

Significance
Twister is a small self-cleaving ribozyme similar in size to the hammerhead ribozyme but uses an orthogonal fold for a similar catalytic rate constant. However, the mechanistic source of the catalytic rate increase generated by twister was unknown. We present crystal structures of twister from Orzyza sativa as well as a twister sequence from an organism that has not been cultured in isolation and identify RNA nucleotides that are vital for self-cleavage, suggest their catalytic roles, and update twister’s conserved secondary structure model.
Keywords: X-ray crystallography, RNA structure, weak derivative phasing, samarium derivatives, cesium derivatives
Abstract
Twister is a recently discovered RNA motif that is estimated to have one of the fastest known catalytic rates of any naturally occurring small self-cleaving ribozyme. We determined the 4.1-Å resolution crystal structure of a twister sequence from an organism that has not been cultured in isolation, and it shows an ordered scissile phosphate and nucleotide 5′ to the cleavage site. A second crystal structure of twister from Orzyza sativa determined at 3.1-Å resolution exhibits a disordered scissile phosphate and nucleotide 5′ to the cleavage site. The core of twister is stabilized by base pairing, a large network of stacking interactions, and two pseudoknots. We observe three nucleotides that appear to mediate catalysis: a guanosine that we propose deprotonates the 2′-hydroxyl of the nucleotide 5′ to the cleavage site and a conserved adenosine. We suggest the adenosine neutralizes the negative charge on a nonbridging phosphate oxygen atom at the cleavage site. The active site also positions the labile linkage for in-line nucleophilic attack, and thus twister appears to simultaneously use three strategies proposed for small self-cleaving ribozymes. The twister crystal structures (i) show its global structure, (ii) demonstrate the significance of the double pseudoknot fold, (iii) provide a possible hypothesis for enhanced catalysis, and (iv) illuminate the roles of all 10 highly conserved nucleotides of twister that participate in the formation of its small and stable catalytic pocket.
The twister RNA motif was identified by bioinformatic searches and then validated biochemically to be a small self-cleaving ribozyme (1). This recently discovered class of ribozymes is called twister because its conserved secondary structure resembles the ancient Egyptian hieroglyph “twisted flax.” Representatives of the twister ribozyme class are found in all domains of life, but its biological role has yet to be determined. In addition to twister, the small self-cleaving ribozyme family includes the hammerhead, hairpin, hepatitis delta virus (HDV), Varkud satellite (VS), and glmS ribozymes (the glmS ribozyme is upsteam of the the glmS gene that codes for the enzyme that catalyzes glucosamine-6-phosphate production) (1–6).
The small self-cleaving ribozyme family can be split into two groups based on whether their active site is formed by an irregular helix (hammerhead, hairpin, and VS) or a double pseudoknot (PK) structure (HDV and glmS) (7). The structures of HDV and glmS are known, whereas twister was predicted from representative sequences to use two PKs to form its active site. It was expected that twister would be smaller in size than either HDV or glmS and more comparable in size and complexity to hammerhead (1).
The self-cleavage rate constant of twister is estimated to be as rapid as or slightly more rapid than the hammerhead ribozyme. The estimated rate constants (kobs) for twister is 1,000 per minute, and the experimental kobs for hammerhead is 870 per minute (1, 8). These two ribozymes are ∼100- to 500-fold faster than other small self-cleaving ribozymes (2–10 per minute under the similar in vitro reaction conditions) (1, 8–10). Twister constructs previously tested exhibited a maximum cleavage rate at 1 mM Mg2+ and pH 7.4 (1). However, twister does not require magnesium or other divalent cations for catalysis; thus, magnesium is important only for structure formation (1).
Biochemical in-line probing experiments and bioinformatics suggested that the consensus secondary structure of twister contains three to six stems, of which P1, P2, and P4 are required, whereas P0, P3, and P5 are optional; that the RNA can be circularly permutated; and that it contains internal and terminal loops that form two PKs (1, 11). Mutations in any of the highly conserved nucleotides or mutations that disrupt the P1 stem, P2 stem, P4 stem, or the two PKs significantly decrease the catalytic rate (1). Other mutational analysis indicated that several nucleotides are important for self-cleavage, but these nucleotides were not expected to be involved in canonical Watson–Crick (WC) base pairing or displayed any covariation (1).
All small self-cleaving ribozymes undergo a specific internal transesterification reaction in which the ribose 2′-oxygen, phosphorus, and 5′-ribose oxygen are aligned for an SN2-like reaction, yielding products with a 2′,3′-cyclic phosphate and a 5′-hydroxyl termini. This single-step reaction is analogous to the reaction catalyzed by RNase A, except that the protein enzyme undergoes a second step to remove the cyclic phosphate (12). There are four general strategies contributing to RNA self-cleavage via internal phosphoester transfer: (i) orientation of the reactive atoms for in-line nucleophilic attack; (ii) neutralization of the negative charge on the nonbridging oxygen atoms of the cleavage site phosphate; (iii) deprotonation of the 2′ oxygen nucleophile; and (iv) neutralization of the developing negative charge on the 5′ leaving group (9). First, all small self-cleaving ribozymes likely use the first strategy for phosphoester transfer, including twister (1, 9). Second, a part of the rate constant enhancement of twister is likely due to the base catalysis because the rate constant has a pH dependency suggesting that the shifts the pKA of the 2′-hydroxyl group at the cleavage site (1, 9). However, it is unknown whether twister uses the transition-state stabilization and/or general acid strategies (1, 9).
Here, we present the 4.1-Å resolution crystal structure of a twister ribozyme sequence from an organism found in the environment that has not been isolated of likely of prokaryotic origin (twister A) and the 3.1-Å resolution X-ray crystal structure of an Orzyza sativa (twister B) twister ribozyme in which the scissile phosphate and nucleotide 5′ to the cleavage site are ordered in the first and disordered in the second. These two crystal structures provide insights into twister’s catalytic mechanism and structural motifs used for formation of its active site. We identify groups that are involved in general base catalysis, transition state stabilization, and provide information about tertiary interactions that form the active site of twister. These structures show variations about how an RNA achieves site-specific self-cleavage and suggest a physical basis for how twister is able to rapidly self-cleave.
Results
Determination of Twister Structure.
To obtain the 3D structure of twister and the structural basis for its large catalytic rate enhancement, we solved X-ray crystal structures of a precatalytic state of twister at 4.1- and 3.1-Å resolution with and without the scissile phosphate and nucleotide 5′ at the cleavage site being ordered (Fig. 1 and Tables S1 and S2). Chemical modification of the catalytically active 2′-hydroxyl has previously been used to trap self-cleaving ribozymes in a precatalytic state (13, 14). To crystallize twister, we created bimolecular constructs consisting of a synthetic “substrate strand” containing the cleavage site with a 2′-deoxy substitution to trap a precatalysis state and an appropriate “enzyme strand” (1). Twister constructs were selected for those that contain the optional P3 stem, have a 5′-guanosine present to facilitate in vitro transcription by T7 RNA polymerase (or constructed with a 5′-guanosine), and contain endogenous G–U base pairs to use a cation-binding site for iridium (III) hexamine trichloride or cesium sulfate to make heavy-atom derivatives and solve the phase problem (Fig. S1) (15). Additionally, five adenosine nucleotides were designed in place of the P5 stem region.
Fig. 1.
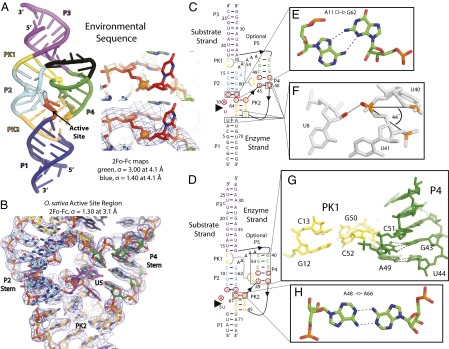
Overall twister structure, electron density map of the O. sativa twister’s active-site region secondary structures of the twister ribozyme constructs crystallized, and unexpected base-pairing and tertiary interactions. (A) The crystal structure of twister A. The P1 stem is dark blue, P2e stem is cyan, the P3 stem is magenta, the P4e stem is green, the five adenosines substituting for the optional P5 stem are black, PK1 is yellow, PK2e is orange, and the active site is red. The substrate strand is outlined in black. The electron density around the scissile phosphate is contoured at 3.0 σ to illustrate the phosphate positions and contoured at 1.4 σ for the entire region at 4.1-Å resolution. (B) 2FO − FC electron density map centered on the active site region of the O. sativa twister crystal structure and contoured at 1.3 σ at 3.1-Å resolution. Coloring for the carbon atoms is the same as in A. The nucleotide 5′ to the cleavage site is shown in magenta. The secondary structure is constructed upon the tertiary interactions revealed in the crystal structures of the (C) the environmental sequence and (D) O. sativa sequence. The substrate strand had a 2′-deoxy substitution to prevent cleavage between the nucleotides indicated with the large arrowhead. The red circles indicate highly conserved nucleotides based on bioinformatics analysis (1). Leontis and Westhof symbols indicate non-WC base pairs. The interaction of PK1 is indicated by a thick gray line. The black lines with internal arrows show strand connectivity. The P1 stem for O. sativa is modeled after the environmental sequence’s P1 stem because this part formed a biologically irrelevant intermolecular dimer that was the result of a crystal artifact. (E) The position of catalytically active G62 is held in position by A11 by forming a trans interaction between the sugar edge of G62 and the Hoogsteen-edge of A11 in the environmental sequence. (F) A sharp bend (44°) occurs in the phosphodiester backbone between the phosphates of U40, U41, and A42 in twister from the environmental sequence and is required to position U40 in the P4 stem and U41 in the PK2. This interaction is stabilized by a hydrogen bond (solid gray line) between a nonbridging oxygen atom of U41 (red) and the 2′-hydroxyl of U8 (red). (G) An unexpected tertiary interaction is seen around the predicted P4 stem shown from O. sativa twister. (H) A trans Watson–Watson base pair interaction was found between nucleotides A48 and A66 in the O. sativa crystal structure.
Overall Structure.
Our twister structures have a similar size and shape as the hammerhead ribozyme (Fig. S2) (16). Both twister ribozymes studied here belong to type P3, in which the optional P3 stem is present but not the optional P0 or P5 stems. The ordered and rigid structure is made up of four helical stems (P1, P2, P3, and P4), two PKs (PK1 and PK2), and a network of stacking interactions.
The substrate strand, in complex with the enzyme strand, base pairs with the enzyme strand and adopts a single-stranded A-form helical conformation nearly along the entire structure (Fig. S3). The two nucleotides of the substrate strand that make up the active site deviate from the A-form helix and are discussed below. Details of data processing and structure refinement statistics along experimental electron density maps at intermediate steps of structure determination are described in Tables S1–S3.
The Conserved P1, P2, and P4 Stems.
The P1 stem forms the platform of the active site when the substrate strand base pairs with the enzyme strand. Sequence analysis of twister revealed that the P1 stem is highly variable in length (1). The P1 stem is eight base pairs and forms a stable, continuous helix in twister A. However, the shorter four-base pair P1 stem designed in the twister B resulted in crystals containing an intermolecular dimer artifact (Fig. S4B). For this reason, the twister A structure is likely the biological-relevant structure poised for catalysis with an ordered scissile phosphate and nucleotide 5′ to the cleavage site; whereas the twister B structure does have the rest of the active site well ordered, it does not have the scissile phosphate and nucleotide 5′ to the cleavage site ordered in the active site (Fig. 1B). The nucleotide numbering for twister A is used, whereas the numbering for twister B is listed in parentheses, unless otherwise stated.
The crystal structures reveal that four base pairs form the P2 stem, which is consistent with the bioinformatics model. In addition, the crystal structure reveals a trans Hoogsteen-sugar edge base pair between A11 and G62 (A7 and G65), which stacks with the rest of the P2 stem (Fig. 1E). The P2 helix is wedged in between the active site/PK2 on one side and PK1/A5 (five adenosines that replace the P5 stem) on the other. Any addition or deletion in the number of base pairs is likely to significantly disrupt the formation of the active site.
The P4 stem identified by bioinformatics was proposed to form a three-base pair stem (Fig. 2A) (1). However, the crystal structure reveals that the P4 stem contains five base pairs including the three that were predicted by bioinformatics (Fig. 2B). The highly conserved nucleotides G39 and C48 form a canonical WC base pair, but the highly conserved nucleotides U40 and A45 arrange to make a noncanonical base pair with some variation. This last base-pairing interaction appears to be critical, because the two nucleotides between A45 and C48 bulge and form one-half of PK1 (Fig. 1G). There is an unexpected variability in the A–U base pairing revealed in two of the four twister B copies, where the adenosine base is in the anti-conformation, so that its Hoogsteen-edge interacts with the Crick side of the uridine base. It remains to be established whether the G–C or A–U pairings are required individually for twister to fold correctly. Nevertheless, a double mutation of both the guanine and the uridine resulted in a mutant that was functionally inactive (1).
Fig. 2.

Comparison of predicted and structurally confirmed conserved consensus secondary structure of twister. (A) Predicted conserved consensus secondary structure and (B) update contacts based on the tertiary structure from the crystal structures. The P2 stem, the P4 stem, and PK2 were found to be extended compared with the original report. Nucleotide conservation and coloring is the same as described by Roth et al. (1). Gray, black, and red are representative of at least 75%, 90%, and 97% conservation, respectively. R and Y indicate purine and pyrimidine, respectively, whereas the red circles indicate positions with less than 75% conservation. The green and orange shading denotes covaried base pairing and base pairing that extends the stems and pseudoknots found in the crystal structures, respectively. Percentages in parentheses indicate the incidence stem structures are present for the P0, P3, and P5 stems along with the minimal length listed.
Between the P4 stem and PK2, there is a very sharp transition in the phosphodiester backbone to position U40 in the P4 stem and U41 in the PK2 (Fig. 1F). The dihedral angles between the phosphates of nucleotides U40, U41, and A42 (U44, C45, and C46) are ∼44°, which is drastically different from 152° in an A-form RNA helix. This junction between the P4 stem and PK2 results in the helices being approximately orthogonal to each other. This junction is on one side of the active site and is stabilized by a hydrogen bond (HB) between a nonbridging oxygen atom of U41 and the 2′-hydroxyl of U8, the penultimate nucleotide 5′ to the cleavage site. It is unclear whether this HB is important for positioning and stabilizing the nucleotide 5′ to the cleavage site or not because it is one of many HBs in the region. All nucleotides in the active site except the nucleotides 5′ and 3′ to the cleavage site are base paired.
Pseudoknots.
Twister was predicted by bioinformatics and confirmed by these two crystal structures to have two PKs just like HDV and glmS (Fig. 1). In all three cases, the PKs are important structural components of the active site. In twister, PK1 and PK2 are the lynchpins that stabilize the active site, as they form part of the walls of the active site and help position nucleotides critical for catalysis. PK1 has two base pairs, whereas PK2 was predicted to have three base pairs; in addition to those three, an additional non-WC base pair occurs in the crystals structures. The added interaction is between the Watson faces (WW) of A44 and A63 (A48 and A66), forming trans twofold symmetric HBs between the exocyclic amine and N1 of the opposite base (Fig. 1H). Of the 12 total nucleotides involved in PK formation, 6 reside within the P4 stem and its connecting loop, and 4 are located on the enzyme strand between the P1 and P2 stems. The PKs form close interactions with nucleotides in the active site and appear to be important in both rigidity of the active site and stability of specific structures of the nucleotides involved in catalysis.
Validation of the Previous Model and Additional Interactions Resulting in Extended Helices and Stacking Interactions.
The structures confirm all expected interactions based on the bioinformatics and reveal additional interactions that extended the lengths of three secondary structure elements, P2, P4, and PK2 (Fig. 2). In the P2 stem, a Hoogsteen-sugar edge base pair between A11 and G62 (A7 and G65) acts to position G62 for catalysis. Within the P4 stem, the WC base pairing between U40 and A45 (U44 and A49) appears to be an important interaction at the base of the P4 and PK1. In PK2, the trans WW base pair interaction of A44 and A63 (A48 and A66) is important in positioning of A63 for catalysis. These previously unpredicted base pairings are critical for either stabilization of the active site or positioning nucleotides for catalysis.
Stacking interactions dominate the overall structure of twister and contribute to its substantial stability. Nearly all bases are stacked except the nucleotide 5′ to the cleavage site and a bulged nucleotide in the P2 stem. A continuous series of stacking interactions span throughout the entire structure, starting with the 3′ end of the enzyme strand containing the P1 stem and PK2, and continues to the substrate strand at conserved nucleotide A11 (A7), and then throughout the P2 stem, PK1, and the P3 stem. Comparison of the three copies in the asymmetric unit of twister A shows that the active site is well defined and rigid at the scissile phosphate and nucleotide 5′ to the cleavage site are ordered, whereas the four copies in twister B in the asymmetric unit of the scissile phosphate and nucleotide 5′ to the cleavage site are disordered (Fig. 1B and Fig. S5 A and B). The active sites in both crystal forms are held together by the same base pairs and base-stacking interactions, including specific interactions by the highly conserved nucleotides. Together, these suggest a rigid structure. The only highly conserved nucleotide that does not form a base pair is the conserved A10 (A6), but this nucleotide is stabilized by stacking on A45 (A49) at the base of the P4 stem.
Discussion
A major question about twister is which strategies and nucleotides are responsible for its rapid catalysis. The twister A structure suggests that it uses three major catalytic strategies available to enzymes that cleave RNA by internal phosphoester transfer (Fig. 3). All self-cleaving ribozymes will likely use the in-line nucleophilic attack strategy, which is a result of forming the active site (9). We propose that twister uses two highly conserved nucleotides to achieve the general base and transition-state stabilization strategies as well (Fig. 4A). The general base strategy could be achieved by the N6 of A63 (A66), which is within HB distance for charge neutralization of one of the nonbridging oxygen atoms. The transition-state stabilization strategy could be promoted by the N1 of G62 (G65) that resides within HB distance of the 2′-hydroxyl to act as a general base. Some of these interactions are unique to twister, whereas others are more common to all small self-cleaving ribozymes.
Fig. 3.

Transesterification cleavage reaction. RNA cleavage by internal phosphoester transfer by in-line nucleophilic attack (twister A numbering).
Fig. 4.

Comparison of the active sites for twister, hairpin, and RNase A. (A) The active site of twister with the conserved nucleotides suggested to be involved in catalysis are shown (environmental sequence). The 2′-hydroxyl is colored pink, and it was computationally modeled and not present in the crystallization construct. (B) Active site of hairpin precatalysis in white (1M5K) and transition state in green (1M5O). A9 and A38 move by about 2 Å each to cradle the phosphate, which also moves into position for in-line phosphoester transfer. (C) Active site of RNase A transition state (6RSA). The arrows are drawn to show the attack of the 2′-oxygen to the phosphate (orange spheres). Groups suggested to be involved in catalysis are shown with their proposed mechanistic strategies labeled. See Discussion for further explanation.
Neutralization of the nonbridging phosphate oxygen atoms is not always a catalytic strategy used by small self-cleaving ribozymes. A structure of the hairpin that was designed to be a transition state (TS) mimic by using vanadate to substitute for the scissile phosphate [Protein Data Bank (PDB) ID code 1M5O] provides the best evidence for a ribozyme to use this strategy (17). Upon formation of the TS-like complex, two adenosines are each displaced by ∼2 Å to be brought within HB distance to the nonbridging oxygen atoms (Fig. 4B). Biochemical evidence from the hammerhead suggests that metal ions are able to help neutralize the charge of the nonbridging oxygen atoms, although they have not yet been observed in a crystal structure (18, 19). It has been proposed but not experimentally validated that glmS would neutralize the developing charge on the nonbridging oxygen atoms if the glucosamine-6-phosphate (GlcN6) is protonated (7). HDV has no specific group(s) that are characterized or suggested to act by the transition-state stabilization strategy. Here, we show that twister likely uses the transition-state stabilization strategy by using the N6 of A63 to neutralize the developing charge of one of the nonbridging oxygen atoms (Fig. 4A).
Only hairpin, and now twister, have structural evidence that suggests specific groups are involved in the transition-state stabilization strategy. An interesting aspect is that the groups involved in catalysis for hairpin are only observable in the TS-like complex, but in the precatalytic state the two adenosines relax by ∼2 Å. In contrast, A63 in twister is prepositioned within HB range in the precatalytic state. The rigidity of the preorganized twister active site, particularly the position of A63, may contribute significantly to the ribozyme’s speed, contributing to a rate constant enhancement of up to 105 (9). The orientation of A63 is stabilized by two interactions. First, A63 stacks with A11, and second, A63 forms a WW base pair with A44, another highly conserved nucleotide. Mutating either of these nucleotides to another base would misorient the N6 of A63.
The most often-used chemical group to be used for the general base strategy is a G among small self-cleaving ribozymes. To prevent catalysis, we removed the attacking group by using a 2′-deoxy substitution in the substrate strand, which also eliminated the normal HB partner of the chemical group that performs general base catalysis. However, when the 2′-hydroxyl is computationally modeled, in at least one copy of twister in crystal form A, G62 is within HB distance. This interaction is similar to interactions found within structures of hairpin, hammerhead, and glmS. HDV is the only small self-cleaving ribozyme that uses a cytosine rather than a guanosine to carry out the general base strategy (7). The originally proposed secondary structure of twister contained only two other highly conserved nucleotide bases that could be proposed to carry out the general base strategy, one guanine and one cytosine. Both of these were predicted to be unpaired, but the two are actually base paired in our structures as part of the P4 stem discussed above, thus leaving G62 as the most likely nucleotide to be used in general base catalysis (1). The position of G62 is stabilized by its forming a Hoogsteen–Hoogsteen base pair with A11 and stacking within the P2 stem.
All of the small self-cleaving ribozymes that use the general acid strategy do so by different mechanisms. Hairpin uses the N1 of an adenosine; hammerhead uses a 2′-hydroxyl of a ribose moiety; glmS uses an amine group on the GlcN6 cofactor; and HDV employs a metal-coordinated water (13, 19–21). We found that twister does not have a HB donor with good geometry and a chemical group with a reasonable pKA to act as a general acid. The only HB donor with reasonable geometry is the N3 of A10, but this nitrogen has a very low pKA of about –4.2 to 1.0 (22, 23). Although this would be a great chemical group to interact with the 5′-OH leaving group, it would require a highly perturbed pKA to act as a general acid. Although studies have illustrated pKa shifts by chemical groups within an RNA molecule being dependent on its local environment, N3 of an adenosine is least likely to be shifted to neutrality (24, 25). The A10 is held in place by two interactions. First, A10 stacks on A45 within the P4 stem, and second, the N7 of A10 forms HBs with the 2′-OH of A44 and a nonbridging oxygen atom of A45. This means that twister is the only small self-cleaving ribozyme not to have a group to be suggested biochemically or structural to act as a general acid.
To summarize, the nucleotides that are important for catalysis identified from our structures are G62 and A63. We predict that catalysis would be severely affected if either A63 or G62 were mutated. It would be interesting to find the role A10 may play. If it is only involved in leaving group stabilization, it is difficult to explain why it is highly conserved. A10 is in a syn-conformation with a C2′-endo sugar pucker. It is possible that a correlation of A10 being in the syn-conformation changes the sugar pucker and thereby changes the A10 phosphate (scissile phosphate) to A11 phosphate distance for appropriate self-cleavage (26). This distance is longer than most other phosphate–phosphate distances (6.8 Å). A10 may then be acting to aid for in-line nucleophilic attack. Hammerhead, HDV, glmS, and hairpin ribozymes all have nucleotides in syn-conformation at critical positions around their active site structures (26).
The two enzymes most closely related to twister in structure and mechanism are hairpin and RNase A. All of these enzymes unstack and splay apart the bases that are 5′ and 3′ to the scissile phosphate in the active site and use all at least three strategies, but they use different chemical moieties to achieve RNA cleavage and bind the substrates in different orientations.
We suggest that twister uses A63 to neutralize the developing negative charge on the nonbridging oxygen atoms, whereas hairpin uses G8, A9, and A38, and RNase A employs K41 (Fig. 4C) (12, 17). Twister and RNase A have preformed active sites that are ready for catalysis. Hairpin ribozymes have two adenosines that are repositioned by ∼2 Å to cradle the phosphate as it proceeds into the TS with G8 forming a HB (17). It is possible that G62 of twister is analogous to G8 of hairpin in its TS.
The twister and hairpin both use a guanosine, whereas RNase A uses a histidine as a general base. The histidine is an excellent general base for RNase A, because it contributes 700,000-fold to the cleavage rate, whereas, due to the pKA of G, this strategy contributes less to catalysis in hairpin, as an abasic G8 mutation results in a 350-fold decrease in the cleavage rate (27, 28). It is likely that the effect of mutating G62 in twister will be similar to the effect of mutating G8 of hairpin. We propose that the preformed and rigid active sites in twister and RNase A contribute to their rate constants.
Hairpin and RNase A use an endocyclic nitrogen to act as a general acid and stabilize the 5′-hydroxyl in the general acid strategy. We cannot assign a chemical group to act as a general acid for twister. The lack of a general acid is a troubling problem for twister to achieve a relatively high catalytic rate. H119 accounts for a 400,000-fold enhancement in the catalytic rate of RNase A, and it was suggested that H119 has a concerted motion with the 5′-oxygen during catalysis (12, 27). It will be interesting to determine a transition state structure of twister to determine whether the active site rearranges to present its self as a general acid.
It has been suggested that all three of these enzymes use three or four of the strategies discussed, although their rate constants are immensely different. Twister has an estimated cleavage rate of 1,000 per minute, which is over 500 times faster than 1.9 per minute for hairpin, but RNase A is 80 times faster than twister with an outstanding rate constant of 80,000 per minute (1, 29, 30). Detailed mechanistic probing of twister’s catalysis is needed to elucidate why the speeds of these three enzymes differ by orders of magnitude, while using the three or four strategies.
Conclusion
The structures of twister reported here suggest that much of the robust catalytic activity is achieved by a rigid active site formed by two uncommon base pairings and extensive base-stacking interactions. The key mechanistic consequence of the twister structures determined here show that two highly conserved nucleotides G62, and A63 (G65, and A66) are situated to act as a general base and to stabilize the transition state, respectively. These likely contribute to the enhanced rate of catalysis of twister compared with many other small self-cleaving ribozymes. First, in the general base strategy, A63 is positioned within HB range before the reaction precedes to the TS. Second, the active site is preformed as evidenced by comparing the two crystal structures in regards to the scissile phosphate and nucleotide 5′ to the cleavage site. We suggest that these two structural insights are important for twister to achieve an enhanced catalytic rate compared with most other small self-cleaving ribozymes.
Materials and Methods
All chemicals except for those listed were purchased from Sigma-Aldrich. Oligonucleotides were obtained from Integrated DNA Technologies. FastStart High Fidelity PCR System kits were purchased from Roche Applied Sciences. Sparse matrix crystallization screens were obtained from Qiagen and Hampton Research.
The twister sequences with the most common isoform (P3) were selected for structure determination. Constructs were designed in a similar manner as the substrate and enzyme experiments in twister’s initial report (1).
Crystallization of Twister Constructs.
Samples for crystallization were mixed in a 1:1 ratio of substrate to enzyme (ES complex), incubated at 70 °C, and immediately allowed to slow cool to 25 °C. Twister A from the environmental sequence crystallizes at 25 °C as hexagonal rods within 1–14 d by sitting-drop vapor diffusion by mixing 3 µL of the ES complex at a final concentration of 89 μM diluted in buffer A with 2 µL of well solution containing 160 mM trisodium citrate (pH 4.6), 700 mM (NH4)2SO4, 1 M Li2SO4, 2.8–3.8% (vol/vol) pentaerythriotol ethoxylate (3:4 EO/OH), and 2.1–4% (wt/vol) 6-aminohexanoic acid. Twister from O. sativa crystallizes at 25 °C as plates within 2–4 wk by sitting-drop vapor diffusion by mixing 1–4 µL of the ES complex at a final concentration of 121 μM diluted in buffer B with 1–5 µL of well solution containing 200 mM (NH4)2SO4, 100 mM sodium acetate (pH 4.5), and 30% (wt/vol) PEG 4000. Crystals of the twister construct derived from the O. sativa (rice) sequence were not reproducible after we collected data from 15 crystals that diffracted at best to 2.9 Å (I/σ = 1.00). We now attribute this to the unexpected intermolecular dimer that crystallized, possibly aided by a degradation in the 5′ end of the substrate strand.
Crystals from the environmental sequence grew to maximum dimensions of 400 × 200 × 200 μm and were stabilized and cryogenically protected by increasing the lithium sulfate or lithium acetate to a final concentration of 1.25 or 3.2 M, respectively. Crystals of the O. sativa sequence grew to maximum dimensions of 200 × 100 × 25 μm and were stabilized and cryogenically protected by increasing the PEG 4000 concentration to 33% (wt/vol) and adding PEG 400 in a stepwise manner to a final concentration of 10% (vol/vol). Crystals were flash frozen by plunging into liquid nitrogen.
Diffraction data were collected at 100 K using synchrotron X-ray radiation at beam line 24ID-C at Advanced Photon Source in Argonne National Laboratory (Argonne, IL). The data were processed and scaled using X-ray Detector Software (XDS) (31). General handling of scaled data was completed using Collaborative Computational Project programs (32).
Structure Determination and Refinement.
See SI Materials and Methods for structure determination procedures. The models were built using COOT (33) and refined using REFMAC5 (32). The structure derived from the environmental sequence was refined against a dataset collected from a higher resolution native crystal using resolution scaled to an overall I/σ of 1.0 at the highest resolution shell (HRS) and then to higher resolution by decreasing the I/σ stepwise by 0.1 increments down to an overall I/σ of 0.5 at the HRS. This dramatically improved the R/Rfree from 26.3/33.3 to 19.7/24.4 and improved the quality of the electron density maps. The structure of the higher resolution model derived from O. sativa was used to rebuild areas of the lower resolution model where there was some ambiguity in syn- or anti-conformations of the bases (Fig. S6). Final crystallographic statistics can be found in SI Materials and Methods.
The crystal structures of the O. sativa and environmental sequence were deposited as PDB ID codes 4QJD and 4QJH, respectively. All structural figures were prepared using PyMOL (www.pymol.org).
Supplementary Material
Acknowledgments
We thank staff at Advanced Photon Source in Argonne National Laboratory (NECAT 24ID-C) for facilitating X-ray data collection, J. Watson for help with preparing the RNA samples, and members of the Breaker Laboratory at Yale for helpful discussion. This work was supported by National Institutes of Health Grant GM022778. D.E. was supported in part by National Institutes of Health Predoctoral Program in Biophysics Grant 5 T32 GM 8283-25.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4QJD and 4QJH).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414571111/-/DCSupplemental.
References
- 1.Roth A, et al. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat Chem Biol. 2014;10(1):56–60. doi: 10.1038/nchembio.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prody GA, Bakos JT, Buzayan JM, Schneider IR, Bruening G. Autolytic processing of dimeric plant virus satellite RNA. Science. 1986;231(4745):1577–1580. doi: 10.1126/science.231.4745.1577. [DOI] [PubMed] [Google Scholar]
- 3.Buzayan JM, Gerlach WL, Bruening G. Nonenzymatic cleavage and ligation of RNAs complementary to a plant-virus satellite RNA. Nature. 1986;323(6086):349–353. [Google Scholar]
- 4.Sharmeen L, Kuo MYP, Dinter-Gottlieb G, Taylor J. Antigenomic RNA of human hepatitis delta virus can undergo self-cleavage. J Virol. 1988;62(8):2674–2679. doi: 10.1128/jvi.62.8.2674-2679.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saville BJ, Collins RA. A site-specific self-cleavage reaction performed by a novel RNA in neurospora mitochondria. Cell. 1990;61(4):685–696. doi: 10.1016/0092-8674(90)90480-3. [DOI] [PubMed] [Google Scholar]
- 6.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428(6980):281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 7.Ferré-D’Amaré AR, Scott WG. Small self-cleaving ribozymes. Cold Spring Harb Perspect Biol. 2010;2(10):a003574. doi: 10.1101/cshperspect.a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canny MD, et al. Fast cleavage kinetics of a natural hammerhead ribozyme. J Am Chem Soc. 2004;126(35):10848–10849. doi: 10.1021/ja046848v. [DOI] [PubMed] [Google Scholar]
- 9.Emilsson GM, Nakamura S, Roth A, Breaker RR. Ribozyme speed limits. RNA. 2003;9(8):907–918. doi: 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breaker RR, et al. A common speed limit for RNA-cleaving ribozymes and deoxyribozymes. RNA. 2003;9(8):949–957. doi: 10.1261/rna.5670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinberg Z, et al. Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 2010;11(3):R31. doi: 10.1186/gb-2010-11-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards FM, et al. Protein structure, ribonuclease-S and nucleotide interactions. Cold Spring Harb Symp Quant Biol. 1972;36:35–43. doi: 10.1101/sqb.1972.036.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Ferré-D’Amaré AR, Zhou K, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395(6702):567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 14.Rupert PB, Ferré-D’Amaré AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410(6830):780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 15.Keel AY, Rambo RP, Batey RT, Kieft JS. A general strategy to solve the phase problem in RNA crystallography. Structure. 2007;15(7):761–772. doi: 10.1016/j.str.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126(2):309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rupert PB, Massey AP, Sigurdsson ST, Ferré-D’Amaré AR. Transition state stabilization by a catalytic RNA. Science. 2002;298(5597):1421–1424. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- 18.Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG. The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem Biol. 1998;5(10):587–595. doi: 10.1016/s1074-5521(98)90116-8. [DOI] [PubMed] [Google Scholar]
- 19.Scott WG. RNA structure, metal ions, and catalysis. Curr Opin Chem Biol. 1999;3(6):705–709. doi: 10.1016/s1367-5931(99)00029-0. [DOI] [PubMed] [Google Scholar]
- 20.Klein DJ, Ferré-D’Amaré AR. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313(5794):1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 21.Ke A, Zhou K, Ding F, Cate JH, Doudna JA. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature. 2004;429(6988):201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 22.Kapinos LE, Operschall BP, Larsen E, Sigel H. Understanding the acid-base properties of adenosine: The intrinsic basicities of N1, N3 and N7. Chemistry. 2011;17(29):8156–8164. doi: 10.1002/chem.201003544. [DOI] [PubMed] [Google Scholar]
- 23.Muth GW, Ortoleva-Donnelly L, Strobel SA. A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science. 2000;289(5481):947–950. doi: 10.1126/science.289.5481.947. [DOI] [PubMed] [Google Scholar]
- 24.Wilcox JL, Bevilacqua PC. pKa shifting in double-stranded RNA is highly dependent upon nearest neighbors and bulge positioning. Biochemistry. 2013;52(42):7470–7476. doi: 10.1021/bi400768q. [DOI] [PubMed] [Google Scholar]
- 25.Wilcox JL, Bevilacqua PC. A simple fluorescence method for pK(a) determination in RNA and DNA reveals highly shifted pK(a)’s. J Am Chem Soc. 2013;135(20):7390–7393. doi: 10.1021/ja3125299. [DOI] [PubMed] [Google Scholar]
- 26.Sokoloski JE, Godfrey SA, Dombrowski SE, Bevilacqua PC. Prevalence of syn nucleobases in the active sites of functional RNAs. RNA. 2011;17(10):1775–1787. doi: 10.1261/rna.2759911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raines RT. Ribonuclease A. Chem Rev. 1998;98(3):1045–1066. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 28.Lebruska LL, Kuzmine II, Fedor MJ. Rescue of an abasic hairpin ribozyme by cationic nucleobases: Evidence for a novel mechanism of RNA catalysis. Chem Biol. 2002;9(4):465–473. doi: 10.1016/s1074-5521(02)00130-8. [DOI] [PubMed] [Google Scholar]
- 29.delCardayré SB, Raines RT. Structural determinants of enzymatic processivity. Biochemistry. 1994;33(20):6031–6037. doi: 10.1021/bi00186a001. [DOI] [PubMed] [Google Scholar]
- 30.Nesbitt S, Hegg LA, Fedor MJ. An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem Biol. 1997;4(8):619–630. doi: 10.1016/s1074-5521(97)90247-7. [DOI] [PubMed] [Google Scholar]
- 31.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.