058-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EG, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina) Nat Biotechnol. 2008;26:553–560. doi: 10.1038/nbt1403. [DOI] [PubMed] [Google Scholar]
- 39.Lai JS, Cheng CW, Sung TY, Hsu WL. Computational comparative study of tuberculosis proteomes using a model learned from signal peptide structures. PLoS One. 2012;7:e35018. doi: 10.1371/journal.pone.0035018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braaksma M, Martens-Uzunova ES, Punt PJ, Schaap PJ. An inventory of the Aspergillus niger secretome by combining in silico predictions with shotgun proteomics data. BMC Genomics. 2010;11:584. doi: 10.1186/1471-2164-11-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abaev I, Foster-Frey J, Korobova O, Shishkova N, Kiseleva N, Kopylov P, Pryamchuk S, Schmelcher M, Becker SC, Donovan DM. Staphylococcal phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site. Appl Microbiol Biotechnol. 2013;97:3449–3456. doi: 10.1007/s00253-012-4252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helsens K, Van Damme P, Degroeve S, Martens L, Arnesen T, Vandekerckhove J, Gevaert K. Bioinformatics analysis of a Saccharomyces cerevisiae N-terminal proteome provides evidence of alternative translation initiation and post-translational N-terminal acetylation. J Proteome Res. 2011;10:3578–3589. doi: 10.1021/pr2002325. [DOI] [PubMed] [Google Scholar]
- 43.Smollett KL, Fivian-Hughes AS, Smith JE, Chang A, Rao T, Davis EO. Experimental determination of translational start sites resolves uncertainties in genomic open reading frame predictions - application to Mycobacterium tuberculosis. Microbiology. 2009;155:186–197. doi: 10.1099/mic.0.022889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chabregas SM, Luche DD, Van Sluys MA, Menck CF, Silva-Filho MC. Differential usage of two in-frame translational start codons regulates subcellular localization of Arabidopsis thaliana THI1. J Cell Sci. 2003;116:285–291. doi: 10.1242/jcs.00228. [DOI] [PubMed] [Google Scholar]
- 45.Fujita M, Kinoshita T. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta. 1821;2012:1050–1058. doi: 10.1016/j.bbalip.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Pittet M, Conzelmann A. Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2007;1771:405–420. doi: 10.1016/j.bbalip.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 47.Maddi A, Fu C, Free SJ. The Neurospora crassa dfg5 and dcw1 genes encode alpha-1,6-mannanases that function in the incorporation of glycoproteins into the cell wall. PLoS One. 2012;7:e38872. doi: 10.1371/journal.pone.0038872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi CJ, Ju HJ, Park BH, Qin R, Jahng KY, Han DM, Chae KS. Isolation and characterization of the Aspergillus nidulans eglC gene encoding a putative beta-1,3-endoglucanase. Fungal Genet Biol. 2005;42:590–600. doi: 10.1016/j.fgb.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, Zhu J, Ying SH, Feng MG. The GPI-anchored protein Ecm33 is vital for conidiation, cell wall integrity, and multi-stress tolerance of two filamentous entomopathogens but not for virulence. Appl Microbiol Biotechnol. 2014;98:5517–5529. doi: 10.1007/s00253-014-5577-y. [DOI] [PubMed] [Google Scholar]
- 50.Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18:292–298. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- 51.Cohen-Gihon I, Sharan R, Nussinov R. Processes of fungal proteome evolution and gain of function: gene duplication and domain rearrangement. Phys Biol. 2011;8:035009. doi: 10.1088/1478-3975/8/3/035009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S, Schwartz DC, Freitag M, Ma LJ, Danchin EG, Henrissat B, Coutinho PM, Nelson DR, Straney D, Napoli CA, Barker BM, Gribskov M, Rep M, Kroken S, Molnar I, Rensing C, Kennell JC, Zamora J, Farman ML, Selker EU, Salamov A, et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5:e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasan RK, Bidochka MJ. The insect-pathogenic fungus Metarhizium robertsii (Clavicipitaceae) is also an endophyte that stimulates plant root development. Am J Bot. 2012;99:101–107. doi: 10.3732/ajb.1100136. [DOI] [PubMed] [Google Scholar]
- 54.Wyrebek M, Huber C, Sasan RK, Bidochka MJ. Three sympatrically occurring species of Metarhizium show plant rhizosphere specificity. Microbiology. 2011;157:2904–2911. doi: 10.1099/mic.0.051102-0. [DOI] [PubMed] [Google Scholar]
- 55.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo Y, Li CI, Ye F, Shyr Y. Evaluation of read count based RNAseq analysis methods. BMC Genomics. 2013;14(8):S2. doi: 10.1186/1471-2164-14-S8-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Lima Morais DA, Fang H, Rackham OJ, Wilson D, Pethica R, Chothia C, Gough J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011;39:D427–D434. doi: 10.1093/nar/gkq1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bagga S, Hu G, Screen SE, St Leger RJ. Reconstructing the diversification of subtilisins in the pathogenic fungus Metarhizium anisopliae. Gene. 2004;324:159–169. doi: 10.1016/j.gene.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 59.Irie T, Matsumura H, Terauchi R, Saitoh H. Serial Analysis of Gene Expression (SAGE) of Magnaporthe grisea: genes involved in appressorium formation. Mol Genet Genomics. 2003;270:181–189. doi: 10.1007/s00438-003-0911-6. [DOI] [PubMed] [Google Scholar]
- 60.Laronde-Leblanc N, Guszczynski T, Copeland T, Wlodawer A. Structure and activity of the atypical serine kinase Rio1. FEBS J. 2005;272:3698–3713. doi: 10.1111/j.1742-4658.2005.04796.x. [DOI] [PubMed] [Google Scholar]
- 61.St Leger RJ, Joshi L, Bidochka MJ, Rizzo NW, Roberts DW. Biochemical characterization and ultrastructural localization of two extracellular trypsins produced by Metarhizium anisopliae in infected insect cuticles. Appl Environ Microbiol. 1996;62:1257–1264. doi: 10.1128/aem.62.4.1257-1264.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.St Leger RJ, Joshi L, Roberts D. Ambient pH is a major determinant in the expression of cuticle-degrading enzymes and hydrophobin by Metarhizium anisopliae. Appl Environ Microbiol. 1998;64:709–713. doi: 10.1128/aem.64.2.709-713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Screen SE, St Leger RJ. Cloning, expression, and substrate specificity of a fungal chymotrypsin. Evidence for lateral gene transfer from an actinomycete bacterium. J Biol Chem. 2000;275:6689–6694. doi: 10.1074/jbc.275.9.6689. [DOI] [PubMed] [Google Scholar]
- 64.Lopez-Berges MS, Rispail N, Prados-Rosales RC, Di Pietro A. A nitrogen response pathway regulates virulence in plant pathogenic fungi: role of TOR and the bZIP protein MeaB. Plant Signal Behav. 2010;5:1623–1625. doi: 10.4161/psb.5.12.13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sevim A, Donzelli BG, Wu D, Demirbag Z, Gibson DM, Turgeon BG. Hydrophobin genes of the entomopathogenic fungus, Metarhizium brunneum, are differentially expressed and corresponding mutants are decreased in virulence. Curr Genet. 2012;58:79–92. doi: 10.1007/s00294-012-0366-6. [DOI] [PubMed] [Google Scholar]
- 66.Garcia-Santamarina S, Boronat S, Calvo IA, Rodriguez-Gabriel M, Ayte J, Molina H, Hidalgo E. Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid Redox Signal. 2013;18:1549–1556. doi: 10.1089/ars.2012.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK, Kredics L, Alcaraz LD, Aerts A, Antal Z, Atanasova L, Cervantes-Badillo MG, Challacombe J, Chertkov O, McCluskey K, Coulpier F, Deshpande N, von Dohren H, Ebbole DJ, Esquivel-Naranjo EU, Fekete E, Flipphi M, Glaser F, Gomez-Rodriguez EY, Gruber S, et al. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 2011;12:R40. doi: 10.1186/gb-2011-12-4-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Srivastava SK, Huang X, Brar HK, Fakhoury AM, Bluhm BH, Bhattacharyya MK. The genome sequence of the fungal pathogen Fusarium virguliforme that causes sudden death syndrome in soybean. PLoS One. 2014;9:e81832. doi: 10.1371/journal.pone.0081832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- 70.Cerqueira GC, Arnaud MB, Inglis DO, Skrzypek MS, Binkley G, Simison M, Miyasato SR, Binkley J, Orvis J, Shah P, Wymore F, Sherlock G, Wortman JR. The Aspergillus Genome Database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 2014;42:D705–D710. doi: 10.1093/nar/gkt1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Druzhinina IS, Seidl-Seiboth V, Herrera-Estrella A, Horwitz BA, Kenerley CM, Monte E, Mukherjee PK, Zeilinger S, Grigoriev IV, Kubicek CP. Trichoderma: the genomics of opportunistic success. Nat Rev Microbiol. 2011;9:749–759. doi: 10.1038/nrmicro2637. [DOI] [PubMed] [Google Scholar]
- 72.St Leger RJ, Joshi L, Roberts DW. Adaptation of proteases and carbohydrates of saprophytic, phytopathogenic and entomopathogenic fungi to the requirements of their ecological niches. Microbiology. 1997;143(Pt 6):1983–1992. doi: 10.1099/00221287-143-6-1983. [DOI] [PubMed] [Google Scholar]
- 73.Cuomo CA, Guldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, Walton JD, Ma LJ, Baker SE, Rep M, Adam G, Antoniw J, Baldwin T, Calvo S, Chang YL, Decaprio D, Gale LR, Gnerre S, Goswami RS, Hammond-Kosack K, Harris LJ, Hilburn K, Kennell JC, Kroken S, Magnuson JK, Mannhaupt G, Mauceli E, Mewes HW, Mitterbauer R, Muehlbauer G, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317:1400–1402. doi: 10.1126/science.1143708. [DOI] [PubMed] [Google Scholar]
- 74.Behie SW, Bidochka MJ. Ubiquity of insect-derived nitrogen transfer to plants by endophytic insect-pathogenic fungi: an additional branch of the soil nitrogen cycle. Appl Environ Microbiol. 2014;80:1553–1560. doi: 10.1128/AEM.03338-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Behie SW, Padilla-Guerrero IE, Bidochka MJ. Nutrient transfer to plants by phylogenetically diverse fungi suggests convergent evolutionary strategies in rhizospheric symbionts. Commun Integr Biol. 2013;6:e22321. doi: 10.4161/cib.22321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Behie SW, Zelisko PM, Bidochka MJ. Endophytic insect-parasitic fungi translocate nitrogen directly from insects to plants. Science. 2012;336:1576–1577. doi: 10.1126/science.1222289. [DOI] [PubMed] [Google Scholar]
- 77.Wang H, Xu Z, Gao L, Hao B. A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol Biol. 2009;9:195. doi: 10.1186/1471-2148-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cove DJ. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim Biophys Acta. 1966;113:51–56. doi: 10.1016/S0926-6593(66)80120-0. [DOI] [PubMed] [Google Scholar]
- 79.Gomez-Alvarez V, Teal TK, Schmidt TM. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3:1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- 80.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordon D, Green P. Consed: a graphical editor for next-generation sequencing. Bioinformatics. 2013;29:2936–2937. doi: 10.1093/bioinformatics/btt515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Almeida LG, Paixao R, Souza RC, Costa GC, Barrientos FJ, Santos MT, Almeida DF, Vasconcelos AT. A System for Automated Bacterial (genome) Integrated Annotation–SABIA. Bioinformatics. 2004;20:2832–2833. doi: 10.1093/bioinformatics/bth273. [DOI] [PubMed] [Google Scholar]
- 83.Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–W312. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 85.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 86.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 87.Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformatics. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gattiker A, Gasteiger E, Bairoch A. ScanProsite: a reference implementation of a PROSITE scanning tool. Appl Bioinformatics. 2002;1:107–108. [PubMed] [Google Scholar]
- 90.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winnenburg R, Urban M, Beacham A, Baldwin TK, Holland S, Lindeberg M, Hansen H, Rawlings C, Hammond-Kosack KE, Kohler J. PHI-base update: additions to the pathogen host interaction database. Nucleic Acids Res. 2008;36:D572–D576. doi: 10.1093/nar/gkm858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roure B, Rodriguez-Ezpeleta N, Philippe H. SCaFoS: a tool for selection, concatenation and fusion of sequences for phylogenomics. BMC Evol Biol. 2007;7(1):S2. doi: 10.1186/1471-2148-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Willis LG, Winston ML, Honda BM. Phylogenetic relationships in the honeybee (genus Apis) as determined by the sequence of the cytochrome oxidase II region of mitochondrial DNA. Mol Phylogenet Evol. 1992;1:169–178. doi: 10.1016/1055-7903(92)90013-7. [DOI] [PubMed] [Google Scholar]
- 99.Kumar S, Stecher G, Peterson D, Tamura K. MEGA-CC: computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics. 2012;28:2685–2686. doi: 10.1093/bioinformatics/bts507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 101.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 102.le Vinh S, Fuehrer A, von Haeseler A. Random Tree-Puzzle leads to the Yule-Harding distribution. Mol Biol Evol. 2011;28:873–877. doi: 10.1093/molbev/msq212. [DOI] [PubMed] [Google Scholar]
- 103.Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 104.Creevey CJ, McInerney JO. Clann: investigating phylogenetic information through supertree analyses. Bioinformatics. 2005;21:390–392. doi: 10.1093/bioinformatics/bti020. [DOI] [PubMed] [Google Scholar]
- 105.Pattemore JA, Hane JK, Williams AH, Wilson BAL, Stodart BJ, Ash GJ. The genome sequence of the biocontrol fungus Metarhizium anisopliae and comparative genomics of Metarhizium species. BMC Genomics. 2014;15:660. doi: 10.1186/1471-2164-15-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: M. anisopliae E6 predicted proteins that exhibit matches in the PHI (Pathogen-Host Interaction) database proteins. (DOCX 34 KB)
Additional file 2: The automated refined secretome prediction pipeline used in this work. (TIFF 188 KB)
Additional file 3: The primary functional categories and PFAM structures of the refined predicted fungal secretomes. (DOC 57 KB)
Additional file 4: Enrichment analysis performed with the GO annotations of the predicted fungal secretomes. Comparisons were conducted with GO annotations of differente lifestyles to entomopathogens employing Fischer enrichment test of the Blast2GO software. (XLSX 46 KB)
Additional file 5: Predicted GPI-Ps coded by the M. anisopliae E6 genome. (DOCX 115 KB)
Additional file 6: Comparative analysis of GPI-Ps coding genes among Metarhizium spp. Each line represents an ortholog, as revealed by the bidirectional best-hit BLAST analysis. Shaded cells represent predicted proteins obtained in the pipeline used in this work to identify secreted proteins. Empty cells represent the lack of an ortholog. (XLSX 16 KB)
Additional file 7: Proportion of gene duplications for sequences predicted as secreted and non-secreted. (DOCX 118 KB)
Additional file 8: RNAseq and alignment analysis statistics. (DOCX 50 KB)
Additional file 9: Principal component analysis of the expression patterns of the three distinct conditions analyzed. (PNG 96 KB)
Additional file 10: Log fold change (logFC) for selected M. anisopliae E6 loci. C-48 h: Control condition with no infection; I-48 h: host cuticle 48 hours infection and I-144 h: host cuticle 144 hours infection. *no significant variation (FDR < 0.05 and logFC ≥ 1 or ≤ -1). (DOC 52 KB)
Additional file 11: Complete edgeR output for the differential expression analysis. (XLS 2 MB)
Additional file 12: Fungal evolutionary history obtained through a phylogenomic approach using Maximum Likelihood Method. Each internal branch indicates the percentage of times the corresponding cluster was found among the 1,000 intermediate trees. The scale bar represents substitutions of amino acids per site. (TIFF 143 KB)
Additional file 13: Description of the proteins identified as potentially responsible for the differential evolution of the secretome and their expression status based on RNA sequencing. (XLS 30 KB)