

Abstract
Apoptosis is a tightly regulated cellular process and faulty regulation of apoptosis is a hallmark of human cancers. Targeting key apoptosis regulators with the goal to restore apoptosis in tumor cells has been pursued as a new cancer therapeutic strategy. XIAP, cIAP1, and cIAP2, members of inhibitor of apoptosis (IAP) proteins, are critical regulators of cell death and survival and are attractive targets for new cancer therapy. The SMAC/DIABLO protein is an endogenous antagonist of XIAP, cIAP1, and cIAP2. In the last decade, intense research efforts have resulted in the design and development of several small-molecule SMAC mimetics now in clinical trials for cancer treatment. In this review, we will discuss the roles of XIAP, cIAP1, and cIAP2 in regulation of cell death and survival, and the design and development of small-molecule SMAC mimetics as novel cancer treatments.
Keywords: SMAC Mimetics, Apoptosis, Inhibitors
Introduction: Apoptosis pathways
Apoptosis is one form of programmed cell-death and is a normal cellular process used by multi-cellular organisms to eliminate damaged or unwanted cells. Apoptosis is a tightly regulated process and faulty regulation of apoptosis is implicated in many human diseases, including cancer, autoimmune diseases, inflammation, and neurogenesis [1–3]. Indeed, resistance to apoptosis is a hallmark of human cancers [4].
Two main apoptotic pathways, the intrinsic and the extrinsic pathways, have been extensively investigated. The intrinsic pathway is also called the mitochondrial pathway and characterized by permeabilization of the mitochondria induced by a variety of stress signals such as chemotherapeutic drugs or radiation. At the molecular level, the intrinsic pathway involves the translocation and oligomerization of BAX or BAK, members of the BCL-2 protein family. BAK or BAX forms a pore in the outer mitochondrial wall leading to the release of cytochrome c and second mitochondrial-derived activator of caspases (SMAC, also known as DIABLO, direct IAP-binding protein with low pI) from mitochondria into the cytosol. In the cytosol, cytochrome c, together with apoptotic protease activating factor 1 (APAF1) and pro-caspase-9, forms a multi-protein complex apoptosome, which cleaves zymogen pro-caspase-9 into active caspase-9. Active caspase-9 then cleaves and activates effector caspases, caspase-3 and caspase-7. Active caspase-3 and caspase-7 cleave down-stream cell-death substrates, ultimately leading to apoptosis.
The extrinsic pathway is initiated by the binding of death ligands such as Fas/Apo-1, TNFα (tumor necrosis factor α), Apo2L/TRAIL (TNF-related apoptosis-inducing ligand) to their corresponding cognate death receptors (CD95/FasR, TNFR1 and DR4/DR5) on the cell-surface. The binding of a death ligand to its receptor(s) results in recruitment of multiprotein death-inducing signaling complex (DISC) at the plasma membrane. The DISC contains an adapter protein, which recruits procaspase-8 into the complex and results in autoactivation of caspase-8. Further cleavage of caspase-8 activates caspase-3 and -7, leading to apoptosis.
There are extensive cross-talks between the intrinsic and extrinsic pathways. For example, caspase-8 also cleaves BID, a BCL-2 member, to generate truncated BID (tBID). tBID translocates to the mitochondrial membrane where it binds to BAX and BAK and stimulates the release of cytochrome c, leading to activation of the intrinsic pathway. In addition, both intrinsic and extrinsic apoptosis pathways converge in the activation of effector caspase-3 and -7, which can further activate caspase-8, creating a positive feedback loop for apoptosis.
IAP proteins as key regulators of apoptosis
Apoptosis is tightly regulated at multiple levels and inhibitor of apoptosis proteins (IAPs) are a class of key negative regulators of both the intrinsic and extrinsic apoptosis pathways.
IAP proteins were first identified in baculoviruses for their ability to inhibit virus-induced apoptosis [5, 6]. IAP family proteins are defined by the presence of one to three of the 70–80 amino acid baculoviral IAP repeat (BIR) domains. The mammalian IAP family is comprised of the following eight members;
neuronal IAP (BIRC1)
cellular IAP1 (cIAP1, BIRC2)
cellular IAP2 (cIAP2, BIRC3)
X chromosome-linked IAP (XIAP, BIRC4)
survivin (BIRC5)
ubiquitin-conjugating BIR domain enzyme apollon (BIRC6)
melanoma IAP (ML-IAP, BIRC7)
IAP-like protein 2 (BIRC8)
Among these, XIAP, cIAP1, cIAP2, and ML-IAP, are known to play a direct role in the regulation of apoptosis [7]. This review will focus on XIAP, cIAP1, and cIAP2, which have predominant roles in the regulation of both intrinsic and extrinsic apoptosis pathways.
Structurally, XIAP contains three BIR domains (BIR1-BIR3), a UBA (ubiquitin-associated domain) [8], and a RING (really interesting new gene) domain (Figure 1). The BIR1 domain mediates XIAP’s interaction with TAK1 (TGFβ-activated kinase) and TAB1 (TAK1 binding protein 1) [9, 10]. The BIR2 domain and the linker region preceding it mediate the interaction of XIAP with active caspase-3 and -7 [11, 12]. The BIR3 domain binds to procaspase-9, blocking its dimerization and activation [13]. The UBA domain mediates XIAP’s association with mono- and polyubiquitin chains [8], and the RING domain confers E3 ubiquitin ligase activity [14]. In addition to these aforementioned functional domains in XIAP, cIAP1 and cIAP2 contain an evolutionary conserved caspase recruitment domain (CARD), and this CARD domain suppresses the activation of the RING domain E3 ligase activity of cIAP1 [15]. Distinct from the BIR1 domain in XIAP, the BIR1 domain of the cIAP1/2 proteins mediates their interactions with TRAF2 (TNF receptor-associated factor 2) [16, 17] (Figure 1).
Figure 1.
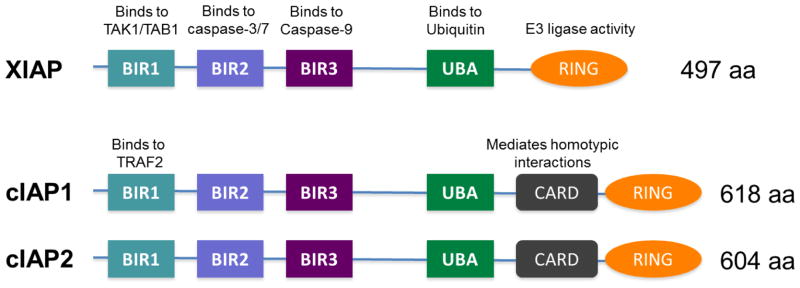
Functional domains of mammalian inhibitor of apoptosis proteins (IAPs). BIR: baculoviral IAP repeat domain; UBA: Ubiquitin-associated domain; CARD: caspase recruitment domain; RING: Really interesting new gene finger domain.
IAP proteins suppress apoptosis by inhibiting the activities and/or the activation of caspases. XIAP is the only IAP protein that inhibits caspase activity through direct binding, whereas cIAP1 and cIAP2 inhibit caspase activity indirectly. The BIR2 domain and the preceding linker region of XIAP bind to the IAP-binding motif and active site of caspase-3 and -7, respectively [12, 18]. The BIR3 domain of XIAP binds to pro-caspase-9, blocking its dimerization and subsequent activation [19]. Since effector caspase-3 and -7 play a key role in the execution of apoptosis in both the extrinsic and intrinsic pathways, and caspase-9 is a critical initiator caspase in the intrinsic pathway, XIAP can effectively inhibit both intrinsic and extrinsic apoptosis pathways [20] (Figure 2). The regulation of apoptosis pathways by cIAP1 and cIAP2 centers on the E3 ubiquitin ligase activity of their RING domain. Dimerization of the RING domain potentiates the ubiquitin ligase activity of IAP proteins -permitting the auto-ubiquitination as well as cross-ubiquitination of other IAP proteins [21, 22]. For example, cIAP1 can mediate the ubiquitination of cIAP2 and XIAP [17, 21, 23]. Other prominent substrates of the E3 ubiquitin ligase of XIAP and/or cIAP proteins include RIP1-4 (receptor-interacting protein) [24, 25], NIK (nuclear factor kB, or NF-kB-inducing kinase) [17], caspases [26], TRAF2 [27], TRAF3/6 [28], SMAC [29], and C-RAF [30] among others [31–33]. Therefore, IAP proteins can modulate the signaling pathways mediated by their substrates. In particular, RIP1 ubiquitination by cIAP1 and cIAP2 is a critical event for TNFα-induced activation of canonical NF-kB signaling [33], which prevents RIP1 from forming a cytosolic complex with caspase-8 and Fas-associated protein with death domain (FADD) (complex II), and subsequent activation of caspases [33, 34].
Figure 2.

IAP proteins play prominent roles in NF-κB signaling and cell death pathways. See the text for details.
IAP proteins as regulators of NF-kB signaling pathways
As described above, the ubiquitin ligase function of cIAP proteins enables them to modulate various signaling pathways, most notably the NF-kB signaling pathways. Numerous studies have established a role of cIAP proteins in regulating both the canonical and non-canonical NF-kB signaling pathways (Figure 2).
NF-kB proteins include RelA (p65), RelB, c-Rel, NF-kB1 (p105/50), and NF-kB2 (p100/52). These proteins function as dimeric transcription factors that regulate the expression of diverse target genes involved in inflammation, proliferation, survival, cell death, angiogenesis, migration, and invasion [35, 36]. In canonical NF-kB signaling, NF-kB proteins are bound to and inhibited by IκB (inhibitor of kB) proteins. For example, the NF-kB p50-RelA dimer is inhibited by IkBα, which blocks the nuclear translocation of NF-kB to activate target gene expression. The ubiquitin ligase activity of cIAP proteins is essential for the recruitment and assembly of the signaling activation complex upstream of NF-kB activation in a number of TNF superfamily receptors, such as TNFR1, LT-βR, and CD40. For instance, the binding of TNF to TNFR1 stimulates the recruitment and formation of a multiprotein complex containing TRADD (TNFR-associated death domain protein), RIP1, TRAF2, and cIAPs [32, 37] (Figure 2). In this complex, cIAP protein promotes the K63-linked polyubiquitination of RIP1 [33, 34]. The ubiquitination of RIP1 serves as a signaling platform for the recruitment of IKK (IκB kinase) complex [IKKα, IKKβ and NEMO (NF-kB essential modulator)], TAK complex (TAK1 and TAB1/2), and LUBAC (linear ubiquitin chain assembly complex), leading to downstream activation of NF-kB and MAPK (mitogen-activated protein kinase) pathways. Notably, XIAP can also promote the activation of TAK1 in TGFβ/BMP signaling and in response to genotoxic stress [9, 38, 39].
In addition to positively regulating canonical NF-kB signaling, cIAP proteins are also key negative regulators of non-canonical NF-kB signaling. At rest, cIAPs control the stability of NIK via ubiquitination, and thus prevent the activation of downstream IKKα. In the absence of cIAPs however, NIK accumulates, leading to the phosphorylation of IKKα. This is followed by the phosphorylation of NF-kB2 p100 and its cleavage to p52. The p52 subunit dimerizes with RelB to activate NF-kB target genes.
NF-kB is frequently activated in human malignancies and plays a critical role in tumorigenesis, tumor progression, and metastasis [40]. In mucosa-associated lymphoid tissue (MALT) lymphoma, the fusion of the BIR domain of cIAP2 with the MALT1 is prevalent, and is associated with constitutive activation of canonical NF-kB signaling [41, 42]. Inactivating mutations of cIAP proteins leads to constitutive activation of the non-canonical NF-kB pathway in multiple myeloma [43, 44]. Meanwhile, XIAP physically associates with survivin to drive NF-kB activation, which promotes tumor cell invasion in vitro and metastasis in vivo [45].
In addition to its most commonly appreciated pro-survival functions, depending on the stimuli and the cellular context, NF-kB can also promote apoptosis through regulating the expression of proteins participating in cell death pathways, including the death-inducing tumor necrosis factor (TNF) superfamily ligands and receptors. As will be discussed in more detail below, the autocrine/paracrine production of TNFα has been shown to mediate SMAC mimetic-induced apoptosis [17, 46–49]. A very recent study has also shown that, in glioblastoma cells, SMAC mimetic stimulates NF-kB-mediated expression of death receptor DR5, followed by the formation of RIP1-containing cell death complex and eventually apoptosis in a death ligand-independent manner [50]. Thus, the SMAC mimetics-stimulated NF-kB activation is central to SMAC mimetic-stimulated apoptosis.
cIAP1 and cIAP2 proteins as negative regulators of RIP1-dependent cell death signaling
RIP1 is a multi-functional signal transducer which mediates adaptive cellular stress responses [51]. Under normal conditions, RIP1, as discussed, is constitutively ubiquitinated by cIAP proteins (Figure 2) and the ubiquitinated RIP1 serves as a signaling platform for the activation of NF-kB and MAPK pathways. In the absence of cIAP proteins or presence of deubiquitinases, ubiquination does not occur and the non-ubiquitinated RIP1 promotes the formation of a cytosolic complex (complex II), which includes the adaptor protein FADD, caspase 8, and RIP1. Complex II mediates the activation of caspase 8, ultimately leading to apoptosis. In response to genotoxic stress and stimulation by TLR3 (toll-like receptor 3), such a cytosolic non-ubiquitinated RIP1-containing caspase-activating complex, ripoptosome, can also be formed, independent of TNFR signaling [52, 53]. If functional caspase-8 is absent, non-ubiquitinated RIP1 interacts with RIP3 through their RIP homotypic interaction motif. The cross-phosphorylation of RIP1 and RIP3 stabilizes their association and activates their pro-necroptotic kinase activity. Activated RIP3 binds to and phosphorylates MLKL (mixed lineage kinase domain-like) to form necrosome, a pro-necroptotic complex, allowing nectoposis (programmed necrosis) to take place [54–58]. Therefore, by promoting the ubiquitination of RIP1, cIAP proteins prevent the recruitment and formation of RIP1-containing cell death activating complexes, thus blocking RIP1-dependent cell death signaling (Figure 2).
IAP proteins and human cancers
Elevated expression of XIAP and cIAP proteins have been reported in a variety of human cancers and their high expression is correlated with chemoresistance and poor prognosis in several types of cancer [59]. In breast carcinoma for example, high nuclear expression of XIAP is associated with poor prognosis [60]. Similarly, elevated levels of XIAP are correlated with poor prognosis in colorectal cancer [61, 62], prostate cancer [63, 64], chronic lymphocytic leukemia [65] and many other types of human cancer. In contrast, XIAP expression is reported to be correlated with good prognosis in non-small cell lung cancer (NSCLC) [66]. The genomic amplification of 11q21-22, which contains genes encoding cIAP1 and cIAP2, occurs at a high frequency in a variety of human cancers, such as hepatocellular carcinoma [67], lung cancer [68], esophageal squamous cell carcinoma [69], and cervical squamous cell carcinoma [70] among many others. In cervical squamous cell carcinoma, elevated levels of cIAP1 are correlated with resistance to radiotherapy [70] and in colorectal and bladder cancer, elevated levels of cIAP proteins are correlated with advanced stages of tumors and poor survival [71, 72]. High expression of cIAP1, cIAP2, and XIAP correlates with poor outcomes in multiple myeloma patients [73]. In MALT lymphoma, the fusion of the BIR domain of cIAP2 with MALT1 is prevalent, and is associated with constitutive activation of NF-kB signaling [41].
IAPs are involved in human cancers not only through direct and indirect regulation of apoptosis pathways but also through modulation of various non-apoptotic pathways, which primarily stem from their E3 ubiquitin ligase activity. Recently studies have reported, albeit controversially, that IAP proteins play a role in cell motility, migration, invasion, and metastasis [74]. Several studies suggest that IAP proteins can promote tumor cell migration and metastasis. Mehrotra et al. report that XIAP, in cooperation with survivin, promotes tumor cell invasion and metastasis by activating NF-kB-integrin β1 signaling and focal adhesion kinase and Src kinase [75]. Members of Rho GTPase family of the Ras superfamily, such as Rac1, RhoA, and Rho B play prominent roles in cell migration through regulating cytoskeleton formation, cellular polarity, and many other metastatic properties [76–78]. In HCT116 colon cancer cell line, XIAP interacts with RhoGDI (Rho GDP dissociation inhibitor) via its RING domain and negatively regulates RhoGDI sumoylation. Genetic depletion of XIAP causes marked reduction in β-actin polymerization and cytoskeleton formation, resulting in decreased cancer cell migration and invasion [79–81]. Furthermore, cIAP1 has been shown to regulate cell migration in a CARD-dependent manner and depletion of cIAP1 suppresses cell migration [15]. On the other hand, there are some studies suggesting that IAPs can regulate tumor cell migration negatively. It has been reported that the stabilities of C-RAF, a key regulator of the MAPK signaling downstream of Ras, are regulated by XIAP and cIAP proteins [30, 82]. Depletion of IAPs enhances the formation of lamellipodia and filopodia in HeLa cells, leading to enhanced cancer cell migration. XIAP and cIAP1 can bind directly to Rac1 in a nucleotide-independent manner to promote its polyubiquitination at Lys147 and proteasomal degradation [83]. Depletion of XIAP or cIAP1 leads to an increase in levels of Rac1 protein in both normal and cancer cells, concomitant with an elongated morphology and enhanced cell migration [83]. The differences in experimental approaches and models employed can contribute to the discrepancies between these reports. Nevertheless, these studies highlight the importance of additional exploration of the non-apoptotic functions of IAP proteins.
SMAC as an endogenous antagonist of IAP proteins
IAP proteins can be antagonized by their endogenous antagonists, such as SMAC/DIABLO [84, 85], Omi/HtrA2 [86], ARTS (apoptosis-related protein in the TGFβ signaling pathway) [87], and XAF1 (XIAP associated factor 1) [88]. SMAC, the best studied natural antagonist of IAP proteins, is released from mitochondria into the cytosol upon apoptotic stimulation [84, 85]. SMAC has a 55-residue mitochondria-targeting sequence at its N-terminus, which is proteolytically removed upon its release from mitochondria, to expose its Ala-Val-Pro-Ile (AVPI) tetrapeptide motif for binding to XIAP, cIAP1 and cIAP2 [84, 85, 89]. Cleaved Smac dimerizes and binds to the BIR2 and BIR3 domains of XIAP via its IAP binding motif and interferes with the interaction of XIAP with caspases [90, 91]. SMAC also binds to the BIR3 domain of cIAP1 and cIAP2 via its AVPI binding motif. cIAP1, cIAP2, and XIAP can all promote the ubiquitination and degradation of SMAC protein [29, 92]. Interestingly, SMAC promotes the auto-ubiquitination and degradation of cIAP1 and cIAP2 upon binding but not XIAP [93]. SMAC functions as an effective cellular antagonist of XIAP, cIAP1 and cIAP2 proteins.
Design of small-molecule SMAC mimetics as antagonists of IAP proteins
The design of small-molecule SMAC mimetics was greatly facilitated by the determination of co-crystal structure of the SMAC protein in a complex with XIAP BIR3 [89] and the solution structure of SMAC peptide complexed with XIAP BIR3 [90]. Based upon these experimentally determined structures, the interaction between SMAC and XIAP BIR3 involves four N-terminal residues (AVPI) in SMAC and a well-defined surface groove on XIAP BIR3 (Figure 3). Biochemical data indicate that a four-residue peptide - AVPI - derived from SMAC binds to the XIAP BIR3 domain protein with the same affinity as SMAC protein and can effectively antagonize the inhibition of caspase activity by the XIAP BIR3 protein. Additionally, the AVPI SMAC peptide also binds to cIAP1 and cIAP2 BIR3 domain proteins with high affinities. These experimental structures and biochemical data form the basis for the design of small molecules (SMAC mimetics) which mimic the binding of SMAC protein to XIAP, cIAP1 and cIAP2. In the last decade, a large number of small-molecule SMAC mimetics have been designed and developed and several of them are currently in clinical development for cancer treatment [20].
Figure 3.
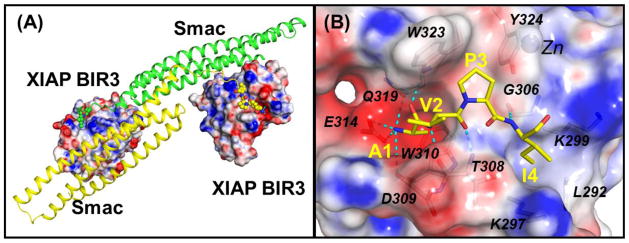
(A) Crystal structure of the dimeric Smac protein in complex with two XIAP BIR3 proteins (PDBID: 1G73). The AVPI motifs are shown in ball models. (B) Detailed interactions between the AVPI binding motif and XIAP BIR3 residues. Oxygen and nitrogen atoms are colored in red and blue colors. Hydrogen bonds are depicted in dash lines. Electrostatic surfaces of XIAP BIR3 are shown where the red, grey and blue colors denote negative, neutral and positive charged regions. The figures are prepared using the PyMOL and APBS programs.
Oost and colleagues at Abbott Laboratories have designed a series of potent and cell-permeable peptidomimetics through extensive chemical modifications of each of the four residues in AVPI [94]. One of the most potent peptidomimetics (compound 1, Figure 4) binds to XIAP BIR3 with a Kd value of 12 nM. Screening a panel of 59 cancer cell lines with diverse tumor types revealed that these peptidomimetics, acting as single agents, effectively inhibit cell growth in a small subset of cancer cell lines, including the MDA-MB-231 breast cancer cell line. Mechanistic studies have shown that these compounds induce robust activation of caspase-3 and caspase-3 dependent cell death in the MDA-MB-231 cell line. Significantly, compound 1 also demonstrated significant antitumor activity in the MDA-MB-231 xenograft tumor model in mice. This study provided the first important proof-of-concept preclinical data that small-molecule SMAC mimetics may have a therapeutic potential for the treatment of a subset of human cancers.
Figure 4.

Chemical structures of representative small-molecule SMAC mimetics.
Employing a structure-based design strategy, our laboratory has designed a large number of SMAC mimetics, including SM-122 [95] and SM-406 [96](Figure 4), and has advanced SM-406 (AT-406/Debio1143) into clinical development. SM-406 has a high binding affinity to XIAP, cIAP1 and cIAP2 BIR3 proteins with Ki values of 66.4, 1.9, and 5.1 nM, respectively [96]. It has an oral bioavailability of 38–55% in rodents and non-rodents. In the MDA-MB-231 xenograft model in mice, SM-406 can completely inhibit tumor growth with an oral dose of 100 mg/kg, administered daily for 2 weeks, while causing no signs of toxicity to the animals. SM-406 is currently in Phase I clinical trials for cancer treatment [97].
Genentech has developed a series of SMAC mimetics two of which have advanced into clinical development. The first clinical candidate molecule is GDC-0152 [98] (Figure 4). GDC-0152 binds to XIAP, cIAP1 and cIAP2 BIR3 domain proteins with Ki values of 28, 17, and 43 nM, respectively. GDC-0152 effectively inhibits tumor growth when administered orally in the MDA-MB-231 breast cancer xenograft model. GDC-0917 (Figure 4) is a second generation of pan-IAP inhibitors that is currently in clinical development [99]. It binds to XIAP, cIAP1 and cIAP2 with Ki values < 60 nM and inhibits tumor growth in the MDA-MB-231 xenograft model in a dose-dependent manner with oral administration [99].
Novartis has developed a class of potent SMAC mimetics [100, 101] and has advanced LCL161 into clinical development (Figure 4). Oral administration of LCL161 inhibits tumor growth in a mouse model of multiple myeloma [100].
Compound 1, SM-122, SM-406, GDC-0152, GDC0917 and LCL161 were all designed to mimic the AVPI tetrapeptide binding motif for interaction with XIAP, cIAP1 and cIAP2. In the co-crystal structure of SMAC in a complex with XIAP BIR3 protein, SMAC protein forms a homodimer, each monomer SMAC interacting with one XIAP BIR3 molecule via its AVPI binding motif [91]. It was subsequently shown that dimeric SMAC protein concurrently interacts with both the BIR2 and BIR3 domains in XIAP, blocking the inhibition of XIAP BIR3 to caspase-9 activity, as well as the inhibition of XIAP BIR2 domain to caspase-3/-7 activity [102]. Consequently, it was proposed that bivalent SMAC mimetics designed to mimic the interaction of dimeric SMAC protein with both BIR2 and BIR3 domains of XIAP could be much more effective antagonists of XIAP and may have a much higher affinity to XIAP [20, 103]. The Harran and Wang Laboratories from the University of Texas Southwestern Medical Center were the first to report such a bivalent SMAC mimetic, compound 2 (Figure 4) [104]. Structurally, this is a symmetrical molecule, containing two identical AVPI mimetics, tethered together with a linker. Compound 2 binds to recombinant XIAP protein containing both BIR2 and BIR3 domains with a Kd value of 0.3 nM and has the same potency as the SMAC protein in activation of caspase-3 in vitro. The single agent activity was not reported in the initial publication but it synergizes with both TNFα and TRAIL to induce caspase activation and apoptosis at concentrations as low as 100 pM in T98G human glioblastoma tumor cells. In addition to its strong binding to XIAP, compound 2 also binds with cIAP1 and cIAP2 in cell lysates. Similar to monovalent SMAC mimetics, it was subsequently shown to be effective as a single agent in cell growth inhibition and apoptosis induction in a subset of human cancer cell lines and capable of inducing tumor regression in xenograft models of non-small cell lung cancer [46]. The impressive potency of this bivalent SMAC mimetic (2) both in biochemical and cell-based assays has provided the inspiration for different research groups to design other classes of bivalent SMAC mimetics.
The first biovalent SMAC mimetic to enter clinical trial is AEG40826 (HSG1029), designed by Aegera Therapeutics but licensed to Human Genome Science for clinical development. (NCT00708006) No structural or preclinical data for AEG40826 have been published.
Our group has designed a series of bivalent SMAC mimetics such as SM-164 [48, 95] (Figure 4). SM-164 binds to XIAP containing BIR2-BIR3 with a Ki value of 500 pM [48, 95]. Using several complementary approaches, we demonstrated that SM-164 achieves its very high affinity by interacting with both the BIR2 and BIR3 domains in XIAP [95]. It also binds to cIAP1 and cIAP2 with low nanomolar affinities [48]. SM-164 induces strong apoptosis in the MDA-MB-231 and other cancer cell lines as a single agent at concentrations as low as 1 nM and is 100-times more potent than corresponding SMAC mimetics that only mimic one AVPI binding motif [48, 95]. SM-164 induces robust apoptosis and achieves tumor regression in the MDA-MB-231 xenograft tumor tissues in mice [48]. More recently, we reported the design and evaluation of SM-1200, which is capable of achieving rapid, complete and permanent tumor regression. An analogue of SM-1200, SM-1387, has been advanced into clinical trials in China and Australia.
TetraLogic Pharmaceuticals has developed a bivalent SMAC mimetic with affinity to cIAP1, birinapant (TL32711). This was shown to bind to XIAP with a Kd value of 45 nM and to cIAP1 with a Kd value of < 1 nM [105]. Similar to other SMAC mimetics, TL32711 has been shown to induce cell death as a single agent in a small subset of human cancer and to achieve synergistic activity when combined with chemotherapeutic agents, TNFα or TRAIL. TL32711 is currently in Phase I/II clinical trials [106–108].
Mechanisms of the antitumor activities of SMAC mimetics
Extensive studies have demonstrated that, as single agents, SMAC mimetics induce TNFα-dependent apoptosis in a small subset of tumor cells [17, 46–49]. On the molecular level, the binding of SMAC mimetics to cIAP1 results in a conformational change in cIAP1, stimulating its dimerization of the RING domain, leading to cIAP1 auto-ubiquitination and subsequent rapid, proteasomal degradation [109]. This transitory activation of cIAP1 promotes the ubiquitination of RIP1 followed by the activation of canonical NF-kB signaling. Following the degradation of cIAP proteins, NIK accumulates and activates the non-canonical NF-κB signaling [17, 47]. The activated NF-κB signaling stimulates the expression of a wide spectrum of NF-kB responsive genes, including TNFα, which activate TNFR1 signaling in an autocrine/paracrine manner [17, 46]. With the degradation of cIAP proteins, non-ubiquitinated RIP1, together with FADD and caspase-8, forms an apoptotic signaling activation platform which activates caspase-8 provoking apoptosis [17]. Therefore, in principle, without TNFα production, SMAC mimetics should be incapable of stimulating apoptosis as single agents. An exception to this is birinapant, which has been reported to promote TNFα-independent apoptosis in inflammatory breast cancer cells [110]. However, the production of TNFα is necessary but not sufficient for induction of apoptosis by SMAC mimetics [49], indicating that additional blockade(s) other than IAP proteins exist for TNFα-induced apoptosis. Similarly, degradation of cIAP1 and cIAP2 by SMAC mimetics is not a predictor for apoptosis induction since SMAC mimetics can effectively induce degradation of cIAP proteins in both sensitive and resistant cells.
Although degradation of cIAP1 and cIAP2 has clearly been established as critical and early events in induction of apoptosis by SMAC mimetics, the role of XIAP is less defined. On one hand, although both caspase-8 and caspase-3 play key roles in induction of apoptosis by SMAC mimetics, caspase-9 plays no or minimal role, suggesting that the binding of SMAC mimetics to XIAP BIR3 is not essential for apoptosis induction as single agents. On the other hand, knock-down of XIAP by siRNA or genetic deletion of XIAP clearly enhances the potency of monovalent SMAC mimetics. Furthermore, bivalent SMAC mimetics that bind to XIAP containing both BIR2 and BIR3 domains with much higher affinities than monovalent SMAC mimetics are much more effective in antagonizing XIAP in cell-free functional assays to activate caspase-3/7 and also >100-times more potent than monovalent SMAC mimetics in induction of apoptosis in tumor cells [48, 111]. Collectively, these studies suggest that while binding to XIAP BIR3 is not required for induction of apoptosis by SMAC mimetics, effective antagonism of full length of XIAP can further enhance the ability of a SMAC mimetic to induce TNFα-dependent apoptosis in tumor cells and XIAP is still an important cellular target for SMAC mimetics.
In addition to promoting TNFα-dependent apoptosis, SMAC mimetics can also prime cancer cells for TNFα-induced necroptosis. Necroptosis, a regulated necrotic cell death pathway controlled by RIP1 and RIP3 kinases, occurs when apoptosis is blocked, and is dependent on RIP1. As described above, in the absence of cIAP proteins, caspase-8 is activated by the RIP1-containing complex II in the TNFα/TNFR signaling. In addition to cleaving downstream effector caspases and BID, activated caspase-8 also cleaves RIP1 and RIP3 [112, 113]. In the absence of functional caspase-8, for example, after inhibition by caspase inhibitors, TNFα-induced apoptosis is blocked, permitting non-ubiquitinated RIP1 to form a necroptosis-activating complex, necrosome. He et al first reported that, upon inhibition of caspase activity, SMAC mimetics, in combination with TNFα, provoke a strong necroptotic response in SMAC mimetic-resistant cancer cells [56]. Similarly, in caspase-8- or FADD-deficient cells, SMAC mimetic promotes cancer cells undergoing TNFα-induced necroptosis [114]. Mechanistically, by promoting the degradation of cIAP proteins, SMAC mimetics stimulate the formation of necrosome and promote necroptosis [115].
Like most other targeted drugs, SMAC mimetics are designed to act directly on tumor cells. Immune cell infiltration, resulting in chronic inflammation, is common in solid tumor lesions. Cytokines such as TNFα and IL-1β produced by innate immune cells (e.g., tumor associated macrophages) play critical roles in the initiation, progression, and metastasis of a variety of human malignancies [116]. Cheung et al. reported that SMAC mimetics synergize with IL-1β to induce caspase-8- and RIP1-dependent apoptosis in cancer cell lines representing diverse tumor types [117]. Thus it is reasonable to postulate that SMAC mimetics exert more robust antitumor activity in vivo, especially against inflammatory tumors. Indeed, Lecis et al. recently reported that the SMAC mimetic, SM83, is active as a single agent in vivo on human and murine cancer cells that are refractory to SM83 in vitro. SM83 exerts its antitumor activity by inducing inflammation and immune cell activation and it stimulates the reversion of the tumor-associated macrophages from a pro-tumoral M2- to a pro-inflammatory M1-like state, resulting in secretion by the host of TNFα, IL-1β, and IFNγ, leading to necroptic cell death and release of HMGB1 (high-mobility group box 1 protein) [118]. It was speculated that, by inducing necrosis, SMAC mimetics can further prime an adaptive immune response [118]. Several other studies have shown that SMAC mimetics possess antitumor activity in vivo against cancer cells that are resistant to SMAC mimetics in vitro [119–121]. Nevertheless, these studies suggest the mechanism of action of SMAC mimetics in the microenvironment in vivo is different from that in 2-dimensional cultured cells in vitro. More robust studies are necessary to evaluate the antitumor activities of SMAC mimetics in clinically more relevant tumor models.
Development of combination therapies with SMAC mimetics for cancer treatment
SMAC mimetics promote TNFα-dependent apoptosis, but the presence of TNFα is not always sufficient for effective induction of apoptosis by SMAC mimetics. Petersen et al. has reported that SMAC mimetic treatment can lead to feedback upregulation of cIAP2 protein [122]. This rebound of cIAP2 can compensate for the removal of cIAP1 by SMAC mimetics thus conferring drug resistance. The feedback upregulation of cIAP2 could be due to the loss of cIAP1-mediated ubiquitination and degradation of cIAP2, activation of NF-kB signaling by SMAC mimetics or alterations in the other signaling pathways, such as the PI3K-Akt pathway, that regulate cIAP2 expression [122]. We have observed that tumor cell lines initially sensitive to SMAC mimetics develop drug resistance when continuously exposed to highly potent SMAC mimetics [123] and we speculated that, in addition to feedback upregulation of cIAP2, other resistant mechanisms exist. Indeed, we discovered that LRIG1, a negative regulator of the stability of RTKs (receptor tyrosine kinase), is downregulated in the resistant cells derived from SMAC mimetic-sensitive cancer cells [123]. Such reduction of LRIG1 is accompanied by upregulation of diverse RTKs in the SMAC mimetic resistant cells. RTKs have been intensively pursued targets for cancer treatment and aberrant RTK activation has been implicated in cancer resistance to molecular-targeted therapies [124]. In fact, combination of SMAC mimetics with inhibitors targeting PDGFR (platelet-derived growth factor receptor), IGF1R (insulin-like growth factor 1 receptor) or EGFR (epidermal growth factor receptor) significantly increases cell death compared with monotherapy in human glioblastoma multiforme [125]. Synergy between SMAC mimetics and inhibitors of FLT3 (FMS-like tyrosine kinase 3) and BCR-ABL was also observed against leukemia in vitro and in vivo [126, 127]. In breast cancer cells, SMAC mimetics increase apoptosis induction by ErbB antagonists in Her2- or EGFR-overexpressing cells [128].
As SMAC mimetics exert death receptor-mediated cell death activity, it is conceivable that the combination of SMAC mimetics with death receptor ligands and agonists would be efficacious in a wide variety of cancer cells. Indeed, strong antitumor activity of SMAC mimetics and death receptor ligands (e.g., TRAIL) and agonists (e.g., monoclonal antibodies against TRAIL receptors) is observed in a large number of preclinical models representing acute lymphocytic leukemia [129], breast cancer [130], fibrosarcoma [131], glioblastoma [104], melanoma [131], neuroblastoma [132], pancreatic carcinoma [133, 134], and rhabdomyosarcoma [135] among others. For example, Abhari et al. recently reported that SMAC mimetic synergizes with mapatumumab and lexatumumab, monoclonal agonist antibodies against TRAIL receptors, to induce RIP1-dependent apoptosis in neuroblastoma cells [132].
Chemotherapy, along with irradiation and surgery, is currently still the mainstay of cancer treatment. The antitumor effects of SMAC mimetics have been extensively examined in combination with chemotherapeutics, and synergistic responses have been reported in numerous studies using in vitro and mouse xenograft models [20]. For example, Probst et al. reported that SMAC mimetics potentiate apoptosis induction by chemotherapeutics, including paclitaxel, etoposide, SN-38, 5-FU, and cisplatin in human cancer cell lines of diverse tumor types [120]. Mechanistic studies revealed that the synergistic activity of SMAC mimetics with chemotherapeutics is due to canonical NF-kB activation, TNFα production, and activation of the extrinsic apoptosis pathway [120]. In other studies, the reported synergy between SMAC mimetics and chemotherapeutics is reported as independent of TNFα, but dependent on the antagonism of XIAP [136, 137]. Recent studies have shown that genotoxic agents such as etoposide can activate cell death pathways via proteasome-dependent depletion of IAP proteins to promote the assembly of a cytoplasmic cell death-activating platform, ripoptosome [52, 53]. Ripoptosome shares the same core components with the complex II originated from death receptor signaling. However, the formation of ripoptosome is independent of death ligands [52, 53]. SMAC mimetic, by promoting the degradation of cIAP proteins, stimulates the formation of ripoptosome and subsequent apoptosis in etoposide-treated SMAC mimetic-resistant cancer cells [53]. SMAC mimetic can also sensitize glioblastoma cells to temozolomide-induced apoptosis through promoting ripoptosome formation [138]. Notably, targeted drugs such as CDK2/cyclin A inhibitor TAT-NBI1 in combination with ERFR inhibitor erlotinib lead to the depletion of IAP proteins, and ultimately ripoptosome-mediated apoptosis in erlotinib-resistant breast tumor cells [139]. Therefore combining SMAC mimetics with standard-of-care chemotherapeutics and other targeted drugs holds clinical promise conceptually.
Taken together, these preclinical data provide evidence that SMAC mimetics may be developed in combination with conventional chemotherapeutics, death receptor ligands and agonists, as well as small molecule targeted drugs. Certain of these combination strategies are under clinical evaluation.
SMAC mimetics in clinical development for cancer treatment and initial clinical findings
To date, four monovalent and two bivalent SMAC mimetics have been tested in clinical trials for their safety, maximum tolerated dose, pharmacokinetics (PK), pharmacodynamics (PD), biomarker identification and initial efficacy in patients with advanced solid tumors and hematological malignancies [140].
GDC-0152 from Genentech was the first of these novel agents to enter a clinical trial [98]. No toxicity or efficacy data were reported, but when administered intravenously to patients with locally advanced or metastatic malignancies, GDC-0152 demonstrated linear pharmacokinetics over doses ranging from 0.049 to 1.48 mg/kg [98].
The first in human phase I trial of GDC-0917 (NCT01226277), a second generation, orally bioavailable SMAC mimetic from Genentech, enrolled 42 patients at doses ranging from 5–600 mg daily on a 2 week on/1 week off dosing schedule [141]. Fatigue, nausea, vomiting, and constipation were the most commonly reported adverse events occurring in approximately of 20% of patients but the maximum tolerated dose (MTD) was not reached. The pharmacokinetics of GDC-0917 was dose-proportional with maximum doses reaching the inhibitory concentrations predicted from preclinical modeling. cIAP1 was rapidly decreased in peripheral blood mononuclear cells (PBMC) at all dose levels, with sequential biopsies in 3 patients showing decreases in tumor cIAP1 and decreases in caspase-3 and PARP. Two patients (1 ovarian cancer, 1 MALT lymphoma) had unconfirmed complete responses and 4 patients had stable disease lasting ≥ 3 months.
SM-406 (AT-406, Debio 1143) is an oral agent currently being evaluated in four Phase I trials. The first, a single agent trial in patients with advanced solid tumors and lymphomas, enrolled 31 patients with doses from 5–900 mg on a 5 day every 3 weeks dosing schedule [97]. The most common adverse events were fatigue, nausea, vomiting, constipation, pruritus and rash, each of which was reported in 10–23% of the study population. One patient had reversible elevation in AST which was the only dose-limiting toxicity observed (DLT) and the MTD was not reached. Pharmacokinetics was dose proportional above 80 mg with no evidence of drug accumulation. At doses above 80 mg drug concentrations reached levels which predicted activity based on preclinical models with prompt degradation of cIAP1 in PBMC, skin biopsies, and tumor biopsies. Five patients (17%) had stable disease at the first evaluation with one patient on study for 196 days. This trial (NCT01078649) is now evaluating an additional dosing schedule (2 weeks on/1 week off). A second phase I trial of AT-406 in combination with daunorubicin and cytarabine in patients with poor-risk acute myelogenous leukemia was initiated but has been terminated. (NCT01265199). Recently, two new phase I trials were initiated. In one trial (NCT01930292), Debio 11430 is being evaluated in combination with both carboplatin and paclitaxel in patients with squamous non-small cell lung cancer (NSCLC), and in patients with platinum-refractory ovarian cancer, or in patients with basal-like/Claudin low triple negative breast cancer. In another trial (NCT02022098), Debio 11430 is being tested in a Phase I/II randomized study to determine the maximum tolerated dose, safety, pharmacokinetics and antitumor activity in combination with concurrent chemoradiation therapy in patients with locally advanced squamous cell carcinoma of the head and neck.
Another orally bioavailable monovalent SMAC mimetic, LCL161 was well tolerated on a weekly dosing schedule in 27 patients with advanced cancer (NCT01098838) and no dose-limiting toxicity was found at doses up to 1800 mg [142, 143]. The most common adverse events were nausea and vomiting. Starting at doses of 320 mg, cIAP1 levels were consistently reduced in skin punch biopsies taken 8 h after the first dose and in a tumor biopsy after 24 h. cIAP1 levels in PMBC decrease 2 h post-dose and recover by the following week. Markers of cell death peak on day 2 and circulating cytokines, including MCP-1 and IL-8, increase 4 h post-dose at higher dose levels (≥900 mg). No objective responses were reported, but a patient with rectal cancer had stable disease through 5 cycles of therapy. A phase Ib trial tested the weekly schedule of LCL161 with 80 mg/m2 of weekly paclitaxel in adults with advanced solid tumors. (NCT01240655) A total of 32 patients were enrolled at LCL161 dose levels of 600, 1200, 1500, and an expansion cohort of 1800 mg weekly. The toxicities were those typical of weekly paclitaxel and there was no pharmacokinetic interaction between the agents. The abstract notes that cytokine release syndrome which was the dose-limiting toxicity (DLT) of the single agent study was not seen with the combination. A randomized, Phase II, neoadjuvant clinical trial of this weekly combination in triple negative breast cancer is in progress. (NCT01617668)
The bivalent SMAC mimetic brinapant (TL-32711) has been studied as weekly intravenous infusion on a 3 week on/1 week off schedule [106]. Doses ranged from 0.18–26 mg/m2 in 27 patients enrolled on the initial phase I trials and there was no DLT. Adverse events included lymphopenia and rash. cIAP1 reduction was demonstrated in PBMC at doses ≥1.44 mg/m2 with increased proportionality with increasing doses which correlated with the pharmacokinetics. Two colon cancer patients had evidence of tumor regression. A second phase I study enrolled 124 patients on 5 arms combining brinapant with either carboplatin/paclitaxel, irinotecan, docetaxel, gemcitabine, or liposomal doxorubicin [107]. Doses of brinapant ranged from 2.8–47 mg/m2. The MTD of brinapant was 47 mg/m2 with both carboplatin/paclitaxel and docetaxel, and 22 mg/m2 with irinotecan. Gemcitabine and brinapant could not be administered in combination in heavily pretreated patients, and drug supply issues prevented evaluation of the combination with liposomal doxorubicin. Reversible Bell’s palsy was seen as a DLT on this study and typically occurred on the first cycle of therapy. In six of the seven patients reported it did not recur with continued therapy. Significant anti-tumor activity was demonstrated with both the brinapant and carboplatin/paclitaxel and irinotecan regimens with activity reported for the combination in patients whose tumors were refractory to prior therapy with irinotecan alone. Based on this activity, a phase II trial of brinapant and irinotecan in patients with irinotecan relapsed/refractory colorectal carcinoma has been conducted. Standard dose irinotecan (350 mg/m2 every three weeks) was administered with brinapant given on a day 1 and 8 schedule using an ascending dose strategy. The treatment was reported to be well-tolerated and in 51 patients enrolled there were two partial responses with 27 patients with stable disease for a clinical benefit rate of 57% [108]. Patients whose tumors had a KRAS mutation appeared to have slightly more benefit than those with KRAS wild-type tumors, although the numbers are small.
Another bivalent SMAC mimetic, HGS1029 (AEG40826), was well tolerated in patients with advanced solid malignancies on two days/week schedules (days 1, 8, and 15 and continuous), and intravenous schedules with an MTD of 3.2 mg/m2. (NCT00708006) [144]. A total of 44 patients were enrolled with DLT in 2/6 patients at 4.8 mg/m2. Dose-limiting toxicities included severe fatigue, elevated amylase and lipase. The most frequent adverse events were nausea, anorexia, fever, vomiting, diarrhea, fatigue, and rash. HGS1029 induces rapid and sustained reduction of cIAP1 levels after a single dose of administration and shows evidence of apoptosis induction in patients. Confirmed tumor regression was reported in a patient with colon cancer and 2 patients (NSCLC, adrenocortical cancer) had stable disease for more than 6 months.
These initial clinical data provide evidence that SMAC mimetics are well-tolerated, induce rapid and sustained cIAP degradation, and have antitumor activity as single agents and in combination in patients with advanced cancer.
Challenges in development of SMAC mimetics for cancer treatment
IAP proteins are prominent regulators of both apoptotic and non-apoptotic signaling pathways. SMAC mimetics, in general, stimulate TNFα-dependent apoptosis. TNFα is a pleiotropic cytokine that provokes various cellular responses, depending on the cellular context. A pertinent concern in the clinical development of SMAC mimetics surrounds the consequences of SMAC mimetic-stimulated NF-κB activation, including the potential adverse effects of elevated levels of cytokines and chemokines on normal tissues. Administration of GDC-0152, for example, causes acute induction of TNFα in the plasma of dogs and rats and GDC-0152 demonstrates a toxicity profile consistent with TNFα-mediated toxicity [145]. The onset and resolution of these acute toxicities generally tracks with the time course of GDC-0152-induced cytokine [145]. Despite these potential toxicity concerns based upon in preclinical data, patients administered intravenous doses of GDC-0152 up to 1.48 mg/kg showed no signs of a severe TNFα-driven systemic inflammatory response [146] and severe cytokine release syndrome, caused by an acute increase in plasma TNFα and other inflammatory cytokines, has not been reported in patients [142–144]. Similarly, phase I studies for other Smac mimetics have not found a severe TNFα-driven systemic inflammatory response or severe cytokine release syndrome.
Bone metastasis is a common complication of many tumor types, including breast, prostate, and lung cancer [147, 148]. RANKL (receptor activator of NF-κB ligand) is the central mediator of the vicious cycle of bone destruction and tumor growth. Cytokines and growth factors produced by tumor cells stimulate osteoblasts to overexpress RANKL, and this in turn drives osteoclast activity and bone resorption. Increased bone resorption releases growth factors from the bone matrix to further promote bone metastasis and perpetuate the cycle. Osteoclast differentiation and function are stimulated by the NIK-mediated non-canonical NF-κB pathway downstream of RANKL/RANK signaling [149]. Employing mouse tumor models of SMAC mimetic-sensitive and -resistant human and murine breast tumors, Yang et al recently reported that SMAC mimetics cause high bone turnover osteoporosis, increase NIK-mediated osteoclastogenesis, enhance tumor-associated osteolysis, and favor bone metastasis [150]. Bisphosphonate, an osteoclast-targeting agent, effectively blocked SMAC mimetic-promoted bone metastasis and this, together with other studies discussed earlier [118], suggests that the effect of SMAC mimetics on the host microenvironment may have an impact on their ultimate efficacy and possibly their side effects.
Concluding remarks
IAP proteins are critical regulators of cell death and survival, and thus attractive targets for the development of novel anticancer drugs. The identification of SMAC as a natural antagonist of IAP proteins promoted the development of small-molecule SMAC mimetics as anticancer drugs and six such compounds have entered human clinical trials. Data from early phase clinical trials have provided evidence for on-target activity for SMAC mimetics and a good toxicity profile in patients with advanced cancer. Consistent with preclinical studies that SMAC mimetics as single agents are effective only in a subset of cancer cell line models, objective clinical responses were observed in a small subset of patients. Therefore, identification and validation of predictive biomarkers for drug response will be critical for the successful development of SMAC mimetics as monotherapies. Since SMAC mimetics have been shown to enhance the antitumor activity of chemotherapeutics and radiation in preclinical studies, a number of clinical trials to evaluate the combination of SMAC mimetics with chemotherapeutics and radiation are being performed, which may prove to be essential.
Table 1.
SMAC mimetics under clinical development
| Drug | Developer | Phase | Study Type | Disease Condition | Identifier | Status (April 2014) |
|---|---|---|---|---|---|---|
| GDC-0152 | Genentech | I | dose escalation | solid cancer | NCT00977067 | completed |
| GDC-0917 (CUDC427) | Genentech Curis | I | dose escalation | solid tumors lymphoma | NCT01908413 | ongoing |
| I | dose escalation | solid tumors lymphoma | NCT01226277 | completed | ||
| SM-406 (AT-406, Debio1143) | University of Michigan Ascenta Therapeutics DebioPharm | I | dose escalation | advanced solid tumors lymphomas | NCT01078649 | recruiting |
| I | with daunorubicin and cytarabine | acute myelogenous leukemia | NCT01265199 | terminated | ||
| I | with chemo-radiation therapy | squamous cell carcinoma of the head and neck | NCT02022098 | recruiting | ||
| I | with carboplatin and paclitaxel | solid tumors | NCT01930292 | recruiting | ||
| LCL161 | Novartis | I | dose escalation | advanced solid cancer | NCT01098838 | completed |
| I | single agent | advanced solid cancer | NCT01968915 | recruiting | ||
| I | with paclitaxel | advanced solid cancer | NCT01240655 | recruiting | ||
| I | with gemcitabine plus nab-paclitaxel | metastatic pancreatic cancer | NCT01934634 | recruiting | ||
| II | with paclitaxel | triple negative breast cancer | NCT01617668 | recruiting | ||
| II | with cyclophosphamide | multiple myeloma | NCT01955434 | recruiting | ||
| II | single agent | leukemia | NCT02098161 | not recruiting yet | ||
| Birinapant (TL32711) | TetraLogic Pharmaceuticals | I | dose escalation | solid tumors or lymphoma | NCT00993239 | completed |
| I | with gemcitabine hydrochloride | advanced solid tumors | NCT01573780 | terminated | ||
| I/II | with 5-azacitidine | myelodysplastic syndrome | NCT01828346 | recruiting | ||
| I/II | with chemotherapy | advanced solid tumors | NCT01188499 | ongoing | ||
| I/II | single agent | acute myelogenous leukemi myelodysplastic syndrome acute lymphoblastic leukemia |
NCT01486784 | recruiting | ||
| I | with conatumumab | ovarian cancer | NCT01940172 | recruiting | ||
| II | single agent | advanced ovarian fallopian tube peritoneal Cancer | NCT01681368 | ongoing | ||
| AEG40826 (HGS1029) | Aegera Human Genome Sciences | I | dose escalation | advanced solid cancer | NCT00708006 | completed |
| I | dose escalation | lymphoid malignancies | NCT01013818 | terminated |
Acknowledgments
We are grateful for the financial support from the Breast Cancer Research Foundation, the Prostate Cancer Foundation, the Department of Defense Prostate Cancer Program (W81XWH-04-1-0213), Ascenta Therapeutics, DebioPharm and the National Cancer Institute, NIH (5R01CA109025 and 5R01CA127551). We like to thank Dr. Liu Liu for generating Figures 1 and Dr. Chao-Yie Yang for generating Figure 3.
Footnotes
Conflict of Interest Statement: Ascenta and Debiopharm have licensed AT-406 for clinical development from the University of Michigan. S. Wang is an inventor of AT-406 and receives research support from Debiopharm and owns stock in Ascenta. D.C. Smith has received research support from Ascenta and Debiopharm.
References
- 1.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21(3):485–95. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 2.Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407(6805):810–6. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- 3.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1(2):111–21. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67(4):2168–74. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birnbaum MJ, Clem RJ, Miller LK. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol. 1994;68(4):2521–8. doi: 10.1128/jvi.68.4.2521-2528.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3(6):401–10. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 8.Gyrd-Hansen M, et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kappaB as well as cell survival and oncogenesis. Nat Cell Biol. 2008;10(11):1309–17. doi: 10.1038/ncb1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu M, et al. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell. 2007;26(5):689–702. doi: 10.1016/j.molcel.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofer-Warbinek R, et al. Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem. 2000;275(29):22064–8. doi: 10.1074/jbc.M910346199. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki Y, et al. X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J Biol Chem. 2001;276(29):27058–63. doi: 10.1074/jbc.M102415200. [DOI] [PubMed] [Google Scholar]
- 12.Riedl SJ, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104(5):791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 13.Shiozaki EN, et al. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell. 2003;11(2):519–27. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 14.Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6(4):287–97. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 15.Lopez J, et al. CARD-mediated autoinhibition of cIAP1’s E3 ligase activity suppresses cell proliferation and migration. Mol Cell. 2011;42(5):569–83. doi: 10.1016/j.molcel.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Samuel T, et al. Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem. 2006;281(2):1080–90. doi: 10.1074/jbc.M509381200. [DOI] [PubMed] [Google Scholar]
- 17.Varfolomeev E, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131(4):669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 18.Chai J, et al. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104(5):769–80. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 19.Srinivasula SM, et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410(6824):112–6. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 20.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–24. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 21.Silke J, et al. Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc Natl Acad Sci U S A. 2005;102(45):16182–7. doi: 10.1073/pnas.0502828102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mace PD, Shirley S, Day CL. Assembling the building blocks: structure and function of inhibitor of apoptosis proteins. Cell Death Differ. 2010;17(1):46–53. doi: 10.1038/cdd.2009.45. [DOI] [PubMed] [Google Scholar]
- 23.Conze DB, et al. Posttranscriptional downregulation of c-IAP2 by the ubiquitin protein ligase c-IAP1 in vivo. Mol Cell Biol. 2005;25(8):3348–56. doi: 10.1128/MCB.25.8.3348-3356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertrand MJ, et al. cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1-4) PLoS One. 2011;6(9):e22356. doi: 10.1371/journal.pone.0022356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Damgaard RB, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46(6):746–58. doi: 10.1016/j.molcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Choi YE, et al. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7 via unique mechanisms at distinct steps in their processing. J Biol Chem. 2009;284(19):12772–82. doi: 10.1074/jbc.M807550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Yang Y, Ashwell JD. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416(6878):345–7. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- 28.Mao AP, et al. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J Biol Chem. 2010;285(13):9470–6. doi: 10.1074/jbc.M109.071043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu S, Yang X. Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J Biol Chem. 2003;278(12):10055–60. doi: 10.1074/jbc.M207197200. [DOI] [PubMed] [Google Scholar]
- 30.Dogan T, et al. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10(12):1447–55. doi: 10.1038/ncb1804. [DOI] [PubMed] [Google Scholar]
- 31.Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22(16):2256–66. doi: 10.1101/gad.1663108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varfolomeev E, et al. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5(216):ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 33.Varfolomeev E, et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283(36):24295–9. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertrand MJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30(6):689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 35.Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12(2):121–32. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 36.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 37.Gentle IE, Silke J. New perspectives in TNF-R1-induced NF-kappaB signaling. Adv Exp Med Biol. 2011;691:79–88. doi: 10.1007/978-1-4419-6612-4_8. [DOI] [PubMed] [Google Scholar]
- 38.Jin HS, et al. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009;69(5):1782–91. doi: 10.1158/0008-5472.CAN-08-2256. [DOI] [PubMed] [Google Scholar]
- 39.Birkey Reffey S, et al. X-linked inhibitor of apoptosis protein functions as a cofactor in transforming growth factor-beta signaling. J Biol Chem. 2001;276(28):26542–9. doi: 10.1074/jbc.M100331200. [DOI] [PubMed] [Google Scholar]
- 40.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 41.Varfolomeev E, et al. The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2. J Biol Chem. 2006;281(39):29022–9. doi: 10.1074/jbc.M605116200. [DOI] [PubMed] [Google Scholar]
- 42.Garrison JB, Samuel T, Reed JC. TRAF2-binding BIR1 domain of c-IAP2/MALT1 fusion protein is essential for activation of NF-kappaB. Oncogene. 2009;28(13):1584–93. doi: 10.1038/onc.2009.17. [DOI] [PubMed] [Google Scholar]
- 43.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annunziata CM, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–30. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10(8):561–74. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- 46.Petersen SL, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12(5):445–56. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vince JE, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131(4):682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 48.Lu J, et al. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 2008;68(22):9384–93. doi: 10.1158/0008-5472.CAN-08-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheung HH, et al. Down-regulation of c-FLIP Enhances death of cancer cells by smac mimetic compound. Cancer Res. 2009;69(19):7729–38. doi: 10.1158/0008-5472.CAN-09-1794. [DOI] [PubMed] [Google Scholar]
- 50.Eckhardt I, Roesler S, Fulda S. Identification of DR5 as a critical, NF-kappaB-regulated mediator of Smac-induced apoptosis. Cell Death Dis. 2013;4:e936. doi: 10.1038/cddis.2013.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229–32. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 52.Feoktistova M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tenev T, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432–48. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Vandenabeele P, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 55.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–11. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 57.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 58.Zhao J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–7. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dubrez L, Berthelet J, Glorian V. IAP proteins as targets for drug development in oncology. Onco Targets Ther. 2013;9:1285–1304. doi: 10.2147/OTT.S33375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, et al. X-linked inhibitor of apoptosis positive nuclear labeling: a new independent prognostic biomarker of breast invasive ductal carcinoma. Diagn Pathol. 2011;6:49. doi: 10.1186/1746-1596-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiang G, et al. Expression of X-linked inhibitor of apoptosis protein in human colorectal cancer and its correlation with prognosis. J Surg Oncol. 2009;100(8):708–12. doi: 10.1002/jso.21408. [DOI] [PubMed] [Google Scholar]
- 62.Moussata D, et al. XIAP as a radioresistance factor and prognostic marker for radiotherapy in human rectal adenocarcinoma. Am J Pathol. 2012;181(4):1271–8. doi: 10.1016/j.ajpath.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 63.Krajewska M, et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin Cancer Res. 2003;9(13):4914–25. [PubMed] [Google Scholar]
- 64.Seligson DB, et al. Expression of X-linked inhibitor of apoptosis protein is a strong predictor of human prostate cancer recurrence. Clin Cancer Res. 2007;13(20):6056–63. doi: 10.1158/1078-0432.CCR-07-0960. [DOI] [PubMed] [Google Scholar]
- 65.Grzybowska-Izydorczyk O, et al. Expression and prognostic significance of the inhibitor of apoptosis protein (IAP) family and its antagonists in chronic lymphocytic leukaemia. Eur J Cancer. 2010;46(4):800–10. doi: 10.1016/j.ejca.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 66.Ferreira CG, et al. Expression of X-linked inhibitor of apoptosis as a novel prognostic marker in radically resected non-small cell lung cancer patients. Clin Cancer Res. 2001;7(8):2468–74. [PubMed] [Google Scholar]
- 67.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125(7):1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dai Z, et al. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum Mol Genet. 2003;12(7):791–801. doi: 10.1093/hmg/ddg083. [DOI] [PubMed] [Google Scholar]
- 69.Imoto I, et al. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 2001;61(18):6629–34. [PubMed] [Google Scholar]
- 70.Imoto I, et al. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 2002;62(17):4860–6. [PubMed] [Google Scholar]
- 71.Krajewska M, et al. Analysis of apoptosis protein expression in early-stage colorectal cancer suggests opportunities for new prognostic biomarkers. Clin Cancer Res. 2005;11(15):5451–61. doi: 10.1158/1078-0432.CCR-05-0094. [DOI] [PubMed] [Google Scholar]
- 72.Che X, et al. Nuclear cIAP1 overexpression is a tumor stage- and grade-independent predictor of poor prognosis in human bladder cancer patients. Urol Oncol. 2012;30(4):450–6. doi: 10.1016/j.urolonc.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 73.Nakagawa Y, et al. IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am J Hematol. 2006;81(11):824–31. doi: 10.1002/ajh.20656. [DOI] [PubMed] [Google Scholar]
- 74.Oberoi-Khanuja TK, Murali A, Rajalingam K. IAPs on the move: role of inhibitors of apoptosis proteins in cell migration. Cell Death Dis. 2013;4:e784. doi: 10.1038/cddis.2013.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mehrotra S, et al. IAP regulation of metastasis. Cancer Cell. 2010;17(1):53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013;25(10):1955–61. doi: 10.1016/j.cellsig.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 77.Baranwal S, Alahari SK. Rho GTPase effector functions in tumor cell invasion and metastasis. Curr Drug Targets. 2011;12(8):1194–201. doi: 10.2174/138945011795906534. [DOI] [PubMed] [Google Scholar]
- 78.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093–101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 79.Liu J, et al. X-linked inhibitor of apoptosis protein (XIAP) mediates cancer cell motility via Rho GDP dissociation inhibitor (RhoGDI)-dependent regulation of the cytoskeleton. J Biol Chem. 2011;286(18):15630–40. doi: 10.1074/jbc.M110.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu J, et al. E3 ligase activity of XIAP RING domain is required for XIAP-mediated cancer cell migration, but not for its RhoGDI binding activity. PLoS One. 2012;7(4):e35682. doi: 10.1371/journal.pone.0035682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu J, et al. RhoGDI SUMOylation at Lys-138 increases its binding activity to Rho GTPase and its inhibiting cancer cell motility. J Biol Chem. 2012;287(17):13752–60. doi: 10.1074/jbc.M111.337469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oberoi-Khanuja TK, et al. Role of melanoma inhibitor of apoptosis (ML-IAP) protein, a member of the baculoviral IAP repeat (BIR) domain family, in the regulation of C-RAF kinase and cell migration. J Biol Chem. 2012;287(34):28445–55. doi: 10.1074/jbc.M112.341297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oberoi TK, et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2012;31(1):14–28. doi: 10.1038/emboj.2011.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Du C, et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 85.Verhagen AM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 86.van Loo G, et al. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002;9(1):20–6. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- 87.Gottfried Y, et al. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23(7):1627–35. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liston P, et al. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3(2):128–33. doi: 10.1038/35055027. [DOI] [PubMed] [Google Scholar]
- 89.Chai J, et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 2000;406(6798):855–62. doi: 10.1038/35022514. [DOI] [PubMed] [Google Scholar]
- 90.Liu Z, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408(6815):1004–8. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 91.Wu G, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408(6815):1008–12. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 92.MacFarlane M, et al. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem. 2002;277(39):36611–6. doi: 10.1074/jbc.M200317200. [DOI] [PubMed] [Google Scholar]
- 93.Yang QH, Du C. Smac/DIABLO selectively reduces the levels of c-IAP1 and c-IAP2 but not that of XIAP and livin in HeLa cells. J Biol Chem. 2004;279(17):16963–70. doi: 10.1074/jbc.M401253200. [DOI] [PubMed] [Google Scholar]
- 94.Oost TK, et al. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J Med Chem. 2004;47(18):4417–26. doi: 10.1021/jm040037k. [DOI] [PubMed] [Google Scholar]
- 95.Sun H, et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc. 2007;129(49):15279–94. doi: 10.1021/ja074725f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cai Q, et al. A Potent and Orally Active Antagonist (SM-406/AT-406) of Multiple Inhibitor of Apoptosis Proteins (IAPs) in Clinical Development for Cancer Treatment. J Med Chem. 2011;54(8):2714–26. doi: 10.1021/jm101505d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hurwitz H, et al. Preliminary Report of a First-in-human, Open-label, Multicenter, Phase I Study of AT-406 (Debio 1143), an Oral Small Molecule Multi-IAP Inhibitor, in Solid Tumors and Lymphomas. European Journal of Cancer. 2012;48(Suppl 6):25. [Google Scholar]
- 98.Flygare JA, et al. Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152) J Med Chem. 2012;55(9):4101–13. doi: 10.1021/jm300060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wong H, et al. Learning and confirming with preclinical studies: modeling and simulation in the discovery of GDC-0917, an inhibitor of apoptosis proteins antagonist. Drug Metab Dispos. 2013;41(12):2104–13. doi: 10.1124/dmd.113.053926. [DOI] [PubMed] [Google Scholar]
- 100.Chauhan D, et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM) Blood. 2007;109(3):1220–7. doi: 10.1182/blood-2006-04-015149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gaither A, et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res. 2007;67 (24):11493–8. doi: 10.1158/0008-5472.CAN-07-5173. [DOI] [PubMed] [Google Scholar]
- 102.Huang Y, et al. Requirement of both the second and third BIR domains for the relief of X-linked inhibitor of apoptosis protein (XIAP)-mediated caspase inhibition by Smac. J Biol Chem. 2003;278(49):49517–22. doi: 10.1074/jbc.M310061200. [DOI] [PubMed] [Google Scholar]
- 103.Wang S. Design of small-molecule Smac mimetics as IAP antagonists. Curr Top Microbiol Immunol. 2011;348:89–113. doi: 10.1007/82_2010_111. [DOI] [PubMed] [Google Scholar]
- 104.Li L, et al. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305(5689):1471–4. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- 105.Benetatos CA, et al. Birinapant(TL32711), a Bivalent Smac Mimetic, Targets TRAF2-associated cIAPs, Abrogates TNF-induced NF-kappaB Activation and is Active in Patient-Derived Xenograft Models. Mol Cancer Ther. 2014 doi: 10.1158/1535-7163.MCT-13-0798. [DOI] [PubMed] [Google Scholar]
- 106.Amaravadi RK, et al. Phase 1 study of the Smac mimetic TL32711 in adult subjects with advanced solid tumors and lymphoma to evaluate safety, pharmacokinetics, pharmacodynamics, and antitumor activity. Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research; 2011 Apr 2–6; Orlando, FL. Philadelphia (PA): AACR; Cancer Res; 2011. p. Abstract nr LB-406. [Google Scholar]
- 107.Amaravadi RK, et al. A phase I study of birinapant (TL32711) combined with multiple chemotherapies evaluating tolerability and clinical activity for solid tumor patients. J Clin Oncol. 2013;31(suppl):abstr 2504. [Google Scholar]
- 108.Senzer NN, et al. Phase II clinical activity and tolerability of the SMAC-mimetic birinapant (TL32711) plus irinotecan in irinotecan-relapsed/refractory metastatic colorectal cancer. J Clin Oncol. 2013;31(suppl):abstr 3621. [Google Scholar]
- 109.Dueber EC, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334(6054):376–80. doi: 10.1126/science.1207862. [DOI] [PubMed] [Google Scholar]
- 110.Allensworth JL, et al. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat. 2013;137(2):359–71. doi: 10.1007/s10549-012-2352-6. [DOI] [PubMed] [Google Scholar]
- 111.Ndubaku C, et al. Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS Chem Biol. 2009;4 (7):557–66. doi: 10.1021/cb900083m. [DOI] [PubMed] [Google Scholar]
- 112.Lin Y, et al. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13(19):2514–26. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Feng S, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19(10):2056–67. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 114.Laukens B, et al. Smac mimetic bypasses apoptosis resistance in FADD- or caspase-8-deficient cells by priming for tumor necrosis factor alpha-induced necroptosis. Neoplasia. 2011;13(10):971–9. doi: 10.1593/neo.11610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vanlangenakker N, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18(4):656–65. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cheung HH, et al. Smac Mimetic Compounds Potentiate Interleukin-1{beta}-mediated Cell Death. J Biol Chem. 2010;285(52):40612–23. doi: 10.1074/jbc.M110.183616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lecis D, et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 2013;4:e920. doi: 10.1038/cddis.2013.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dineen SP, et al. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 2010;70(7):2852–61. doi: 10.1158/0008-5472.CAN-09-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Probst BL, et al. Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-alpha-dependent manner. Cell Death Differ. 2010;17 (10):1645–54. doi: 10.1038/cdd.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krepler C, et al. The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin Cancer Res. 2013;19(7):1784–94. doi: 10.1158/1078-0432.CCR-12-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Petersen SL, et al. Overcoming cancer cell resistance to Smac mimetic induced apoptosis by modulating cIAP-2 expression. Proc Natl Acad Sci U S A. 2010;107(26):11936–41. doi: 10.1073/pnas.1005667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bai L, et al. LRIG1 modulates cancer cell sensitivity to Smac mimetics by regulating TNFalpha expression and receptor tyrosine kinase signaling. Cancer Res. 2012;72 (5):1229–38. doi: 10.1158/0008-5472.CAN-11-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ziegler DS, et al. Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J Clin Invest. 2008;118(9):3109–22. doi: 10.1172/JCI34120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weisberg E, et al. Potentiation of antileukemic therapies by Smac mimetic, LBW242: effects on mutant FLT3-expressing cells. Mol Cancer Ther. 2007;6(7):1951–61. doi: 10.1158/1535-7163.MCT-06-0810. [DOI] [PubMed] [Google Scholar]
- 127.Weisberg E, et al. Smac mimetics: implications for enhancement of targeted therapies in leukemia. Leukemia. 2010;24(12):2100–9. doi: 10.1038/leu.2010.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Foster FM, et al. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 2009;11(3):R41. doi: 10.1186/bcr2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fakler M, et al. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood. 2009;113(8):1710–22. doi: 10.1182/blood-2007-09-114314. [DOI] [PubMed] [Google Scholar]
- 130.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24(49):7381–8. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
- 131.Varfolomeev E, et al. X chromosome-linked inhibitor of apoptosis regulates cell death induction by proapoptotic receptor agonists. J Biol Chem. 2009;284(50):34553–60. doi: 10.1074/jbc.M109.040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abhari BA, et al. RIP1 is required for IAP inhibitor-mediated sensitization for TRAIL-induced apoptosis via a RIP1/FADD/caspase-8 cell death complex. Oncogene. 2013;32(27):3263–73. doi: 10.1038/onc.2012.337. [DOI] [PubMed] [Google Scholar]
- 133.Vogler M, et al. Small molecule XIAP inhibitors enhance TRAIL-induced apoptosis and antitumor activity in preclinical models of pancreatic carcinoma. Cancer Res. 2009;69(6):2425–34. doi: 10.1158/0008-5472.CAN-08-2436. [DOI] [PubMed] [Google Scholar]
- 134.Stadel D, et al. TRAIL-induced apoptosis is preferentially mediated via TRAIL receptor 1 in pancreatic carcinoma cells and profoundly enhanced by XIAP inhibitors. Clin Cancer Res. 2010;16(23):5734–49. doi: 10.1158/1078-0432.CCR-10-0985. [DOI] [PubMed] [Google Scholar]
- 135.Basit F, Humphreys R, Fulda S. RIP1 protein-dependent assembly of a cytosolic cell death complex is required for inhibitor of apoptosis (IAP) inhibitor-mediated sensitization to lexatumumab-induced apoptosis. J Biol Chem. 2012;287(46):38767–77. doi: 10.1074/jbc.M112.398966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Greer RM, et al. SMAC mimetic (JP1201) sensitizes non-small cell lung cancers to multiple chemotherapy agents in an IAP-dependent but TNF-alpha-independent manner. Cancer Res. 2011;71(24):7640–8. doi: 10.1158/0008-5472.CAN-10-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eschenburg G, et al. Smac mimetic LBW242 sensitizes XIAP-overexpressing neuroblastoma cells for TNF-alpha-independent apoptosis. Cancer Res. 2012;72(10):2645–56. doi: 10.1158/0008-5472.CAN-11-4072. [DOI] [PubMed] [Google Scholar]
- 138.Wagner L, et al. Smac mimetic sensitizes glioblastoma cells to Temozolomide-induced apoptosis in a RIP1- and NF-kappaB-dependent manner. Oncogene. 2013;32 (8):988–97. doi: 10.1038/onc.2012.108. [DOI] [PubMed] [Google Scholar]
- 139.Orzaez M, et al. Intrinsic caspase-8 activation mediates sensitization of erlotinib-resistant tumor cells to erlotinib/cell-cycle inhibitors combination treatment. Cell Death Dis. 2012;3:e415. doi: 10.1038/cddis.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bai L, Wang S. Targeting Apoptosis Pathways for New Cancer Therapeutics. Annu Rev Med. 2014;65:20.1–20.17. doi: 10.1146/annurev-med-010713-141310. [DOI] [PubMed] [Google Scholar]
- 141.Tolcher AW, et al. Phase I study of safety and pharmacokinetics (PK) of GDC-0917, an antagonist of inhibitor of apoptosis (IAP) proteins in patients (Pts) with refractory solid tumors or lymphoma. J Clin Oncol. 2013;31(suppl):abstr 2503. [Google Scholar]
- 142.Dienstmann R, et al. A phase Ib study of LCL161, an oral inhibitor of apoptosis (IAP) antagonist, in combination with weekly paclitaxel in patients with advanced solid tumors. Cancer Res. 2012;72(24 Suppl):Abstract nr P6-11-06. [Google Scholar]
- 143.Houghton PJ, et al. Initial testing (stage 1) of LCL161, a SMAC mimetic, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2012;58(4):636–9. doi: 10.1002/pbc.23167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sikic BI, et al. Safety, pharmacokinetics (PK), and pharmacodynamics (PD) of HGS1029, an inhibitor of apoptosis protein (IAP) inhibitor, in patients (Pts) with advanced solid tumors: Results of a phase I study. J Clin Oncol. 2011;29(suppl):abstr 3008. [Google Scholar]
- 145.Erickson RI, et al. Toxicity profile of small-molecule IAP antagonist GDC-0152 is linked to TNF-alpha pharmacology. Toxicol Sci. 2013;131(1):247–58. doi: 10.1093/toxsci/kfs265. [DOI] [PubMed] [Google Scholar]
- 146.Wong H, et al. Dogs are more sensitive to antagonists of inhibitor of apoptosis proteins than rats and humans: a translational toxicokinetic/toxicodynamic analysis. Toxicol Sci. 2012;130(1):205–13. doi: 10.1093/toxsci/kfs235. [DOI] [PubMed] [Google Scholar]
- 147.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11(6):411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Suva LJ, et al. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7(4):208–18. doi: 10.1038/nrendo.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Novack DV, et al. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003;198(5):771–81. doi: 10.1084/jem.20030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang C, et al. Antagonism of inhibitor of apoptosis proteins increases bone metastasis via unexpected osteoclast activation. Cancer Discov. 2013;3(2):212–23. doi: 10.1158/2159-8290.CD-12-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]