

Abstract
Cereal grain contamination by trichothecene mycotoxins is known to negatively impact human and animal health with adverse effects on food intake and growth being of particular concern. The head blight fungus Fusarium graminearum elaborates five closely related 8-ketotrichothecene congeners: (1) deoxynivalenol (DON), (2) 3-acetyldeoxynivalenol (3-ADON), (3) 15-acetyldeoxynivalenol (15-ADON), (4) fusarenon X (FX), and (5) nivalenol (NIV). While anorexia induction in mice exposed intraperitoneally to DON has been linked to plasma elevation of the satiety hormones cholecystokinin (CCK) and peptide YY3–36 (PYY3–36), the effects of oral gavage of DON or of other 8-keotrichothecenes on release of these gut peptides have not been established. The purpose of this study was to (1) compare the anorectic responses to the aforementioned 8-ketotrichothecenes following oral gavage at a common dose (2.5 mg/kg bw) and (2) relate these effects to changes plasma CCK and PYY3–36 concentrations. Elevation of plasma CCK markedly corresponded to anorexia induction by DON and all other 8-ketotrichothecenes tested. Furthermore, the CCK1 receptor antagonist SR 27897 and the CCK2 receptor antagonist L-365,260 dose-dependently attenuated both CCK- and DON-induced anorexia, which was consistent with this gut satiety hormone being an important mediator of 8-ketotrichothecene-induced food refusal. In contrast to CCK, PYY3–36 was moderately elevated by oral gavage with DON and NIV but not by 3-ADON, 15-ADON, or FX. Taken together, the results suggest that CCK plays a major role in anorexia induction following oral exposure to 8-ketotrichothecenes, whereas PYY3–36 might play a lesser, congener-dependent role in this response.
Keywords: mycotoxin, trichothecene, anorexia, deoxynivalenol, 3-acetyldeoxynivalenol, 15-acetyldeoxynivalenol, fusarenon X, nivalenol
Worldwide changes in climate and agricultural practices have led to increased incidence of Fusarium head blight in grains with a concomitant elevation in contamination by 8-ketotrichothecene mycotoxins that include five structurally related congeners: (1) deoxynivalenol (DON), (2) 3-acetyldeoxynivalenol (3-ADON), (3) 15-acetyldeoxynivalenol (15-ADON), (4) fusarenon X (FX), and (5) nivalenol (NIV) (Fig. 1) (Pestka, 2010a). DON and other 8-ketotrichothecenes have been associated with a spectrum of adverse effects that include anorexia, growth suppression, emesis, nausea, neuroendocrine changes, and immunosuppression (Pestka, 2010b). Induction of anorexia and resultant growth suppression by DON is of particular concern from the perspective of human and animal health (Forsell et al., 1986; Iverson et al., 1995; JECFA, 2011). Various countries have established specific regulations for DON based on the risks of this adverse outcome. Despite the importance of anorexia induction by DON and other 8-ketotrichothecenes to growth suppression (Flannery et al., 2013), the pathophysiologic basis for this effect remains poorly understood.
FIG. 1.

Structures of 8-ketotrichothecenes included in this study.
Appetite is regulated by both central and peripheral factors that influence the balance of anorexigenic and orexigenic signaling (Schwartz, 2006). Importantly, DON upregulates the expression of anorexigenic molecules including pro-opiomelanocortin (POMC), melanocortin 4 receptor (MC4R), and amphetamine-regulated transcript (CART) within the hypothalamic neurons of the mouse (Girardet et al., 2011). A possible upstream mechanism for elevated expression of anorexigenic hormones in the hypothalamus is the increased secretion of the gut satiety peptides cholecystokinin (CCK) and peptide YY3–36 (PYY3–36) by intestinal enteroendocrine cells (Steinert et al., 2013). CCK is secreted by I cells within the duodenum and jejunum (Strader and Woods, 2005) and this satiety peptide upregulates POMC and MC4R expression in the hypothalamus (Kohno et al., 2008). Both peripheral and central administration of CCK to animals decrease food intake (Della-Fera and Baile, 1980; Ebenezer et al., 1990; Zhang et al., 1986). PYY3–36 is released from the L cells of the distal ileum and colon and has also been implicated in the upregulation of anorexigenic and downregulation of orexigenic signaling molecules within the brain (Challis et al., 2003). Peripheral administration of PYY3–36 induces satiety in many species including humans, nonhuman primates, and rodents (Batterham et al., 2002, 2003a, b).
To study DON-induced food refusal, our laboratory has developed a mouse model for anorexia predicated on the placement of food and measurement of its consumption during the nocturnal feeding period (Flannery et al., 2011). We have recently observed in this model that both intraperitoneal (IP) and orolingual administration of DON rapidly induce plasma CCK and PYY3–36 in the B6C3F1 female mouse suggesting that these hormones are possible upstream contributors to DON-induced anorexia (Flannery et al., 2012). In support of this contention, direct administration of exogenous PYY or CCK similarly caused reduced food intake in this mouse model. Food intake experiments using the NPY2 receptor antagonist BIIE0246 suggested that PYY mediated DON-induced anorexia. Although the CCK1 receptor antagonist devazepide showed a similar trend in the reversal of DON's hypophagic (i.e., food refusal) effects, the differences were not significant.
The aforementioned mouse food refusal model has also been employed to compare the capacities of the 8-ketotrichothecenes to induce anorexia (Wu et al., 2012). Anorectic responses to DON, 3-ADON, and 15-ADON following both IP exposure and oral gavage were transient with food intake recovering to control levels within 16 h. The no observed adverse effect levels (NOAEL) and lowest observed adverse effect levels (LOAEL) were 0.5 and 1 mg/kg bw following IP exposure, respectively, and 1 and 2.5 mg/kg bw after oral exposure, respectively. In contrast, food refusal persisted from 48 to 96 h following IP and oral exposure to FX and NIV. For both IP and oral FX exposure, the NOAEL was 0.025 mg/kg bw and LOAEL was 0.25 mg/kg bw, whereas the NOAELs and LOAELs for NIV were 0.01 and 0.1 mg/kg bw, respectively, after IP exposure and 0.1 and 1 mg/kg bw, respectively, following oral exposure. Accordingly, these data and the prior DON study (Flannery et al., 2012) suggest that anorectic responses to 8-ketotrichothecenes were always greater or equal when administered IP as compared with oral exposure.
Because ingestion is the primary means of exposure to DON and other 8-ketotrichothecenes for humans and animals, it is important from a public health perspective to understand the mechanisms by which oral administration of these congeners affects plasma CCK and PYY3–36. Here, we utilized the mouse anorexia model to relate effects of oral gavage to a common anorexigenic dose of 2.5 mg/kg bw DON, 3-ADON, 15-ADON, NIV, and FX on food consumption to the release of CCK and PYY3–36. The results suggest that CCK is a major contributor to induction of anorexia after oral exposure to all 8-ketotrichothecenes whereas PYY3–36 might play a lesser, congener-dependent role in this response.
MATERIALS AND METHODS
Toxins and drugs
Sources of 8-ketotrichothecenes and purity (> 98%) verified by elemental analysis and LC-MS as previously described (Wu et al., 2012). CCK (23–33, sulfated), CCK1 receptor antagonist SR 27897 and CCK2 receptor antagonist L-365,260, were purchased from Tocris Biosciences (Ellisville, MO). All toxins and CCK were dissolved in filter-sterilized phosphate buffered saline (PBS). SR 27897 and L-365,260 were dissolved in 5% dimethyl sulfoxide (DMSO) in filter-sterilized PBS.
Assessment of anorexia in the mouse
Female B6C3F1 mice (10–12 weeks old) were obtained from Charles River Breeding (Portage, MI) and housed individually in polycarbonate cages in a room maintained at 21–24°C and 40–55% relative humidity under a 12 h light (6:00–18:00 h)/dark (18:00–6:00 h) cycle. All studies were conducted in accordance with National Institutes of Health guidelines and approved by the Michigan State University Institutional Animal Care and Use Committee (MSU-IACUC). The general experimental design for the anorexia study (Fig. 2A) was based on previously described protocols (Flannery et al., 2011; Wu et al., 2012). Briefly, mice were acclimated a week after arriving and randomly divided into different groups according to body weight 1 day before experiment. On the day of the experiment, groups of mice (n = 5) were fasted but water provided ad lib from 10:00 to 18:00 h and orally gavaged with 0 and 2.5 mg/kg bw DON, 3-ADON, 15-ADON, FX, and NIV in 100 μl PBS, respectively, using a sterile 22 G 1.5′′ disposable feeding tube (Instech Solomon; Plymouth Meeting, PA). This dose was chosen based on previous data from showing significant transient anorectic effects were observed after oral exposure to all of these congeners (Wu et al., 2012). Following 8-ketotrichothenene treatment, mice were then immediately provided two pre-weighted food pellets (≈7 g) and food intake measured at 0.5, 1, 2, 3, 6, and 16 h after toxin exposure.
FIG. 2.
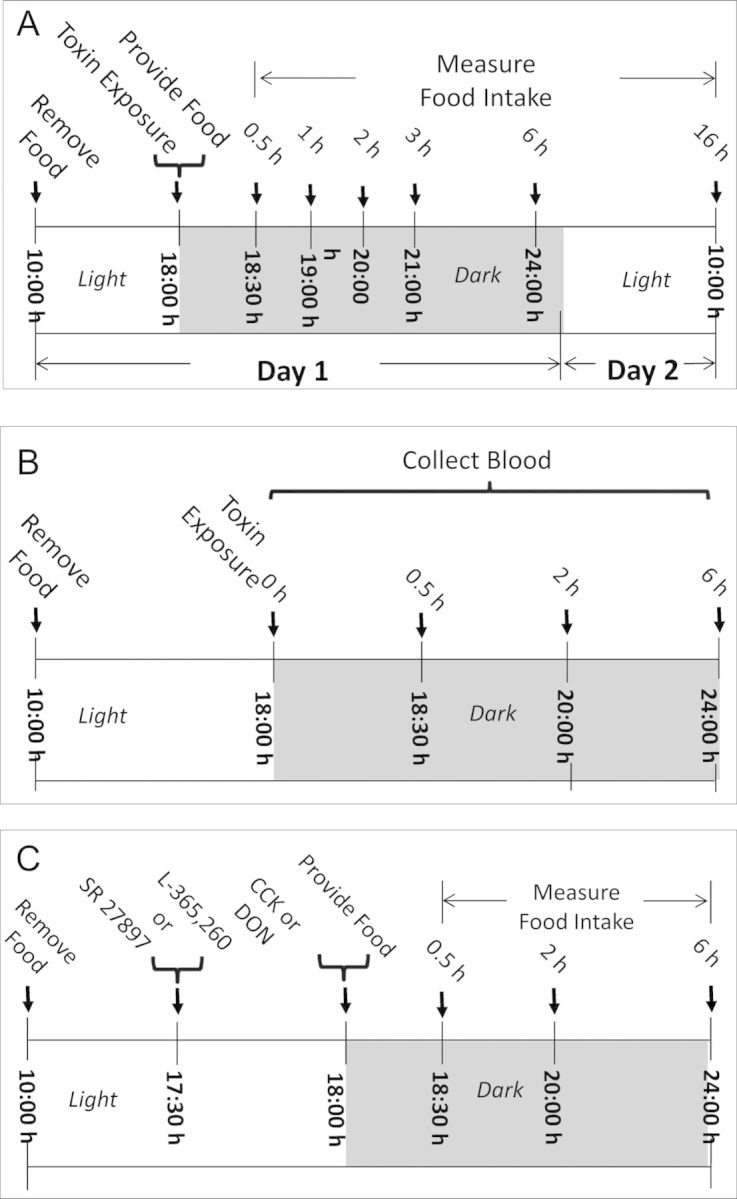
(A) Experimental design for anorexia bioassay in mice. Mice were given oral gavage of 8-ketotrichothecenes immediately before the dark feeding cycle. Food intake was measured at 0.5, 1, 2, 3, 6, and 16 h post administration. (B) Experimental design for 8-ketotrichothecenes-induced plasma CCK and PYY3–36 release in mice. Plasma was analyzed for CCK and PYY3–36 at 0, 0.5, 2, and 6 h post administration. (C) Experimental design for determining the effects of CCK receptor antagonists on CCK- and DON-induced anorexia. Mice were pretreated with SR 27897 or L-365,260 and 30 min later treated with PBS, CCK, or DON immediately before the dark cycle. Food intake was then measured at 0.5, 2, and 6 h post administration.
Measurement of plasma CCK and PYY3–36
Groups of mice (n = 6) were orally gavaged with 0 or 2.5 mg/kg bw DON, 3-ADON, 15-ADON, FX, and NIV in 100 μl PBS, respectively, and cohorts were sacrificed at 0, 0.5, 2, and 6 h after exposure (Fig. 2B). At experiment termination, mice were anesthetized by IP injection with 100 μl 56 mg/ml sodium pentobarbital. Blood was collected with sterile syringes containing 20 μl 1% (vol/vol) EDTA (pH 7.5) from the inferior vena cava and transferred to EDTA-coated tubes. Plasma was separated from blood by centrifugation at 3500 × g for 10 min at 4°C and frozen at −80°C until analysis. Plasma hormones CCK and PYY3–36 were analyzed using enzyme immunoassay kits for CCK (CCK [26–33], nonsulfated; human-, rat-, and mouse-specific) and PYY (PYY [3–36]; mouse-, rat-, porcine-, and canine-specific) (Phoenix Pharmaceuticals, Burlingame, CA).
Effects of CCK receptor antagonists on DON-induced anorexia
The general design for receptor antagonist experiments (Fig. 2C) was identical to anorexia models except for pretreatment with antagonist 30 min before DON exposure. Doses of antagonists were chosen based on the previous demonstrated efficacies in previous in vivo investigations of these receptors (Blandizzi et al., 1999; Cano et al., 2003; Lotti et al., 1986; Poncelet et al., 1993). To establish that CCK1 receptor antagonist SR 27897 and CCK2 receptor antagonist L-365260 could attenuate CCK-induced anorexia, 100 μl of each antagonist at 0, 1, 2.5, and 5 mg/kg bw doses was given by oral gavage. After 30 min, mice were given an IP injection of CCK at 0.01 mg/kg bw in 100 μl. This dose was chosen based on previous study (Flannery et al. 2012). In that study, we found IP injection of CCK at 0.01 mg/kg bw significantly induced anorexia in mouse. Control groups were first gavaged with either vehicle (5% DMSO) or 5 mg/kg bw antagonist (SR 27897 or L-365260), then injected IP with PBS. Food intake was measured at 0.5, 2, and 6 h post-treatment. To ascertain the potential role of CCK in DON-induced anorexia, mice were gavaged with 100 μl of antagonist (SR 27897 or L-365260) at 0, 1, 2.5, and 5 mg/kg bw 30 min before oral exposure to 100 μl of DON at 2.5 mg/kg bw. Controls and food intake measurement times were the same as for the CCK study.
Statistics
Data were plotted and statistically analyzed using SigmaPlot 11 for Windows (Jandel Scientific; San Rafael, CA). Means were considered significantly different at p < 0.05. Food intake at specific time points was analyzed by t-test to determine significant differences between treatment and the respective control. Two-way repeated ANOVA (one-factor) using Holm-Sidak Method was used to analyze significant differences in food consumption as compared with the control over time. A one-way ANOVA using the Student-Newman-Keuls method was used to analyze significant differences between treatment groups in food consumption for CCK1 and CCK2 receptor antagonist study. A two-way ANOVA using Bonferroni t-test was used to assess significant differences in CCK and PYY3–36 concentrations in plasma over time.
RESULTS
When the anorectic effect of oral exposure to DON at 2.5 mg/kg bw was measured, significant reductions in cumulative food intake relative to control were observed in cumulative food consumption at 0.5 h (86%), 1 h (87%), 2 h (85%), 3 h (73%), and 6 h (36%), respectively (Figs. 3 and 4A). From 6 to 16 h, there was a trend toward increased food consumption. After 16 h, no differences being observed between control and DON group. Plasma CCK was significantly increased by DON treatment after 0.5 and 2 h, but recovered to control level by 6 h (Fig. 4B). PYY3–36 was significantly elevated after 0.5 h but returned to control level by 2 h (Fig. 4C). Similar to DON, food intake measurements in mice orally exposed 3-ADON at 2.5 mg/kg bw indicated evoked 82, 74, 72, 54, and 45% reduction in cumulative food intake at 0.5, 1, 2, 3 and 6 h, respectively, with no differences being observed between control and 3-ADON group after 16 h (Figs. 5 and 6A). Plasma CCK was elevated at 0.5 and 2 h, but returned to basal level at 6 h after exposure to 3-ADON (Fig. 6B). Unlike DON, oral 3-ADON exposure had no effect on plasma PYY3–36 (Fig. 6C).
FIG. 3.

Oral exposure to DON impairs cumulative food intake for up to 6 h. Data are mean ± SEM (n = 5/gp) and analyzed using t-test. * indicates significant differences between treatment and the respective control (p < 0.05).
FIG. 4.
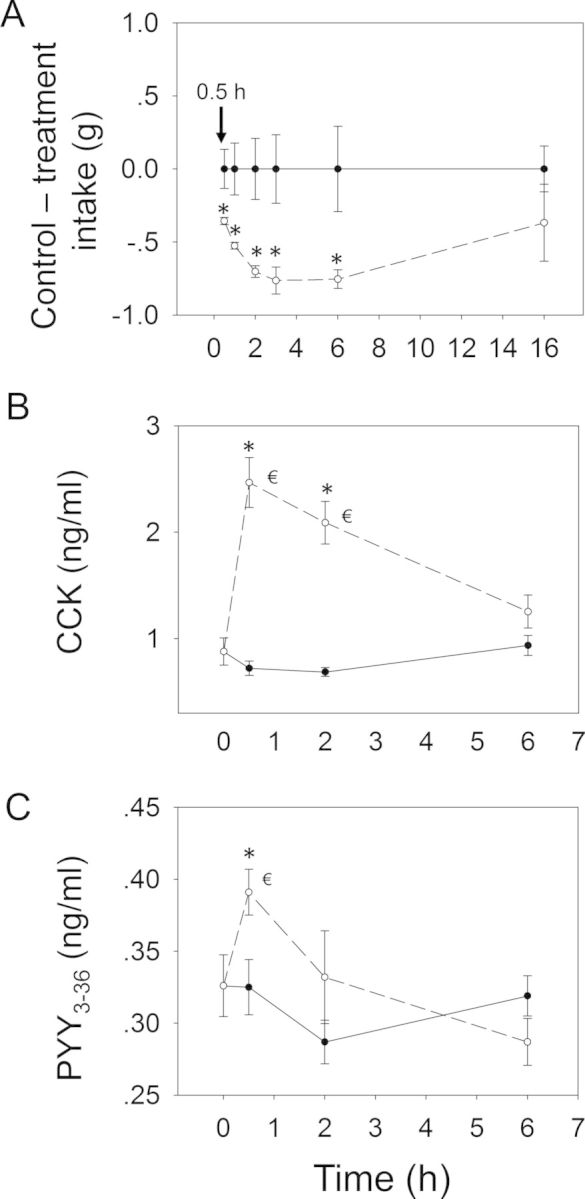
Relationship of DON-induced anorectic response to plasma CCK and PYY3–36. Mice were orally gavaged with either PBS (solid lines) or 2.5 mg/kg bw DON (broken lines). (A) Rapid and transient anorectic effect following oral exposure to DON. Data are mean ± SEM (n = 5/gp). Two-way repeated ANOVA (one factor) using Holm-Sidak Method was used to analyze significant differences in food consumption as compared with the control. * indicates a statistically significant difference in cumulative food consumption relative to the control at specific time point (p < 0.05). Kinetics of DON-induced plasma (B) CCK and (C) PYY3–36 elevation in mice. Data represent mean ± SEM (n = 6/gp). Two-way ANOVA using Bonferroni t-test was used to assess significant differences in kinetics of CCK and PYY3–36 concentrations in plasma as compared with the control. * indicates difference in plasma CCK or PYY3–36 concentration relative to the control at specific time point (p < 0.05) and € indicates difference in plasma CCK or PYY3–36 concentration relative to the 0 h time point (p < 0.05).
FIG. 5.

Oral exposure to 3-ADON impairs cumulative food intake for up to 6 h. Experiment conducted and data analyzed as described in Figure 3 legend.
FIG. 6.
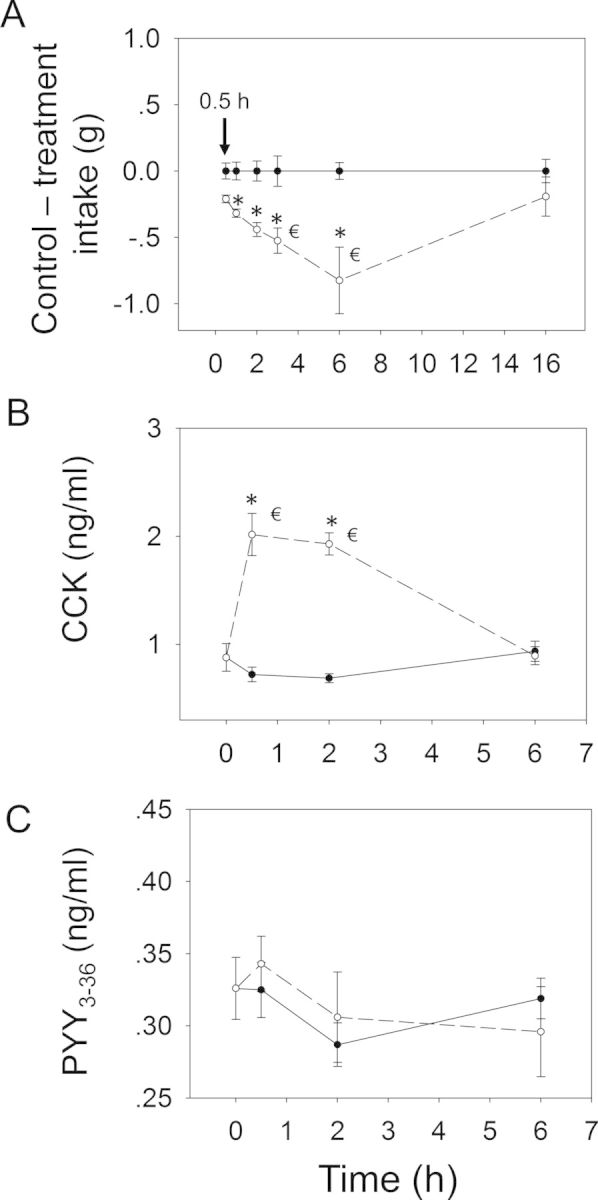
Relationship of 3-ADON-induced (A) anorectic response to plasma (B) CCK and (C) PYY3–36. Mice were orally gavaged with either PBS (solid lines) or 2.5 mg/kg bw DON (broken lines). Experiment conducted and data analyzed as described in Figure 4 legend except that symbol € in panel (A) indicates a significant difference in cumulative food consumption relative to the 0.5 h time point within a given dose (p < 0.05).
Oral exposure to 15-ADON at 2.5 mg/kg bw also caused a similar decreased food consumption in mice during the first 6 h (Figs. 7 and 8A). Significant reduction in cumulative food intake was observed at 0.5 h (94%), 1 h (69%), 2 h (79%), 3 h (64%), 6 h (45%), and 16 h (17%). The rate of food intake by the 15-ADON group paralleled the control mice during 6–16 h, suggesting that anorexia did not occur after 6 h. Exposure to 15-ADON significantly elevated CCK concentrations at 0.5 h, which peaked at 2 h (Fig. 8B). Similar to DON and 3-ADON, plasma CCK returned to basal levels at 6 h. As observed for 3-ADON, plasma PYY3–36 was not affected during the experimental period (Fig. 8C).
FIG. 7.

Oral exposure to 15-ADON impairs cumulative food intake for up to 6 h. Experiment was conducted and data analyzed as described in Figure 3 legend.
FIG. 8.
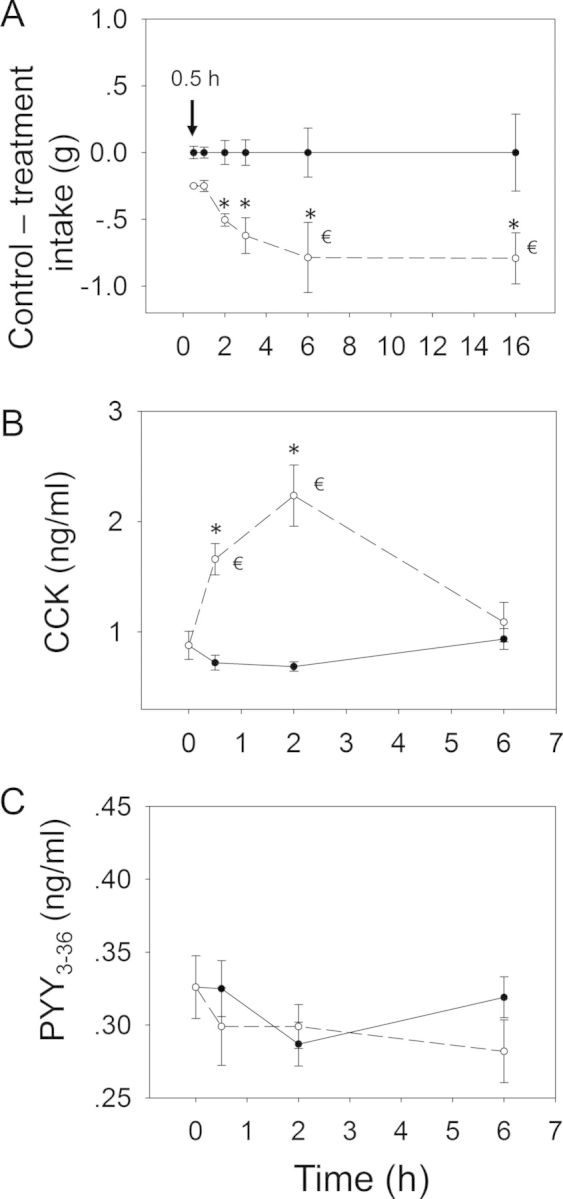
Relationship of 15-ADON-induced (A) anorectic response to plasma (B) CCK and (C) PYY3–36. Mice were orally gavaged with either PBS (solid lines) or 2.5 mg/kg bw DON (broken lines). Experiment was conducted and data analyzed as described in Figures 4 and 6 legends.
Following oral administration with FX at 2.5 mg/kg bw, marked reductions in food intake occurred at 0.5 h (92%), 1 h (72%), 2 h (78%), 3 h (72%), 6 h (71%), and 16 h (39%), respectively (Figs. 9 and 10A). A general trend toward reduced food consumption was observed over the entire 16 h period. Following oral exposure to FX, plasma CCK concentrations were elevated at 0.5 h, peaked at 2 h, and still markedly raised at 6 h (Fig. 10B), suggesting the prolonged elevation of CCK correlated with persistent anorexia. Again, FX had no effect on plasma PYY3–36 concentrations (Fig. 10C).
FIG. 9.

Oral exposure to FX impairs cumulative food intake for up to 16 h. Experiment was conducted and data analyzed as described in Figure 3 legend.
FIG. 10.
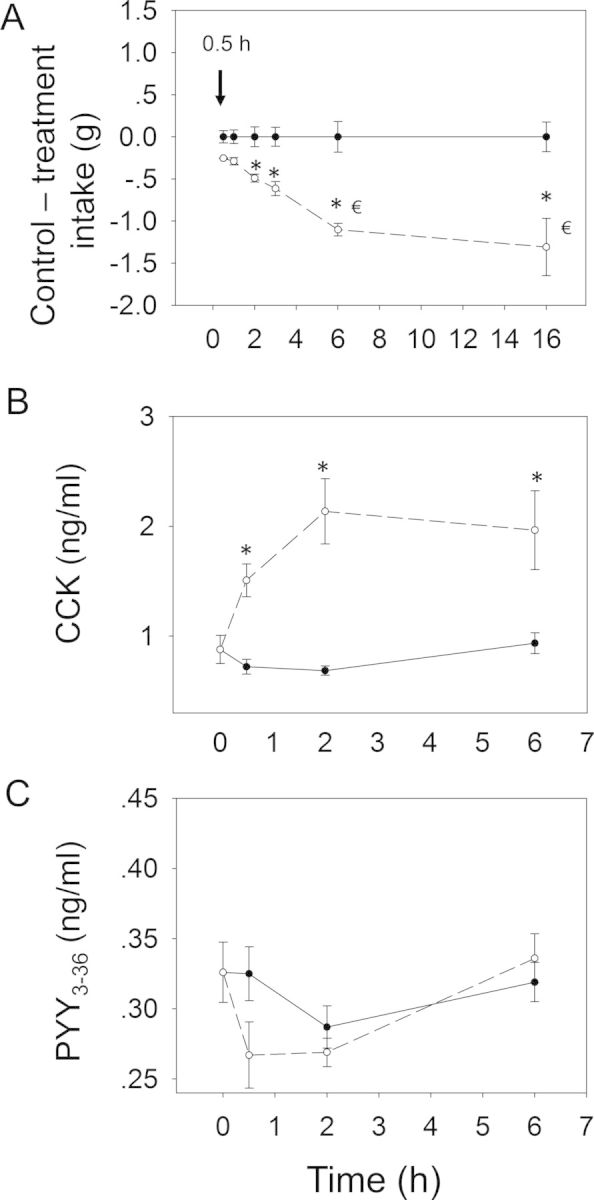
Relationship of FX-induced (A) anorectic response to plasma (B) CCK and (C) PYY3–36. Mice were orally gavaged with either PBS (solid lines) or 2.5 mg/kg bw DON (broken lines). Experiment was conducted and data analyzed as described in Figures 4 and 6 legends.
Contrary to the aforementioned DON congeners, oral gavage with NIV at 2.5 mg/kg bw only modestly decreased food intake during the 16 h experimental period (Figs. 11 and 12A). Significant reduction in cumulative food consumption was observed only at 2 h (40%) and 3 h (38%) after exposure. Although differences between NIV exposure group and control were not evident thereafter, food intake was still depressed. Exposure to NIV significantly raised plasma CCK concentrations only at 2 h (Fig. 12B). Interestingly, moderate elevation of PYY3–36 was observed at 2 and 6 h (Fig. 12C), suggesting that both CCK and PYY3–36 might contribute to NIV-induced anorexia.
FIG. 11.
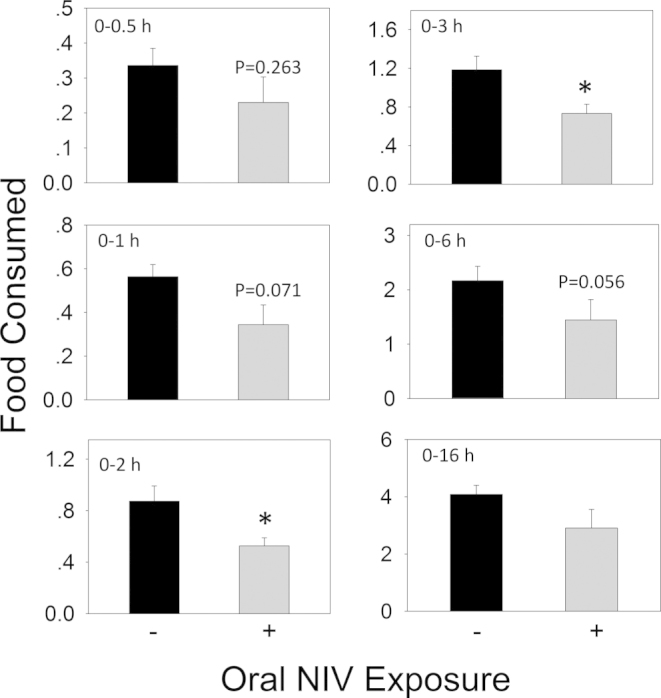
Oral exposure to NIV impairs cumulative food intake for up to 3 h. Experiment was conducted and data analyzed as described in Figure 3 legend.
FIG. 12.
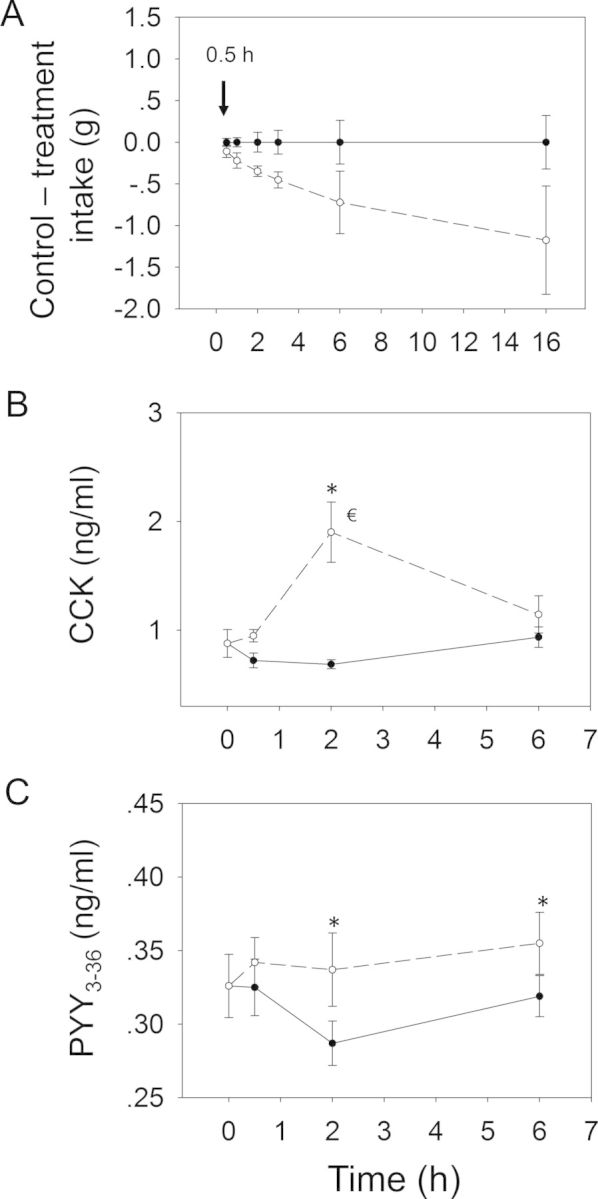
Relationship of NIV-induced (A) anorectic response to plasma (B) CCK and (C) PYY3–36. Mice were orally gavaged with either PBS (solid lines) or 2.5 mg/kg bw DON (broken lines). Experiment was conducted and data analyzed as described in Figures 4 and 6 legends.
The effects of CCK1 and CCK2 receptor antagonists on both CCK- and DON-induced anorexia were evaluated (Fig. 2C). IP administration of CCK at 0.01 mg/kg bw decreased food intake by 76 and 69% relative to the control at 0.5 and 2 h, respectively (Figs. 13A and 14A). After 6 h, no differences were observed when compared with control. These effects were dose-dependently attenuated with the CCK1R antagonist SR 27897 and the CCK2R antagonist L-365,260 at 2 h post-treatment. Exposure to the highest dose (5 mg/kg bw) of either antagonist alone had no effect on food intakes therefore excluding the possibility of functional antagonism. These results suggested that, in this mouse model, CCK evoked anorexia by activating both CCKR1 and CCKR2.
FIG. 13.

CCK1 receptor antagonist SR 27897 inhibits (A) CCK- and (B) DON-induced anorectic response. Mice were pretreated with 0, 1, 2.5, and 5 mg/kg bw SR 27897 and 30 min later treated with PBS, 0.01 mg/kg bw CCK or 2.5 mg/kg bw DON. Food intake was then measured at 0.5, 2, and 6 h post administration. Data are mean ± SEM (n = 6/gp) and analyzed by one way ANOVA using Student-Newman-Keuls Method. Bars without the same letter are significantly different (p < 0.05).
FIG. 14.

CCK2 receptor antagonist L-365,260 inhibits (A) CCK- and (B) DON-induced anorectic response. (A) Mice were pretreated with 0, 1, 2.5, and 5 mg/kg bw L-365,260 and 30 min later treated with PBS, 0.01 mg/kg bw CCK or 2.5 mg/kg bw DON. Food intake was then measured at 0.5, 2, and 6 h post administration. Data are mean ± SEM (n = 6/gp) and were analyzed as described in Figure 13 legend. Bars without the same letter are significantly different (p < 0.05).
Consistent with the earlier experiment (Fig. 3), mice treated with DON exhibited a significant reduction in food intake relative to control (Figs. 13B and 14B). DON-exposed mice that were pretreated with SR 27985 or L-365,260 exhibited a dose-dependent trend of increased food intake. Following pretreatment of SR 27897 with 1 mg/kg bw, mice consumed 19 and 15% more food at 0.5 and 2 h, respectively, than DON alone (Fig. 13B). After 6 h, no differences were observed compared with control. Mice pretreated with 2.5 and 5 mg/kg bw SR 27897 consumed 25 and 61% more food at 0.5 h than DON alone, respectively, and no differences were observed when compared with control after 2 h. Mice receiving 1, 2.5, and 5 mg/kg bw L-365,260 consumed 29, 37, and 54% more food than DON alone at 0.5 h, respectively (Fig. 14B). After 2 h, no differences in cumulative food intake were evident between L-365,260-treated and control mice. Collectively, these strong, dose-dependent antagonistic effects strongly suggest that CCK plays an essential role in the anorexia induction by DON, and operated via two distinct receptors.
DISCUSSION
Although anorexia and concomitant growth suppression have long been a major concern for human and animal food exposure by 8-ketotrichothecenes, the underlying mechanisms for initiating these effects are poorly understood. Gut satiety hormones play an important upstream role in regulating appetite and appear to be important targets of this mycotoxin class. These hormones mediate complex physiological processes that sequentially involve (1) hormone secretion in gastrointestinal (GI) tract after meal consumption, (2) signaling satiation via brain-gut axis, (3) differential expression of anorexigenic and orexigenic signaling factors within the hypothalamus, and (4) altered regulation of appetite and GI tract motility. One of the most studied gut satiety hormones, CCK, has been demonstrated to be an effective short-term, meal-reducing signal in both human and animals (Dockray, 2009; Strader and Woods, 2005). Herein, we demonstrate for the first time that (1) oral gavage with five major 8-ketotrichothecenes caused robust increases in plasma CCK that correlated with anorexia induction and (2) both DON and CCK could mediate anorexia via CCK1R- and CCK2R-dependent mechanisms.
Our results are highly consistent with induction of CCK exocytosis by DON from I cells in the GI tract that normally occurs after meal consumption. When CCK is elicited in intestinal tract, it can act in a paracrine manner, and activate CCK receptors located on the abdominal vagal afferents, which relay to the hypothalamus and eventually cause anorexia (Reidelberger et al., 2004). Another site of paracrine action for CCK is the spinal afferent nerves of the intestinal tract that feed into splanchnic nerve (Brown et al., 2011). In addition to these paracrine effects, there is growing evidence that CCK can act in an endocrine fashion on the central nervous system (CNS), most notably at the nucleus tractus solitarius and dorsal motor nucleus of the vagus (Baptista et al., 2007; Hao et al., 2008; Holmes et al., 2009; Reidelberger et al., 2004; Rogers and Hermann, 2008). It is also important to note that, in addition to being synthesized by I cells in GI tract, CCK is also produced within CNS including various brain regions (Beinfeld et al., 1981; Rehfeld et al., 1985). We previously reported that DON distributes into the brain of mice within 5 min following oral exposure (Pestka et al., 2008). It is possible that 8-ketotrichothecenes directly target the CNS to modulate anorectic neurocircuitry by stimulating CCK secretion in the brain. The finding that DON induces c-Fos in the circumventricular organs of the brain and surrounding structures are consistent with CNS being a direct target of DON (Girardet et al., 2011).
CCK acts via two different receptor subtypes—CCK1R and CCK2R. CCK1R is located throughout the GI tract and CNS, and there is wide consensus that this receptor plays an essential role in the CCK-induced satiety response (Hao et al., 2008; Reidelberger et al., 2004). It is notable that, in a prior mouse study (Flannery et al., 2012), we found robust increases in plasma CCK following IP administration of DON in mice. Although a trend was observed suggesting that DON-induced anorexia was reversed of by prior IP treatment with the CCK1R antagonist devezapide, the differences were not statistically significant. There were two critical differences between the present and aforementioned studies that could explain the conflicting results. First, the CCK1R antagonist used here, SR 27897, is 7 to 10 times more effective at inhibiting CCK-induced anorexia and reduction of gut motility than devazepide in rats (Poncelet et al., 1993). Second, in this study we delivered the SR 27897 via oral gavage whereas in the former study the devezapide was administered IP. Because it is likely that DON induces CCK secretion from I cells in the small intestine and that some of this elicited hormone acts in a paracrine fashion on vagal afferents, oral administration of a CCK1R antagonist might be more efficacious at attaining the inhibitory concentrations necessary for countering CCK-mediated hypophagia.
It is known that CCK2R is expressed in the brain where it integrates signaling of pain, anxiety, sexual behavior, learning, and memory (Crawley and Corwin, 1994). Besides the brain, CCK2R can be expressed in GI tissues (stomach, pancreas, and liver), vagal afferent fibers, adipocytes, adrenal gland, and mononuclear cells (Dufresne et al., 2006; Guilloteau et al., 2006). Studies of the role of CCK2R on appetite and feeding behavior have yielded conflicting results. Subcutaneous administration of L-365,260 increases food intake in partially satiated rats and postpones the onset of satiety (Dourish et al., 1989). In analogous fashion, Intracerebroventricular (ICV) injection of a CCK2R antagonist into rats decreased the satiating potency of the ingested food during the meal and decreased time required for re-initiation of eating during the postprandial inter-meal interval (Dorre and Smith, 1998). Genetic studies also suggest the involvement of CCK2R in the modulation of energy metabolism and food intake. CCK2R receptor-deficient mice exhibit significantly increased food intake compared with wild-type mice (Clerc et al., 2007; Miyasaka et al., 2002; Weiland et al., 2004), elevated body weight (Chen et al., 2006), signs of obesity (Weiland et al., 2004) as well as increased plasma levels of insulin and leptin (Cano et al., 2003; Clerc et al., 2007). In contrast to the above-mentioned studies, ICV injection with specific CCK2R antagonists inhibits food intake in rats (Corp et al., 1997; Frommelt et al., 2013). Also, in another genetic investigation, CCK2R-deficient mice exhibited normal eating behavior and body weight gain (Kopin et al., 1999). Clearly, results of studies of the involvement of CCK2R in eating behavior are not as clear-cut as those of CCK1R and seem to be model-dependent. Nevertheless, the demonstration here that administration of exogenous CCK evoked anorexia and that this could be inhibited by L-365,260 suggests that, in our mouse anorexia model, CCK2R indeed mediated both CCK- and DON-driven feed refusal.
The data presented herein suggest that hypophagia induction by other 8-ketotrichothecenes is also likely to involve CCK. It was notable that DON, 3-ADON, 15-ADON, and FX evoked plasma CCK elevations within 0.5 h, a time that coincided with the initiation of significant food refusal. Interestingly, elevation of CCK induced by NIV did not occur until 2 h when anorexia was first detected. Clearances of DON, FX, and NIV in the mouse follow two-compartment kinetics with an initial rapid phase of elimination (t1/2α) and a slower terminal elimination phase (t1/2β) (Azcona-Olivera et al., 1995; Poapolathep et al., 2003). Previous studies indicate t1/2α = 0.36 or 0.29 h for DON and 0.878 h for FX, respectively (Azcona-Olivera et al., 1995; Pestka et al., 2008; Poapolathep et al., 2003). However, a longer initial phase of elimination was observed for NIV (t1/2α = 2.5 h) (Poapolathep et al., 2003). The possible reason for delayed CCK elevation and anorectic effect might be the slower absorption of NIV by oral exposure. Except for FX-treated group, plasma CCK returned to basal level after 6 h, and food intake recovered at 16 h after exposure. Consistent with CCK's satiety-inducing effects, FX caused persistent anorexia lasting more than 16 h, which has also been observed previously (Wu et al., 2012). FX's longer elimination half-life might explain the prolonged CCK elevation and anorectic effects observed here.
Unlike CCK, our results indicated that the satiety hormone PYY3–36 was not robustly elevated following oral exposure to 8-ketotrichothecenes. Although we have reported DON causes anorexia following IP exposure by inducing PYY3–36 secretion (Flannery et al., 2012), we found here that CCK elevation was more consistent with anorexia induction by DON and other 8-ketotrichothecenes following oral gavage. The possible reason for differences in secretion of these satiety hormones following IP versus oral gavage might result from the different relative distributions of CCK and PYY3–36 secreting cells in gut. Most CCK-producing I-cells are located in the upper GI tract including duodenal and jejunal mucosa (Polak et al., 1975), whereas PYY3–36 secreting L-cells are particularly numerous in the lower GI tract including the distal ileum and colon, with only a few cells presented in the upper GI tract (Hörsten et al., 2004). It might be speculated that following oral gavage exposure toxins can reach the upper GI tract to induce CCK release, but are absorbed before reaching the lower GI tract and causing secretion of PYY3–36. IP exposure might evoke uniform DON concentrations in the small and large intestines from the blood, activating exocytosis of both CCK and PYY.
It is important to note that when we previously compared exposures to DON (2.5 mg/kg bw) by the IP route to the orolingual route, we observed nearly identical anorectic effects as well as similarly robust PYY and CCK responses (Flannery et al., 2012). This finding could possibly relate to DON's capacity to be rapidly absorbed and distributed by either of these routes of exposure as compared with oral gavage as used in the present study. There are several inherent differences between orolingual and oral gavage exposure routes. It is likely that much the DON delivered by the orolingual route is rapidly absorbed sublingually via the reticulated vein that lies underneath the oral mucosa, and transported through the facial veins, internal jugular vein, and braciocephalic vein and, finally, then drained into systemic circulation (Nibha and Pancholi, 2012). Another difference is the sublingual route would avoid first-pass metabolism that is encountered after oral gavage. The picture is further complicated by the recent discovery that PYY is also synthesized by taste cells in the taste buds and be detectable in saliva (Acosta et al., 2011). How this would affect plasma PYY is unknown. It has also been demonstrated that orally administered PYY can induce anorexia via metabolic circuit associated with Y2R-positive cells in the oral cavity and extending through brainstem nuclei into hypothalamic satiety centers (Hurtado et al., 2013). Accordingly, when an experimental animal consumes a DON-contaminated food, it will be exposed to the toxin sublingually for a short time during mastication but upon swallowing, the compound would will enter the GI tract which is best mimicked by gavage exposure. In the natural setting, DON is likely taken up both sublingually and intestinally evoking both CCK and PYY elevation. Thus, future studies should focus on the effects of consuming DON-contaminated food on satiety hormone levels in plasma and saliva as well as their functional roles in hypophagia.
To summarize, the results presented herein indicate that satiety hormone CCK was robustly induced upon oral administration of 8-ketotrichothecenes, whereas PYY3–36 was not. Furthermore, CCK elevation was consistent with the induction of food refusal by these congeners. From public and veterinary health perspectives, studies such as this will improve our understanding of how 8-ketotrichothecenes adversely affect humans and animals. Future investigations should focus on elucidating how and where 8-ketotrichothecenes activate exocytosis of CCK and other satiety hormones in the intestinal tract and CNS as well as how these hormones resulted in appetite signaling via the peripheral and CNSs.
FUNDING
USDA NIFA Award (2011–0635); USDA Wheat and Barley SCAB Initiative Award (59-0206-9-058); National Institutes of Health (Public Health Service Grant ES03553).
Acknowledgments
We would like to acknowledge the assistance of Felicia Wu, Erica Clark, Melissa A. Bates, and Mary Rosner.
Footnotes
These authors made an equal contribution to this manuscript.
REFERENCES
- Acosta A., Hurtado M. D., Gorbatyuk O., La Sala M., Duncan D., Aslanidi G., Campbell-Thompson M., Zhang L., Herzog H., Voutetakis A., et al. Salivary PYY: A putative bypass to satiety. PloS one. 2011;6:e26137. doi: 10.1371/journal.pone.0026137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcona-Olivera J. I., Ouyang Y., Murtha J., Chu F. S., Pestka J. J. Induction of cytokine mRNAs in mice after oral exposure to the trichothecene vomitoxin (deoxynivalenol): Relationship to toxin distribution and protein synthesis inhibition. Toxicol. Appl. Pharmacol. 1995;133:109–120. doi: 10.1006/taap.1995.1132. [DOI] [PubMed] [Google Scholar]
- Baptista V., Browning K. N., Travagli R. A. Effects of cholecystokinin-8s in the nucleus tractus solitarius of vagally deafferented rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R1092–R1100. doi: 10.1152/ajpregu.00517.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham R., Le Roux C., Cohen M., Park A., Ellis S., Patterson M., Frost G., Ghatei M., Bloom S. Pancreatic polypeptide reduces appetite and food intake in humans. J. Clin. Endocrin. Metab. 2003a;88:3989–3992. doi: 10.1210/jc.2003-030630. [DOI] [PubMed] [Google Scholar]
- Batterham R. L., Cohen M. A., Ellis S. M., Le Roux C. W., Withers D. J., Frost G. S., Ghatei M. A., Bloom S. R. Inhibition of food intake in obese subjects by peptide YY3–36. N. Engl. J. Med. 2003b;349:941–948. doi: 10.1056/NEJMoa030204. [DOI] [PubMed] [Google Scholar]
- Batterham R. L., Cowley M. A., Small C. J., Herzog H., Cohen M. A., Dakin C. L., Wren A. M., Brynes A. E., Low M. J., Ghatei M. A. Gut hormone PYY3–36 physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- Beinfeld M., Meyer D., Brownstein M. Cholecystokinin in the central nervous system. Peptides. 1981;2:77–79. doi: 10.1016/0196-9781(81)90015-2. [DOI] [PubMed] [Google Scholar]
- Blandizzi C., Lazzeri G., Colucci R., Carignani D., Tognetti M., Baschiera F., Del Tacca M. CCK1 and CCK2 receptors regulate gastric pepsinogen secretion. Eur. J. Pharmacol. 1999;373:71–84. doi: 10.1016/s0014-2999(99)00212-5. [DOI] [PubMed] [Google Scholar]
- Brown T. A., Washington M. C., Metcalf S. A., Sayegh A. I. The feeding responses evoked by cholecystokinin are mediated by vagus and splanchnic nerves. Peptides. 2011;32:1581–1586. doi: 10.1016/j.peptides.2011.06.024. [DOI] [PubMed] [Google Scholar]
- Cano V., Ezquerra L., Ramos M. P., Ruiz-Gayo M. Regulation of leptin distribution between plasma and cerebrospinal fluid by cholecystokinin receptors. Brit. J. Pharmacol. 2003;140:647–652. doi: 10.1038/sj.bjp.0705477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis B., Pinnock S., Coll A., Carter R., Dickson S., O'rahilly S. Acute effects of PYY3–36 on food intake and hypothalamic neuropeptide expression in the mouse. Biochem. Biophys. Res. Com. 2003;311:915–919. doi: 10.1016/j.bbrc.2003.10.089. [DOI] [PubMed] [Google Scholar]
- Chen H., Kent S., Morris M. J. Is the CCK2 receptor essential for normal regulation of body weight and adiposity? Eur. J. Neurosci. 2006;24:1427–1433. doi: 10.1111/j.1460-9568.2006.05016.x. [DOI] [PubMed] [Google Scholar]
- Clerc P., Coll Constans M. G., Lulka H., Broussaud S., Guigne C., Leung-Theung-Long S., Perrin C., Knauf C., Carpene C., Penicaud L., et al. Involvement of cholecystokinin 2 receptor in food intake regulation: Hyperphagia and increased fat deposition in cholecystokinin 2 receptor-deficient mice. Endocrinology. 2007;148:1039–1049. doi: 10.1210/en.2006-1064. [DOI] [PubMed] [Google Scholar]
- Corp E. S., Curcio M., Gibbs J., Smith G. P. The effect of centrally administered CCK-receptor antagonists on food intake in rats. Physiol. Behav. 1997;61:823–827. doi: 10.1016/s0031-9384(96)00561-6. [DOI] [PubMed] [Google Scholar]
- Crawley J. N., Corwin R. L. Biological actions of cholecystokinin. Peptides. 1994;15:731–755. doi: 10.1016/0196-9781(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Della-Fera M. A., Baile C. A. CCK-octapeptide injected in CSF decreases meal size and daily food intake in sheep. Peptides. 1980;1:51–54. doi: 10.1016/0196-9781(80)90035-2. [DOI] [PubMed] [Google Scholar]
- Dockray G. J. Cholecystokinin and gut-brain signalling. Regul. Pept. 2009;155:6–10. doi: 10.1016/j.regpep.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Dorre D., Smith G. P. Cholecystokinin(B) receptor antagonist increases food intake in rats. Physiol. Behav. 1998;65:11–14. doi: 10.1016/s0031-9384(98)00080-8. [DOI] [PubMed] [Google Scholar]
- Dourish C. T., Rycroft W., Iversen S. D. Postponement of satiety by blockade of brain cholecystokinin (CCK-B) receptors. Science. 1989;245:1509–1511. doi: 10.1126/science.2781294. [DOI] [PubMed] [Google Scholar]
- Dufresne M., Seva C., Fourmy D. Cholecystokinin and gastrin receptors. Physiol. Rev. 2006;86:805–847. doi: 10.1152/physrev.00014.2005. [DOI] [PubMed] [Google Scholar]
- Ebenezer I., De La Riva C., Baldwin B. Effects of the CCK receptor antagonist MK-329 on food intake in pigs. Physiol. Behav. 1990;47:145–148. doi: 10.1016/0031-9384(90)90053-7. [DOI] [PubMed] [Google Scholar]
- Flannery B. M., Amuzie C. J., Pestka J. J. Evaluation of insulin-like growth factor acid-labile subunit as a potential biomarker of effect for deoxynivalenol-induced proinflammatory cytokine expression. Toxicology. 2013;304:192–198. doi: 10.1016/j.tox.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery B. M., Clark E. S., Pestka J. J. Anorexia induction by the trichothecene deoxynivalenol (vomitoxin) is mediated by the release of the gut satiety hormone peptide yy. Toxicol. Sci. 2012;130:289–297. doi: 10.1093/toxsci/kfs255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery B. M., Wu W., Pestka J. J. Characterization of deoxynivalenol-induced anorexia using mouse bioassay. Food Chem. Toxicol. 2011;49:1863–1869. doi: 10.1016/j.fct.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsell J. H., Witt M. F., Tai J. H., Jensen R., Pestka J. J. Effects of 8-week exposure of the B6C3F1 mouse to dietary deoxynivalenol (vomitoxin) and zearalenone. Food Chem. Toxicol. 1986;24:213–219. doi: 10.1016/0278-6915(86)90231-0. [DOI] [PubMed] [Google Scholar]
- Frommelt L., Lembke V., Hofmann T., Goebel-Stengel M., Monnikes H., Wiedenmann B., Klapp B. F., Stengel A., Kobelt P. The CCKB antagonist CI988 reduces food intake in fasted rats via a dopamine mediated pathway. Peptides. 2013;39:111–118. doi: 10.1016/j.peptides.2012.11.012. [DOI] [PubMed] [Google Scholar]
- Girardet C., Bonnet M. S., Jdir R., Sadoud M., Thirion S., Tardivel C., Roux J., Lebrun B., Wanaverbecq N., Mounien L. The food-contaminant deoxynivalenol modifies eating by targeting anorexigenic neurocircuitry. PloS one. 2011;6:e26134. doi: 10.1371/journal.pone.0026134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilloteau P., Le Meuth-Metzinger V., Morisset J., Zabielski R. Gastrin, cholecystokinin and gastrointestinal tract functions in mammals. Nutr. Res. Rev. 2006;19:254–283. doi: 10.1017/S0954422407334082. [DOI] [PubMed] [Google Scholar]
- Hao S., Sternini C., Raybould H. E. Role of CCK1 and Y2 receptors in activation of hindbrain neurons induced by intragastric administration of bitter taste receptor ligands. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;294:R33–R38. doi: 10.1152/ajpregu.00675.2007. [DOI] [PubMed] [Google Scholar]
- Holmes G. M., Tong M., Travagli R. A. Effects of brain stem cholecystokinin-8s on gastric tone and esophageal-gastric reflex. Am. J. Physiol. Gastro. Liver Physiol. 2009;296:G621–G631. doi: 10.1152/ajpgi.90567.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörsten S., Hoffmann T., Alfalah M., Wrann C., Karl T., Pabst R., Bedoui S. PP, PYY and NPY: Synthesis, storage, release and degradation. Neuropept. Y Relat. Pept. 2004;162:23–44. [Google Scholar]
- Hurtado M. D., Sergeyev V. G., Acosta A., Spegele M., La Sala M., Waler N. J., Chiriboga-Hurtado J., Currlin S. W., Herzog H., Dotson C. D., et al. Salivary peptide tyrosine-tyrosine 3–36 modulates ingestive behavior without inducing taste aversion. J. Neurosci. (2013);33:18368–18380. doi: 10.1523/JNEUROSCI.1064-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson F., Armstrong C., Nera E., Truelove J., Fernie S., Scott P., Stapley R., Hayward S., Gunner S. Chronic feeding study of deoxynivalenol in B6C3F1 male and female mice. Teratog. Carcinog. Mutagen. 1995;15:283–306. doi: 10.1002/(SICI)1520-6866(1996)15:6<283::AID-TCM5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- JECFA. Deoxynivalenol. WHO Technical Report Series No. 959. Geneva, Switzerland: WHO Press; 2011. Evaluation of Certain Contaminants in Food: 72nd Report of the Joint FAO/WHO Expert Committee on Food Additives; pp. 37–48. [Google Scholar]
- Kohno D., Nakata M., Maejima Y., Shimizu H., Sedbazar U., Yoshida N., Dezaki K., Onaka T., Mori M., Yada T. Nesfatin-1 neurons in paraventricular and supraoptic nuclei of the rat hypothalamus coexpress oxytocin and vasopressin and are activated by refeeding. Endocrinology. 2008;149:1295–1301. doi: 10.1210/en.2007-1276. [DOI] [PubMed] [Google Scholar]
- Kopin A. S., Mathes W. F., McBride E. W., Nguyen M., Al-Haider W., Schmitz F., Bonner-Weir S., Kanarek R., Beinborn M. The cholecystokinin-A receptor mediates inhibition of food intake yet is not essential for the maintenance of body weight. J. Clin. Invest. 1999;103:383–391. doi: 10.1172/JCI4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti V. J., Chang R. S., Kling P. J., Cerino D. J. Evidence that cholecystokinin octapeptide (CCK-8) acts as a potent, full agonist on gastrin receptors for acid secretion in the isolated mouse stomach: Lack of antagonism by the specific CCK antagonist asperlicin. Digestion. 1986;35:170–174. doi: 10.1159/000199363. [DOI] [PubMed] [Google Scholar]
- Miyasaka K., Ichikawa M., Ohta M., Kanai S., Yoshida Y., Masuda M., Nagata A., Matsui S., Noda T., Takiguchi S., et al. Energy metabolism and turnover are increased in mice lacking the cholecystokinin-B receptor. J. Nutri. 2002;132:739–741. doi: 10.1093/jn/132.4.739. [DOI] [PubMed] [Google Scholar]
- Nibha K. P., Pancholi S. S. An overview on: Sublingual route for systemic drug delivery. Int. J. Res. Pharmaceut. Biomed. Sci. 2012;3:913–923. [Google Scholar]
- Pestka J. J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010a;84:663–679. doi: 10.1007/s00204-010-0579-8. [DOI] [PubMed] [Google Scholar]
- Pestka J. J. Toxicological mechanisms and potential health effects of deoxynivalenol and nivalenol. World Mycotoxin J. 2010b;3:323–347. [Google Scholar]
- Pestka J. J., Islam Z., Amuzie C. J. Immunochemical assessment of deoxynivalenol tissue distribution following oral exposure in the mouse. Toxicol. Letts. 2008;178:83–87. doi: 10.1016/j.toxlet.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poapolathep A., Sugita-Konishi Y., Doi K., Kumagai S. The fates of trichothecene mycotoxins, nivalenol and fusarenon-X, in mice. Toxicon. 2003;41:1047–1054. doi: 10.1016/s0041-0101(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Polak J. M., Bloom S., Rayford P., Pearse A., Buchan A., Thompson J. Identification of cholecystokinin-secreting cells. The Lancet. 1975;306:1016–1018. doi: 10.1016/s0140-6736(75)90297-4. [DOI] [PubMed] [Google Scholar]
- Poncelet M., Arnone M., Heaulme M., Gonalons N., Gueudet C., Santucci V., Thurneyssen O., Keane P., Gully D., Le Fur G., et al. Neurobehavioural effects of SR 27897, a selective cholecystokinin type A (CCK-A) receptor antagonist. Naunyn Schmiedebergs Arch. Pharmacol. 1993;348:102–107. doi: 10.1007/BF00168544. [DOI] [PubMed] [Google Scholar]
- Rehfeld J. F., Hansen H. F., Marley P. D., Stengaardpedersen K. Molecular-forms of cholecystokinin in the brain and the relationship to neuronal gastrins. Ann. N.Y. Acad. Sci. 1985;448:11–23. doi: 10.1111/j.1749-6632.1985.tb29902.x. [DOI] [PubMed] [Google Scholar]
- Reidelberger R. D., Hernandez J., Fritzsch B., Hulce M. Abdominal vagal mediation of the satiety effects of CCK in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;286:R1005–R1012. doi: 10.1152/ajpregu.00646.2003. [DOI] [PubMed] [Google Scholar]
- Rogers R. C., Hermann G. E. Mechanisms of action of CCK to activate central vagal afferent terminals. Peptides. 2008;29:1716–1725. doi: 10.1016/j.peptides.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M. W. Central nervous system regulation of food intake. Obesity. 2006;14:1S–8S. doi: 10.1038/oby.2006.275. [DOI] [PubMed] [Google Scholar]
- Steinert R. E., Feinle-Bisset C., Geary N., Beglinger C. Secretion of gastrointestinal hormones and eating control. J. Anim. Sci. 2013;91:1963–1973. doi: 10.2527/jas.2012-6022. [DOI] [PubMed] [Google Scholar]
- Strader A. D., Woods S. C. Gastrointestinal hormones and food intake. Gastroenterology. 2005;128:175–191. doi: 10.1053/j.gastro.2004.10.043. [DOI] [PubMed] [Google Scholar]
- Weiland T. J., Voudouris N. J., Kent S. The role of CCK2 receptors in energy homeostasis: Insights from the CCK2 receptor-deficient mouse. Physiol. Behav. 2004;82:471–476. doi: 10.1016/j.physbeh.2004.04.065. [DOI] [PubMed] [Google Scholar]
- Wu W., Flannery B. M., Sugita-Konishi Y., Watanabe M., Zhang H., Pestka J. J. Comparison of murine anorectic responses to the 8-ketotrichothecenes 3-acetyldeoxynivalenol, 15-acetyldeoxynivalenol, fusarenon X and nivalenol. Food Chem. Toxicol. 2012;50:2056–2061. doi: 10.1016/j.fct.2012.03.055. [DOI] [PubMed] [Google Scholar]
- Zhang D.-M., Bula W., Stellar E. Brain cholecystokinin as a satiety peptide. Physiol. Behav. 1986;36:1183–1186. doi: 10.1016/0031-9384(86)90498-1. [DOI] [PubMed] [Google Scholar]