

Abstract
• Background and Aims Genetic variability was estimated for Atriplex tatarica from 25 populations in the Czech Republic. Since its north-western range margin is in central Europe, a relationship between marginality and low within-population genetic diversity was tested in accordance with the Central–Marginal Model.
• Methods Population genetic diversity was expressed by assessing patterns of variation at 13 putatively neutral allozyme loci (comprising 30 putative alleles) within and between 25 natural populations of A. tatarica along a north-west–south-east transect in the Czech Republic.
• Key Results Atriplex tatarica is a species of human-made habitats with a mixed mating system and wide geographic distribution. Overall, A. tatarica displayed moderate levels of genetic diversity in comparison with other herbaceous plants. The percentage of loci that were polymorphic was 47·1 %, with average values of 1·55, 0·151 and 0·155 for the average number of alleles per polymorphic locus (A), observed heterozygosity (Ho) and expected heterozygosity (He), respectively. There was only weak evidence of inbreeding within populations (FIS = 0·031) and significant population differentiation (FST = 0·214). Analysis of the data provides no evidence for isolation-by-distance for the whole study area. However, Mantel tests were highly significant for the marginal Bohemian region and non-significant for the central Moravian region. While northern populations of A. tatarica showed significantly lower allelic richness (A = 1·462) than populations from the southern part of the study area (A = 1·615), they did not differ in observed heterozygosity (Ho), gene diversity (HS), inbreeding within populations (FIS) or population differentiation (FST), despite generally lower values of particular genetic measurements in the marginal region.
• Conclusions Genetic diversity, with the exception of allelic richness, was not significantly lower at the margins of the species' range. This, therefore, provides only weak support for the predictions of the Central–Marginal Model.
Keywords: Allozyme, Atriplex, Central–Marginal Model, Chenopodiaceae, founder effect, inbreeding, invasion, population genetic structure
INTRODUCTION
Genetic variation and population structure reflect both the influence of present-day evolutionary forces (e.g. natural selection, genetic drift, and gene flow) and historical processes associated with patterns of colonization and migration (Barrett, 1982). The study of historical plant invasions reveals differing levels of genetic variation within colonizing species, mainly due to founder effects, number of population bottlenecks, the mating system or ability to hybridize (Barrett, 1982).
Both the founder effect, when numbers of immigrants are important, and population bottlenecks lead to lowered amounts of genetic information present in the new population, as compared with the source population. This sampling error causes random fluctuations in allele frequencies called genetic drift. The extent of the loss of genetic variation in comparison to the source population depends on the size of the founder population, the number of times a founder event occurs and the severity of a population bottleneck (Nei et al., 1975; Goodnight, 1987, 1988). For example, multiple introductions of ornamental species such as Lathyrus latifolius (Godt and Hamrick, 1991), Lonicera japonica (Schierenbeck et al., 1995) or Pueraria lobata (Pappert et al., 2000) resulted in higher levels of genetic variability compared with single introductions such as for Bromus tectorum (Novak and Mack, 1993) or Reynoutria japonica var. japonica (Hollingsworth and Bailey, 2000; Mandák et al., 2003, 2005).
The importance of the mating system has been repeatedly documented for a variety of invasive species. For example, species with a predominantly autogamous mode of reproduction usually exhibit low levels of genetic variation and often form highly homozygous populations composed of a few genotypes, e.g. Capsella bursa-pastoris (Bosbach and Hurka, 1981), Lolium temulentum (Hayward and Zaruk, 1982), Polygonum pensylvanicum (Kubetin and Schaal, 1979) and Senecio viscosus (Koniuszek and Vereij, 1982). On the other hand, invading species with predominantly allogamous modes of reproduction may possess a high level of genetic diversity, e.g. Echium plantagineum (Brown and Burdon, 1983).
Hybridization can increase genetic variability and generate both novelty and variation (Rieseberg et al., 2003). In alien plants, it can occasionally be followed by the spread of hybrid gene combinations (Ellstrand and Schierenbeck, 2000; Vila et al., 2000). Hence, hybridization can lead to adaptive evolution in a number of ways, for example, through the generation of novel genotypes and variation, fixed heterosis stabilized by apomixis or clonality and dumping of genetic load (Ellstrand and Schierenbeck, 2000). However, not all hybridization events lead to increased fitness or adaptive evolution (Arnold, 1997). The hybridization process is important not only at the interspecific level but it can also act as a stimulus for the evolution of invasiveness within plant species (Ellstrand and Schierenbeck, 2000), i.e. hybridization between previously isolated populations of the same species, in the case of multiple introductions from various sites within the native distribution range.
In plant species with continuous distributions, genetic variation among geographic regions may result from the presence of barriers to gene flow among populations (e.g. a mountain range) or as a result of historical events such as glaciations (Hewitt, 1996, 1999; Ibrahim et al., 1996; Chauvet et al., 2004; Persson et al., 2004). On the basis of the concept of recolonization of new areas typically constructed for patterns of genetic variation in a colonized area after glaciations, two models for recently spreading species can be introduced. The ‘gradual expansion scenario’, when migration proceeds from a gradually expanding continuous front, assumes that most genetic diversity will be retained through the course of an expansion. In contrast, the ‘satellite population scenario’ assumes many spatially isolated and initially small marginal populations, which in turn become the source of new founding events. In the latter case, an enhanced potential for founder effects that reduce allelic diversity within populations (Nei et al., 1975) will occur and, as a consequence, these marginal populations will constitute a number of small demes representing only part of the genetic variation of the central populations. The classical view, often referred to as the Central–Marginal Model (Da Cunha and Dobzhansky, 1954), shows that patterns of genetic variation between central and marginal populations may thus depend on the degree to which markers are under selection, rates of gene flow into marginal populations, and the amount of habitat variability and rates of population growth in central and marginal areas (Franks et al., 2004).
Species that have increased their range recently are particularly appropriate for addressing questions regarding patterns of spatial genetic structuring and comparison of genetic variation between central and marginal areas. This is due to their fragmented distribution in the marginal areas, when populations are often restricted to the most ecologically suitable habitats only, as opposed to the naturally continuous distribution in the central area. Here, using polymorphism at allozyme loci, the genetic diversity of A. tatarica in the Czech Republic was investigated to address the following specific questions: (a) what is the pattern of genetic variation within and among populations of A. tatarica and is there evidence of inbreeding and population differentiation; and (b) is there evidence of reduced genetic diversity in the north-western marginal populations of A. tatarica compared with the south-eastern central populations, due to the likely migration north-west from the south-eastern area of continuous distribution?
MATERIALS AND METHODS
The plant studied
Atriplex tatarica L. (syn. A. laciniata L., A. sinuata Hoffm., A. veneta Willd.) (Chenopodiaceae) is one of two annual, heterocarpic, diploid species of the section Sclerocalymma Aschers in the Czech Republic (Mandák, 2003a). Atriplex tatarica has spread from its native Central Asia, Asia Minor and eastern Europe (Aellen, 1960) to central Europe (Fig. 1) where it is abundant in urban areas, on disturbed sites, along railways and roads mainly in the warmer areas of the Czech Republic, with a tendency towards spreading further (Mandák, 2003a). It grows well in nitrogen-rich soils and tolerates a high content of NaCl (Mandák, 2003b). The north-west border of its continuous European distribution lies partly in the Czech Republic. The species is very common in the south-eastern part of the Czech Republic (south Moravia), which probably represents part of the native continuous area of distribution from south-eastern Europe through the Pannonian lowland. In the rest of the Czech Republic, i.e. the western part (Bohemia), the species is only found in several localities geographically isolated from the Moravian sites and from one another (Fig. 1).
Fig. 1.
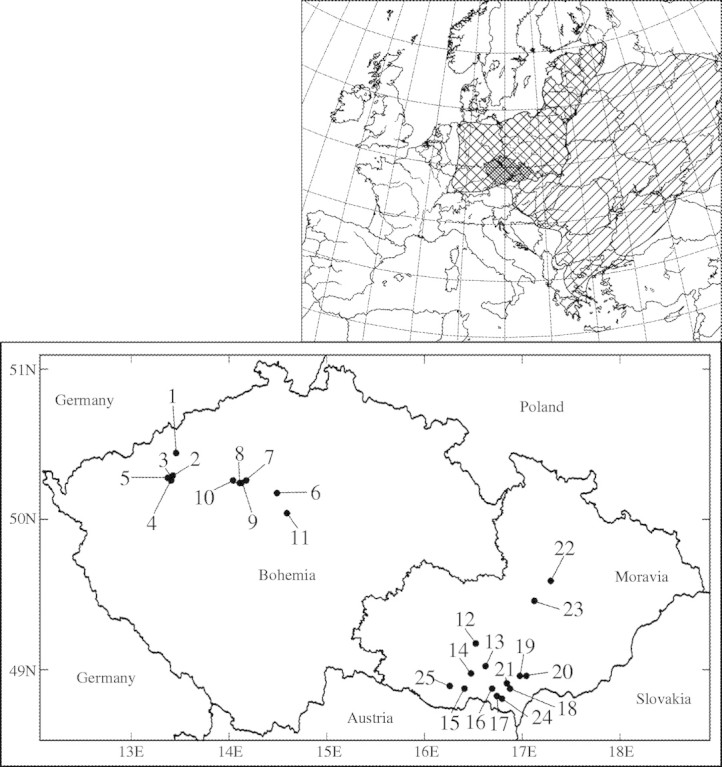
Map showing the total distribution of Atriplex tatarica in Europe (redrawn from Jalas and Suominen, 1988). The shaded area indicates native area distribution of A. tatarica and the cross-hatched area indicates secondary area distribution in Europe (Jalas and Suominen, 1988). The detailed map of the Czech Republic shows the location of the 25 populations of A. tatarica used in this study.
The populations studied
Twenty-five populations were studied along a geographical gradient through the Czech Republic; 11 populations from Bohemia and 14 from Moravia (Table 1 and Fig. 1). Samples for genetic analysis were collected from June to August in 2002–2003. A 50-m transect was located in each of the selected populations and ten different individuals were collected in each population at 5-m intervals. Samples were transported in a cool box and the youngest expanded leaf of each plant analysed within 24 h.
Table 1.
Summary of genetic diversity within 25 populations of Atriplex tatarica based on nine putative allozyme loci (enzyme systems AAT, LAP, MDH, SKDH and SOD) and mean species values
| Population* |
Latitude |
Longitude |
n |
PL |
A |
Ae |
Ho |
He |
f (=FIS) |
|---|---|---|---|---|---|---|---|---|---|
| B1 | 50°25′85″ | 13°26′68″ | 6 | 53·9 | 1·54 | 1·31 | 0·192 | 0·195 | 0·013 |
| B2 | 50°16′75″ | 13°24′65″ | 3 | 46·2 | 1·46 | 1·26 | 0·169 | 0·168 | −0·005 |
| B3 | 50°15′83″ | 13°23′21″ | 6 | 53·9 | 1·54 | 1·22 | 0·162 | 0·150 | −0·080 |
| B4 | 50°14′79″ | 13°24′17″ | 4 | 53·9 | 1·54 | 1·32 | 0·208 | 0·201 | −0·034 |
| B5 | 50°16′25″ | 13°21′69″ | 6 | 23·1 | 1·23 | 1·16 | 0·077 | 0·088 | 0·135 |
| B6 | 50°10′35″ | 14°29′06″ | 5 | 30·8 | 1·31 | 1·10 | 0·054 | 0·070 | 0·241 |
| B7 | 50°15′25″ | 14°09′64″ | 1 | 38·5 | 1·38 | 1·26 | 0·139 | 0·157 | 0·124 |
| B8 | 50°14′25″ | 14°07′18″ | 2 | 38·5 | 1·46 | 1·24 | 0·131 | 0·136 | 0·044 |
| B9 | 50°14′15″ | 14°05′80″ | 5 | 30·8 | 1·38 | 1·18 | 0·131 | 0·111 | −0·191 |
| B10 | 50°15′07″ | 14°02′08″ | 3 | 46·2 | 1·54 | 1·28 | 0·162 | 0·178 | 0·096 |
| B11 | 50°02′43″ | 14°35′18″ | 2 | 38·5 | 1·69 | 1·32 | 0·123 | 0·135 | 0·094 |
| M12 | 49°10′40″ | 16°30′76″ | 2 | 69·2 | 1·85 | 1·27 | 0·169 | 0·185 | 0·092 |
| M13 | 49°01′44″ | 16°36′75″ | 4 | 46·2 | 1·46 | 1·25 | 0·177 | 0·161 | −0·107 |
| M14 | 48°58′18″ | 16°27′86″ | 4 | 61·5 | 1·77 | 1·42 | 0·231 | 0·253 | 0·094 |
| M15 | 48°52′09″ | 16°24′04″ | 2 | 38·5 | 1·46 | 1·10 | 0·085 | 0·075 | −0·138 |
| M16 | 48°52′05″ | 16°41′14″ | 6 | 38·5 | 1·38 | 1·24 | 0·123 | 0·139 | 0·122 |
| M17 | 48°49′09″ | 16°44′52″ | 6 | 69·2 | 1·85 | 1·29 | 0·162 | 0·166 | 0·026 |
| M18 | 48°51′69″ | 16°52′23″ | 2 | 53·9 | 1·62 | 1·27 | 0·162 | 0·156 | −0·038 |
| M19 | 48°56′84″ | 16°58′51″ | 2 | 53·9 | 1·62 | 1·32 | 0·154 | 0·186 | 0·182 |
| M20 | 48°57′57″ | 17°02′43″ | 5 | 38·5 | 1·38 | 1·14 | 0·108 | 0·097 | −0·115 |
| M21 | 48°54′02″ | 16°50′34″ | 4 | 46·2 | 1·77 | 1·45 | 0·192 | 0·189 | −0·018 |
| M22 | 49°35′39″ | 17°17′59″ | 6 | 30·8 | 1·31 | 1·16 | 0·108 | 0·103 | −0·050 |
| M23 | 49°26′80″ | 17°06′86″ | 6 | 69·2 | 1·77 | 1·35 | 0·177 | 0·226 | 0·228 |
| M24 | 48°47′78″ | 16°46′84″ | 6 | 53·9 | 1·69 | 1·31 | 0·208 | 0·177 | −0·182 |
| M25 | 48°53′14″ | 16°15′23″ | 6 | 53·9 | 1·69 | 1·31 | 0·169 | 0·180 | 0·062 |
| Mean | 47·1 | 1·55 | 1·26 | 0·151 | 0·155 | 0·024 | |||
| (±SD) | (12·6) | (0·18) | (0·09) | (0·043) | (0·046) | (0·120) |
Number of individual sampled from each population was ten.
PL = percentage of polymorphic loci; A = average number of alleles per polymorphic locus; Ae = effective allele number; Ho = observed heterozygosity; He = expected heterozygosity; f = Weir and Cockerham's estimate (Weir and Cockerham, 1984) of FIS (Wright's fixation index) per population over loci.
Populations were assigned to six partly arbitrary classes, in which n = estimated total number of individuals: 1, n ≤ 50; 2, 50 < n ≤ 100; 3, 100 < n ≤ 200; 4, 200 < n ≤ 300; 5, 300 < n ≤ 400; 6, n > 400.
The first letter of each population name refers to the province in which the population was found, i.e. B = Bohemia and M = Moravia.
To examine the relationship between population size and population genetic structure, populations were assigned to six partly arbitrary classes, ranging from populations with <50 individuals to populations with >400 individuals (Table 1).
Allozyme procedures
Twelve enzymatic systems were tested and those which provided the best results in the given group were selected for further analysis, i.e. AAT (EC 2.6.1.1), LAP (EC 3.4.11.1), MDH (EC 1.1.1.37), SKDH (EC 1.1.1.25) and SOD (EC 1.15.1.1).
Electrophoresis was performed on crude protein extracts of leaf material. The tissue was ground in ice-cold TRIS–HCl extraction buffer [0·1 m TRIS–HCl, pH 8·0; 70 mm 2-mercaptoethanol, 26 mm sodium metabisulfite, 11 mm ascorbic acid, 4 % (w/v) polyvinylpyrrolidone]. Roughly 80 mg of fresh leaf material, along with Dowex.Cl (1-X8), was homogenized on ice in 0·75 mL of extraction buffer. Extracts were centrifuged for 10 min at 21 150 g and the clear supernatants were stored at −75 °C. Isozymes were separated on native-PAGE and 30 μL of each sample were employed for electrophoresis in a Hoefer vertical unit.
All enzyme systems (AAT, LAP, MDH, SKDH and SOD) were investigated on polyacrylamide gels [8 % acrylamide, discontinuous TRIS–glycine buffer system (pH 8·3)]. The staining procedures followed Vallejos (1983) for LAP and AAT and Wendel and Weeden (1989) for SKDH, MDH, ME and SOD, with certain modifications. Two staining solutions were prepared for AAT (20 mL 0·1 m TRIS–HCl (pH 8·4), 240 mg aspartic acid, 40 mg α-ketoglutaric acid and 20 mL 0·1 m TRIS–HCl (pH 8·4), 50 mg Fast Blue BB Salt, 50 mg Fast Violet B, 25 mg pyridoxal-5-phosphate). The gel was rinsed in water and then in TRIS–HCl pH 7 buffer. The solutions were then mixed and poured on the gel. The gel was incubated in the dark at 32 °C until bands appeared, and then rinsed and fixed with a 1 : 1 : 3 : 5 solution of glycerine, acetic acid, H2O and methanol. The gel stained for LAP was rinsed in buffer [0·2 m TRIS–maleate (pH 6)] and incubated for 10 min with 40 mg l-leucyl-β-naphthylamide.HCl (in 50 % acetone) and 60 mg MgCl2 (both dissolved in 30 mL buffer). Afterwards, a solution of 25 mg Fast Black K Salt in 30 mL buffer was added. For SKDH ingredients, 30 mg of shikimic acid, 5 mg of NADP, 6 mg of MTT and 2 mg of PMS were combined and dissolved in 30 mL of 0·1 m TRIS–HCl (pH 8·4). A standard staining solution for ME was prepared by dissolving 150 mg malic acid in 25 mL 0·05 m TRIS–HCl (pH 8·0) and adjusted to pH 7·5 with 1 n NaOH; to this was added a solution of 10 mg of MTT, 5 mg of NADP and 2 mg of PMS in 25 mL of 0·05 m TRIS–HCl (pH 8·0), and the resulting staining solution was poured over the gel. A parallel gel was stained for MDH [50 mL 0·1 m TRIS–HCl (pH 7·5), 150 mg malic acid, 15 mg NAD, 10 mg MTT, 2 mg PMS], as some MDH isozymes were active with NADP as a co-factor and, hence, were visualized with this stain. Best results were obtained with ME staining. All gels were incubated in the dark at 32 °C until bands appeared. For SOD ingredients, 50 ml of 0·05 m TRIS–HCl (pH 8·2), 5 mg of EDTA, 5 mg of NBT and 2 mg of riboflavin were combined and poured over the gel. This was incubated for 20 min in the dark at 32 °C then removed and illuminated under a lamp until bands appeared on the blue background. Afterwards, all gels were thoroughly rinsed in distilled water, dried between two cellophane sheets and stored.
Statistical analyses
To estimate genetic diversity and genetic structure, a locus was considered polymorphic if the frequency of the most common allele did not exceed 0·95. Genetic diversity parameters, i.e. percentage polymorphic loci (PL), average number of alleles per locus (A), effective number of alleles (Ae), observed heterozygosity (Ho) and Nei's unbiased heterozygosity (He), were estimated using the POPGEN program (Yeh et al., 1999). In addition, Weir and Cockerham's parameter f(FIS), a measure of inbreeding within populations (Weir and Cockerham, 1984), was calculated for each population with FSTAT (Goudet, 1995).
Genetic variation among populations was analysed in several ways. First, genetic structure was investigated via Nei's measures of genetic diversity (Nei, 1973), which include total genetic diversity (i.e. total expected heterozygosity) at a polymorphic locus (HT), mean genetic diversity within populations (HS), and the proportion of genetic diversity occurring among populations [GST = (HT – HS)/HT] (Culley et al., 2002). Secondly, Weir and Cockerham's estimates (Weir and Cockerham, 1984) of Wright's F statistics (Wright, 1965) were generated for each polymorphic locus. Significant deviations from the null expectation of F = 0 were determined by 5000 bootstrap replicates, calculations being undertaken using the FSTAT program (Goudet, 1995). In the bootstrap analysis, F (corresponding to Wright' FIT) was estimated by alleles permutated among populations, f(FIS) was estimated by the permutation of alleles within samples, and f(FST) was estimated by the permutation of alleles among samples. The average gene flow among populations (Nm) was estimated from f-values as f = 1/(4Nmα + 1) where α = [n/(n – 1)]2 and n is number of populations (Crow and Aoki, 1984). Measures of within-population variation were related to latitude and population size using Spearman rank correlation (rs). Probability values for differences between regions are given for the two-sided t-test, after 10 000 permutations. All analyses were performed using FSTAT software (Goudet, 1995).
A Mantel test was used to assess the model of isolation-by-distance using the genetic distance for pairs of populations (Nei, 1978) and geographic distance among these populations. The Nei's genetic distance was also employed to obtain a UPGMA phenogram, after 1000 bootstrap samples (calculated using TFPGA; Miller, 1997).
RESULTS
Allozyme polymorphism
Altogether, five enzyme systems (AAT, LAP, MDH, SKDH and SOD) were consistently resolved and scored for 13 putative loci (Aat-1, Aat-2, Lap-1, Lap-2, Mdh-1, Mdh-2, Mdh-3, Skdh-1, Sod-1, Sod-2, Sod-3, Sod-4 and Sod-5). Four loci (Aat-1, Sod-1, Sod-2 and Sod-5) turned out to be monomorphic across the study area. Nine polymorphic (at the 0·05 level) loci (Aat-2, Lap-1, Lap-2, Mdh-1, Mdh-2, Mdh-3, Skdh-1, Sod-3 and Sod-4), with a total of 30 alleles, were evaluated further. The allelic frequencies of each polymorphic locus (for all populations) are given in Appendix 1. Enzyme systems ADH, EST, G-6-PDH, IDH, ME, PGI, PGM, 6-PGDH and locus Skdh-2 were not consistently resolved.
Variation in allele frequencies and levels of inbreeding
The average number of alleles per polymorphic locus (A) ranged from 1·23 (locality 5) to 1·85 (localities 12 and 17), with a mean of 1·55. The effective number of alleles per locus (Ae) was 1·26 and the mean percentage of polymorphic loci (PL), evaluated at the 95 % confidence level, was 47·1 %. The interpopulation differences in observed heterozygosity (Ho) are due to deviations from the Hardy–Weinberg equilibrium, as shown by the average fixation indices per populations (Table 1). The Ho ranged from 0·054 (locality 6) to 0·231 (locality 14), with a mean of 0·151, while the expected heterozygosity (He) ranged from 0·070 (locality 6) to 0·253 (locality 14), with a mean of 0·155.
The value of f(FIS) was highest in population 6 (0·241) and lowest in population 9 (–0·191), indicating the presence of both populations with an excess of heterozygotes (f < 0) compared with expected Hardy–Weinberg allelic frequencies and populations showing an excess of homozygous individuals, suggesting high levels of inbreeding (Table 1).
Frequency maps for representative loci (Fig. 2) revealed little variation in allele frequencies between regional populations, a majority of the alleles occurring in all regions and with few alleles completely absent from the Bohemian marginal region. While there was only weak geographical structure in allele frequencies, the average number of alleles per polymorhic locus (A) was negatively correlated with latitude (rs = −0·40, P = 0·045). Other genetic diversity measures, i.e. percentage of polymorphic locus (PL), observed heterozygosity (Ho), expected heterozygosity (He) and Weir and Cockerham's estimate (Cockerham, 1984) of FIS(f), were not correlated with latitude (rs = −0·28, P = 0·183; rs = −0·06, P = 0·758; rs = −0·03, P = 0·870; rs = −0·18, P = 0·384, respectively). When the population size was taken into account, no significant correlation was found between population size and A, PL, Ho, He and FIS (f) (data not shown).
Fig. 2.


Allele frequencies at four polymorphic loci (Aat-2, Mdh-3, Lap-1 and Skdh-1) for samples taken from the 25 localities in the Czech Republic.
Hierarchical partitioning of diversity
Estimates of the total genetic diversity (HT) reached a mean value of 0·196, ranging from 0·103 to 0·434, and the within-population genetic diversity (HS) reached a mean value 0·156, ranging between 0·053 and 0·347 (Table 2). The between-population component of diversity showed a mean value of 0·208, ranging from 0·051 to 0·789 (Table 2).
Table 2.
Statistics for genetic variation and structure for nine polymorphic loci (enzyme systems AAT, LAP, MDH, SKDH and SOD) identified in 25 Atriplex tatarica populations
| Nei's |
Wright's |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus (alleles) |
HT |
HS |
GST |
f(FIT) |
f(FIS) |
f(FST) |
Nm |
|||||
| Lap-1 (5) | 0·434 | 0·347 | 0·200 | 0·196* | −0·013 | 0·207* | 0·88 | |||||
| Lap-2 (2) | 0·342 | 0·289 | 0·153 | 0·244* | 0·101 | 0·158* | 1·23 | |||||
| Sod-3 (2) | 0·184 | 0·168 | 0·083 | 0·327* | 0·264* | 0·086* | 2·45 | |||||
| Sod-4 (2) | 0·303 | 0·275 | 0·093 | −0·222 | −0·352 | 0·096* | 2·17 | |||||
| Mdh-1 (2) | 0·367 | 0·314 | 0·146 | 0·080 | −0·084 | 0·151* | 1·30 | |||||
| Mdh-2 (2) | 0·103 | 0·097 | 0·051 | 0·961* | 0·959* | 0·053* | 4·12 | |||||
| Mdh-3 (3) | 0·155 | 0·132 | 0·144 | 0·178* | 0·034 | 0·150* | 1·31 | |||||
| Skdh-1 (5) | 0·413 | 0·346 | 0·161 | 0·191* | 0·029 | 0·167* | 1·15 | |||||
| Aat-2 (3) | 0·253 | 0·053 | 0·789 | 0·832* | 0·175 | 0·796* | 0·06 | |||||
| Over all loci | 0·196 | 0·156 | 0·208 | 0·239* | 0·031 | 0·214* | 0·85 | |||||
HT = total genetic diversity for the species; HS = mean within-population genetic diversity; GST = proportion of total genetic diversity among populations; F, f, f = Weir and Cockerham's estimates of Wright's F statistics (FIT, FIS and FST, respectively), which represents deviations from Hardy–Weinberg expectations over all populations, deviations within individual populations, and the proportion of total genetic diversity partitioned among populations.
Nm was approximated from f using Crow and Aoki's formula (Crow and Aoki, 1984).
Significant deviation (P < 0·05) from the null expectation of F = 0.
The mean overall f(FIS) of 0·031 was not statistically different from zero, only Sod-3 and Mdh-2 exhibiting significant levels of inbreeding (f > 0) (Table 2). f(FST) was high (0·214), and significantly different from zero (Table 2), indicating significant genetic differentiation among populations, i.e. 21·4 % of the total genetic variation was due to differences among populations and 78·6 % of the total genetic variability was found within populations. In this case, all individual loci had significant f(FST) values. As a consequence, F(FIT) = 0·239 was significantly positive (Table 2).
Genetic differentiation
To further understand the pattern of inbreeding and differentiation, the 25 populations were divided into two geographic groups (Table 1): Bohemia (localities 1–11) and Moravia (localities 12–25) (Fig. 1). The mean values of f(FIS) were not significantly different from zero and increased from the geographically marginal populations in Bohemia to populations situated in the area of continuous distribution in Moravia (Table 3). Significant differentiation among populations in both regions was detected. Higher values of f(FST) were reached by populations in the area of continuous distribution (Moravia) and lower values were detected in the marginal populations of Bohemia (Table 3).
Table 3.
Nei's and Wright's statistics and estimates of Nm per regions of Atriplex tatarica from Bohemia (1–11) and Moravia (12–25)
The multi locus estimate of the mean number of migrants per generation (Nm) was low (Table 2, average number 0·85), suggesting that gene flow is probably restricted in A. tatarica. In considering both of our geographic regions separately (Table 3), Nm declines from Moravia to Bohemia, suggesting lower levels of gene flow among Moravian populations.
Comparison of the two regions studied revealed significant differences in allelic richness (A), with populations in the area of continuous distribution (Moravia) containing more alleles per polymorphic locus (Table 4). In contrast, there were no significant differences between regions for the observed heterozygosity, gene diversity, or FIS and FST estimates (Table 4), in spite of generally lower values of particular genetic measurements in the marginal region of Bohemia.
Table 4.
Comparison of population polymorphism between Bohemia (n = 11 populations) and Moravia (n = 14
| Diversity measure |
Bohemia |
Moravia |
P |
|---|---|---|---|
| A | 1·462 | 1·615 | 0·032 |
| Ho | 0·141 | 0·159 | 0·309 |
| Hs | 0·145 | 0·164 | 0·314 |
| FIS | 0·029 | 0·033 | 0·947 |
| FST | 0·186 | 0·225 | 0·629 |
A = average number of alleles per polymorphic locus, Ho = observed heterozygosity, HS = gene diversity, FIS and FST are estimated according to Weir and Cockerham (1984).
Probability values for differences between regions are given for two-sided t-test, after 10 000 permutations. Analysis was performed with FSTAT software.
Cluster analysis (Fig. 3), based on Nei's genetic distances (Nei, 1978), did not show any clear geographic structure. There was no consistent association between genetic and geographic distances, as indicated by the Mantel test (r = 0·017, R2 = 0·03, P = 0·758), for the whole study area. However, Mantel tests were significant for the Bohemian marginal region (r = 0·356, R2 = 0·13, P = 0·009) and non-significant for the Moravian central ones (r = 0·0002, R2 = 0, P = 0·999).
Fig. 3.

UPGMA phenogram constructed on the basis of five enzyme systems (AAT, LAP, MDH, SKDH and SOD) using Nei's genetic distances (Nei, 1978) for 25 populations of Atriplex tatarica in the Czech Republic. Populations were coded by region: B, Bohemia; M, Moravia (see Table 1 and Fig. 1 for population locations).
DISCUSSION
The present study investigated genetic diversity at allozyme loci in an annual herb common in human-made habitats, A. tatarica, from a sample of populations through the north-eastern margin of the species' range. The possibility of comparing genetic variation of A. tatarica with other Atriplex species is limited due to the low number of species examined for allozyme diversity. Indeed, the genus Atriplex has been studied with respect to its physiology and ecology (Osmond et al., 1980) much more than for genetic variability, the only published genetic work being on A. halimus in North Africa (Haddioui and Baaziz, 2001). Unfortunately, Atriplex halimus is a perennial desert halophyte species that has been classified as an outcrossing wind-pollinated species on the basis of allozyme data, making it difficult to compare it with annual species of disturbed habitats. Hamrick and Godt (1996) characterized Chenopodiaceae as the species with the lowest percentage of polymorphic loci and genetic diversity within species and the highest amount of genetic diversity found among populations. Although it was stated that ‘genetic diversity and its distribution are more closely associated with individual species’ life history traits than with their phylogenetic status', genetic variation measures are in agreement with a predominant autogamous reproduction mode in Chenopodiaceae, leading to higher population differentiation and lower within-population variability. Hamrick and Godt (1996) further identified mating system and geographical range as important correlates of the levels of genetic variation. Considering both its wide range, from central Europe to central Asia, and mixed mating system, the annual A. tatarica tends to be less variable (HS = 0·156) than expected based on the estimated mean level of genetic variation in this group (HS = 0·206; Hamrick and Godt, 1996), and the overall GST estimate for A. tatarica (0·208) is higher than that normally found in widespread species with a mixed-mating system (GST = 0·169) (Hamrick and Godt, 1996). Based on the regional analysis (Tables 3 and 4), these patterns can be attributed to the deviating estimates for populations in the central region, rather than consistent differences in the structuring of variation between A. tatarica and other widespread species with mixed-mating systems.
Previous work has shown conflicting results as regards the relationship between marginality and measures of within-species variation. Several authors have found marginal populations to be less variable than populations representing more central regions (Mahy et al., 1997; Broyles, 1998; Jorgensen et al., 2002; Griffin and Barrett, 2004; Persson et al., 2004), whereas a number of studies report no or weak effects (e.g. Levin, 1977; Yeh and O'Malley, 1980; Comps et al., 1990; Betancourt et al., 1991; Schiemann et al., 2000).
The populations in the area of continuous distribution of A. tatarica showed significantly higher allelic richness, but observed heterozygosity, gene diversity, and FIS and FST estimates were not significantly higher (Table 4). This result is not surprising, considering the small sizes of the marginal populations and the typical loss of variability in many small isolated populations (Cole, 2003). The significant reduction of A, but non-significant reduction of HS was predicted by Nei et al. (1975). If population size increases rapidly after going through a bottleneck, the reduction in average heterozygosity is small compared with the reduction in the average number of alleles. This difference occurs mainly because genetic drift eliminates low frequency alleles. However, the pattern of population sub-division showed that populations from central areas are more divergent from each other than those from the marginal area (Table 4). It might be argued that the similarity among the marginal populations has been generated by a single population giving rise to the others in this marginal area. This would seem especially likely if the marginal populations were adjacent to each other and isolated from the central ones as in the case of A. tatarica. The scenario that marginal populations have a greater likelihood of gene flow from central populations than from other adjacent marginal populations has lower probability due to the geographical distance (>200 km) of the central population of A. tatarica. This view is also encouraged by the non-significant correlation between geographical and genetic distances within the whole study area and by the significant result in the case of the Bohemian marginal region as opposed to the Moravian central regions.
Theoretically, repeated founding events associated with metapopulation dynamics may decrease genetic diversity within, and increase genetic differentiation among, local populations (Ingvarsson et al., 1997). These effects will be strongest where the colonists are derived from single rather than multiple demes and where rates of gene exchange among extant demes are low relative to the rates of population turnover (Whitlock and McCauley, 1990).
Atriplex tatarica is a species of early succession stages that does not survive for >3 years due to proceeding ecological succession; therefore, the species has high spatio-temporal variation across its range. The spreading of A. tatarica to new areas, with consequent formation of isolated populations, is probably due to both human building activity, which moves fruits over long distances through transportation of soil, (Mandák, 2003a) and salt treatment of roads in winter, A. tatarica being a facultative halophilic species (Mandák, 2003b) that is able to grow and spread on habitats with higher salt concentrations. Thus, treating roads with salt in winter results in several halophytic species being spread for long distances to climatically less favourable conditions where they survive in salty habitats without surrounding vegetation, forming small isolated populations (Mandák, 2003a). These specific ‘urban’ niches promote the spread of this species and enable it to occupy relatively large areas. The present analysis of population structure of A. tatarica revealed considerable population differentiation at allozyme loci. However, there was no clear geographical clustering of populations in the UPGMA analysis or evidence for isolation-by-distance over the whole study area.
In summary, several conclusions can be drawn from the patterns of genetic variation and structure in A. tatarica. First, A. tatarica, as a species of human-made habitats with a mixed mating system and wide geographic distribution, possesses moderate genetic diversity, which is consistent with the expectation for that type of organism. Secondly, when considering the whole study area, there were no consistent associations between genetic and geographic distances, probably due to occasional long-distance dispersal, disturbance events, or to anthropogenic factors. Thirdly, genetic diversity, with the exception of allelic richness, was not significantly lower at the margins of the species' range, in spite of generally lower values of particular genetic measurements in the marginal region, thus only weakly supporting the predictions of the Central–Marginal Model.
Acknowledgments
We would like to thank Christian Lexer, Jan Štěpánek, Mohammed Baaziz and an anonymous reviewer for helpful comments on the manuscript and Vít Dvořák for assisting in the preparation of the maps. Frederick Rooks kindly improved our English. This study was supported by grant no. IAA6005206 from the Grant Agency of the Academy of Sciences of the Czech Republic.
LITERATURE CITED
- Aellen P. 1960.Atriplex In: Hegi G, ed. Illustrierte Flora von Miteleuropa. München, 3/2: 664–693 [in German]. [Google Scholar]
- Arnold ML. 1997.Natural hybridization and evolution. Oxford: Oxford University Press. [Google Scholar]
- Barrett SCH. 1982. Genetic variation in weeds. In: Charudattan R, Walker H, eds. Biological control of weeds with plant pathogens. New York: John Wiley and Sons, 73–98. [Google Scholar]
- Betancourt JL, Schuster WS, Mitton JB, Anderson RS. 1991. Fossil and genetic history of a pinyon pine (Pinus edulis) isolate. Ecology 72: 1685–1697. [Google Scholar]
- Bosbach K, Hurka H. 1981. Biosystematic studies on Capsella bursa-pastoris (Brassicaceaea): enzyme polymorphism in natural populations. Plant Systematics and Evolution 137: 73–94. [Google Scholar]
- Brown AHD, Burdon JJ. 1983. Multilocus diversity in an outbreeding weed, Echium plantagineum L. Australian Journal of Biological Science 36: 503–509. [Google Scholar]
- Broyles SB. 1998. Postglacial migration and the loss of allozyme variation in northern populations of Asclepias exaltata (Asclepiaceae). American Journal of Botany 85: 202–206. [PubMed] [Google Scholar]
- Chauvet S, van der Velde M, Imbert E, Guillemin ML, Mayol M, Riba M, et al. 2004. Past and current gene flow in the selfing, wind dispersed species Mycelis muralis in western Europe. Molecular Ecology 13: 1391–1407. [DOI] [PubMed] [Google Scholar]
- Cole CT. 2003. Genetic variation in rare and common plants. Annual Review of Ecology, Evolution and Systematics 34: 213–237. [Google Scholar]
- Comps B, Thiébaut B, Paule L, Merzead D, Letouzey J. 1990. Allozyme variability in beechwoods (Fagus sylvatica L.) over central Europe: spatial differentiation among and within populations. Heredity 65: 407–417. [Google Scholar]
- Crow JF, Aoki K. 1984. Group selection for a polygenic behavioral trait: estimating the degree of population subdivision. Proceedings of the National Academy of Sciences of the USA 81: 6073–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culley TM, Wallace LE, Gengler-Nowak KM, Crawford DJ. 2002. A comparison of two methods of calculating GST, a genetic measure of population differentiation. American Journal of Botany 89: 460–465. [DOI] [PubMed] [Google Scholar]
- Da Cunha AB, Dobzhansky T. 1954. A further study of chromosomal polymorphism in Drosophila willistoni in relation to environment. Evolution 8: 119–134. [Google Scholar]
- Ellstrand NC, Schierenbeck KA. 2000. Hybridization as a stimulus for the evolution of invasiveness in plants? In: Ayala FJ, Fitch WM, Clegg MT, eds. Variation and evolution in plants and microorganisms. Washington, DC: National Academy Press, 289–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks SJ, Richards CL, Gonzales E, Cousins JE, Hamrick JL. 2004. Multi-scale analysis of Uniola paniculata (Poaceae): a coastal species with linear, fragmented distribution. American Journal of Botany 91: 1345–1351. [DOI] [PubMed] [Google Scholar]
- Godt MJW, Hamrick JL. 1991. Genetic variation in Lathyrus latifolius (Leguminosae). American Journal of Botany 78: 1163–1171. [Google Scholar]
- Goodnight CJ. 1987. On the effect of founder events on epistatic genetic variance. Evolution 41: 80–91. [DOI] [PubMed] [Google Scholar]
- Goodnight CJ. 1988. Epistasis and the effect of founder events on the additive genetic variance. Evolution 42: 441–454. [DOI] [PubMed] [Google Scholar]
- Goudet J. 1995. Fstat version 1·2: a computer program to calculate F-statistics. Journal of Heredity 86: 485–486. [Google Scholar]
- Griffin SR, Barrett SCH. 2004. Genetic variation in Trillium erectum (Melanthiaceae), a widespread forest herb in eastern North America. Canadian Journal of Botany 82: 316–321. [Google Scholar]
- Haddioui A, Baaziz M. 2001. Genetic diversity of natural populations of Atriplex halimus L. in Marocco: an isoenzyme-based overview. Euphytica 121: 99–106. [Google Scholar]
- Hamrick JL, Godt JW. 1996. Effects of life history traits on genetic diversity in plant species. Philosophical Transactions of the Royal Society of London 351: 1291–1298. [Google Scholar]
- Hayward MD, Zaruk MTM. 1982. Allozyme variation in the inbreeding species Lolium temulentum L. Heredity 49: 255–257. [Google Scholar]
- Hewitt GM. 1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biological Journal of the Linnean Society 58: 247–276. [Google Scholar]
- Hewitt GM. 1999. Post-glacial re-colonization of Europaean biota. Biological Journal of the Linnean Society 68: 87–112. [Google Scholar]
- Hollingsworth ML, Bailey JP. 2000. Evidence for massive clonal growth in the invasive Fallopia japonica (Japanese Knotweed). Botanical Journal of the Linnean Society 133: 463–472. [Google Scholar]
- Ibrahim KM, Nichols RA, Hewitt GM. 1996. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77: 282–291. [Google Scholar]
- Ingvarsson PK, Olsson K, Ericson L. 1997. Extinction–recolonization dynamics in the mycophagous beetle Phalacrus substriatus Evolution 51: 187–195. [DOI] [PubMed] [Google Scholar]
- Jalas J, Suominen J. eds. 1988.Atlas florae Europaeae: distribution of vascular plants in Europe, Volume II. Cambridge: Cambridge University Press. [Google Scholar]
- Jorgensen S, Hamrick JL, Wells PV. 2002. Regional patterns of genetic diversity in Pinus flexilis (Pinaceae) reveal complex species history. American Journal of Botany 89: 792–800. [DOI] [PubMed] [Google Scholar]
- Koniuszek JWJ, Verkeij JAC. 1982. Genetic variation in two related annual Senecio species occurring in the same habitat. Genetica 59: 133–137. [Google Scholar]
- Kubetin WR, Schaal BA. 1979. Apportionment of isozyme variability in Polygonum pemsylvanicum (Polygonaceaea). Systematic Botany 4: 148–156. [Google Scholar]
- Levin DA. 1977. The organisation of genetic diversity in Phlox drummondii Evolution 31: 477–494. [DOI] [PubMed] [Google Scholar]
- Mahy G, Vekemans X, Jacquemart A, De Sloover J. 1997. Allozyme diversity and genetic structure in south-western population of heather (Calluna vulgaris). New Phytologist 137: 325–334. [DOI] [PubMed] [Google Scholar]
- Mandák B. 2003. Distribution of four Atriplex species with different degrees of invasiveness in the Czech Republic. In: Child LE, Brock JH, Brundu G, Prach K, Pyšek P, Wade PM, et al, eds. Plant invasions: ecological threats and management solutions. Leiden: Backhuys Publisher, 313–328. [Google Scholar]
- Mandák B. 2003. Germination requirements of invasive and non-invasive Atriplex species: a comparative study. Flora 198: 45–54. [Google Scholar]
- Mandák B, Bímová K, Pyšek P, Štěpánek J, Plačková I. 2005. Isoenzyme diversity in Reynoutria taxa: escape from sterility by hybridization. Plant Systematics and Evolution 253: 219–230. [Google Scholar]
- Mandák B, Pyšek P, Lysák M, Suda J, Krahulcová A, Bímová K. 2003. Variation in DNA-ploidy levels of Reynoutria taxa in the Czech Republic. Annals of Botany 92: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. 1997.Tools for population genetic analyses (TFPGA) 1·3: a Windows program for the analysis of allozyme and molecular population genetic data http://bioweb.usu.edu/mpmbio/tfpga.asp (6 Dec. 2004). Website now at http://www.marksgeneticsoftware.net [Google Scholar]
- Nei M. 1973. Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences of the USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Maruyama T, Chakraborty R. 1975. The bottleneck effect and genetic variability in populations. Evolution 29: 1–10. [DOI] [PubMed] [Google Scholar]
- Novak GF, Mack RN. 1993. Genetic variation in Bromus tectorum (Poaceae): comparison between native and introduced populations. Heredity 71: 167–176. [Google Scholar]
- Osmond CB, Björkman O, Anderson DJ. 1980.Physiological processes in plant ecology – towards a synthesis with Atriplex. Berlin: Springer Verlag. [Google Scholar]
- Pappert RA, Hamrick JL, Donovan LA. 2000. Genetic variation in Pueraria lobata (Fabaceaea), an introduced, clonal, invasive plant of the southeastern United States. American Journal of Botany 87: 1240–1245. [PubMed] [Google Scholar]
- Persson H, Widén B, Andersson S, Svensson L. 2004. Allozyme diversity and genetic structure of marginal and central populations of Corylus avellana (Betulaceae) in Europe. Plant Systematics and Evolution 244: 157–179. [Google Scholar]
- Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, et al. 2003. Major ecological transitions in wild sunflowers facilitated by hybridisation. Science 301: 1211–1216. [DOI] [PubMed] [Google Scholar]
- Schiemann K, Tyler T, Widén B. 2000. Allozyme diversity in relation to geographic distribution and population size in Lathyrus vernus (L.) Bernh. (Fabaceae). Plant Systematics and Evolution 225: 119–132. [Google Scholar]
- Schierenbeck KA, Hamrick JL, Mack RN. 1995. Comparison of allozyme variability in a native and an introduced species of Lonicera Heredity 75: 1–9. [Google Scholar]
- Vallejos CE. 1983. Enzyme activity staining. In: Tanksley SD, Orton TJ, eds. Isozyme in plant genetics and breeding, Part A. Amsterdam: Elsevier, 469–516. [Google Scholar]
- Vila M, Weber E, D'Antonio CM. 2000. Conservation implications of invasion by plant hybridization. Biological Invasions 2: 207–217. [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370. [DOI] [PubMed] [Google Scholar]
- Wendel JF, Weeden NF. 1989. Visualisation and interpretation of plant isozymes. In: Soltis DE, Soltis PS, eds. Isozymes in plant biology. Portland, OR: Dioscoroides Press, 5–45. [Google Scholar]
- Whitlock MC, McCauley DE. 1990. Some population genetic consequences of colony formation and extinction – genetic correlations within founding groups. Evolution 44: 1717–1724. [DOI] [PubMed] [Google Scholar]
- Wright S. 1965. The interpretation of population structure by F-statistics with special regard to system mating. Evolution 19: 395–420. [Google Scholar]
- Yeh FC, O'Malley DO. 1980. Enzyme variation in natural populations of Douglas fir, Pseudotsuga menziesii (Mibr.) Franco, from British Columbia. 1. Genetic variation in coastal populations. Silvae Genetica 29: 83–92. [Google Scholar]
- Yeh FC, Yang R-C, Boyle T. 1999.POPGENE version 1·32. Microsoft Window-based freeware for population genetic analysis http://www.ualberta.ca/~fyeh/. 6 Dec. 2004. [Google Scholar]